ICML 2024 | PLINDER: 蛋白质配体相互作用数据和评估集

PLINDER 是目前最大、注释最全面的蛋白-配体相互作用(PLI)数据集,包含约45万全配体系统,配备丰富的蛋白质、结合口袋、相互作用及配体多层面相似性指标,并支持未结合态和预测结构。该项目创新性地设计了高质量、低信息泄漏的训练-测试集划分方法,保证测试集的多样性和可靠性。基于不同划分方案重新训练DiffDock模型,验证了数据划分对模型泛化性能评估的关键影响。PLINDER资源公开开源,支持社区持续更新与多任务评测,助力蛋白-配体相互作用预测、药物设计与蛋白质工程等领域的发展。

获取详情及资源:
- 论文:https://doi.org/10.1101/2024.07.17.603955
- 代码:https://github.com/plinder-org/plinder
- 网站:https://www.plinder.sh
0 摘要
蛋白–配体相互作用(Protein-ligand interactions, PLI)是小分子药物设计的核心基础。随着计算方法日益逼近实验精度,对一个高质量且覆盖广泛的PLI数据集的需求愈加迫切。现有数据集在规模和多样性上往往受限,且常用评估集存在训练信息泄漏问题,阻碍了对方法泛化能力的客观评估。为此,该研究构建了PLINDER——迄今为止最大且注释最全面的PLI数据集,涵盖449,383个PLI系统,每个系统配备超过500条详细注释,并提供蛋白质、结合口袋、相互作用和配体多个层面的相似性指标,同时支持未结合态(apo)及预测结构。该研究设计了一套高效的训练集与评估集划分策略,最大限度减少任务相关信息泄漏,提升测试集质量。基于此,该研究系统比较了不同划分方式下重新训练DiffDock模型所产生的性能差异,展示了划分策略对模型泛化能力评估的重要影响。
1 引言
蛋白质-配体研究领域近年来在基于深度学习的预测方法应用上取得了显著进展,尤其体现在以下任务中:刚体对接,即预测配体在给定的刚性蛋白质结构中的构象;柔性口袋对接,允许结合位点残基的侧链发生移动;协同折叠,同时预测蛋白质构象和配体姿势;基于口袋条件的配体生成,在特定蛋白结构和结合口袋中生成新型配体分子;基于配体条件的蛋白质工程,反过来设计新的蛋白质序列以选择性结合配体;以及分子支架设计,通过改造配体以增强其对蛋白质或口袋的亲和力。
这些方法有望通过精准预测配体在蛋白质结构中的结合构象,加速药物发现与蛋白质工程的发展。然而,这些方法的有效性极大依赖于所用的数据集,在训练与评估过程中需要解决以下几个关键问题:
- 训练集的多样性,以实现高数据量训练,学习潜在模式而非简单记忆
- 降低训练集与测试集之间的信息泄漏,以准确评估泛化能力,避免因过拟合而夸大性能
- 测试集质量,确保预测结果与可靠的真实数据进行比较,避免因实验质量不稳定或结合位点周围缺失原子而导致的不确定性
- 测试集的多样性,展示模型在不同复杂类型和应用场景下的性能
- 真实推理场景,超越“重新对接”场景,即预测配体在实验中从未出现过的配体结合蛋白构象中的姿势。
尽管当前已有大量公开报道的蛋白质-配体相互作用(PLI)结构数据集,但多数在上述关键方面表现不佳。例如,BioLip2虽为一个大型数据集,重点在于功能注释,但缺乏适合训练和测试机器学习方法的合理划分。PDBBind及其变体虽然提供了推荐的划分,但数据集规模较小,且划分之间存在信息泄漏。虽然已有部分研究尝试解决 PDBBind 的泄漏问题,但这些方法的数据集仍然较小,且未提供在多种泄漏评估指标下的系统性模型重训练评估。
有研究尝试使用蛋白质结构域的进化分类(ECOD)注释,将先前整理的数据集进行有选择地组合(如 DockGen),从而确保测试集包含新颖结构域以评估泛化能力。然而,这种方法仍受限于数据集规模,且受到 ECOD 手动注释的偏倚影响,难以评估已知结构域下新配体及新结合方式的表现。
PLINDER 提供了迄今为止规模最大、种类最丰富的蛋白质-配体复合物数据集,涵盖多配体系统、寡核苷酸、肽类和糖类等多种类型。它从蛋白质、结合口袋、PLI 和配体四个层面计算复合物间的相似性,以评估多样性并检测信息泄漏。此外,PLINDER 还对复合物的质量和结构域信息进行注释,并提出一种方法,用以优先选择多样性高、质量好且泄漏最小的测试集。它还将结合配体(holo)复合物与相关的非结合状态(apo)和预测结构进行关联,支持更贴近实际的推理场景。通过上述措施,PLINDER 致力于为蛋白质-配体领域的研究者提供一个强大、可靠的数据集,用于训练和评估基于深度学习的预测方法,最终推动新型药物发现与蛋白质工程方法的发展。
2 方法
2.1 数据集整理与注释
该研究获取了截至2024年4月9日的PDB的所有条目,使用了PDB NextGen档案资源中的MMCIF文件。对于通过X射线晶体学解析的条目,该研究从相应的X射线验证报告中提取了条目和残基级别的信息。利用OpenStructure生成了每个PDB条目的每个生物单元组装体,并通过蛋白-配体相互作用分析器(PLIP)获取所有类配体链的蛋白-配体相互作用数据。仅考虑蛋白质与配体原子之间,或配体原子与水分子之间的相互作用。
在PDB条目中,若链符合以下任一条件,则被标记为配体链:链类型为非聚合物;该链关联有生物学兴趣分子参考词典标识符;链类型为多肽、寡糖或寡核苷酸且残基数少于10个;或者链类型为多肽且残基数少于20个且无关联UniProt ID。彼此距离在4埃以内或具有可检测相互作用的配体链会被合并为同一蛋白-配体相互作用系统。每个系统同时关联“相互作用”残基(参与配体链相互作用的残基)和“邻近”残基(距离配体链6埃以内的残基),共同构成该系统的结合口袋。
因此,蛋白-配体相互作用系统由PDB ID、生物单元标识符、配体链的具体实例及相互作用蛋白链的具体实例共同定义。每个系统都配备了分类注释,所有结构域注释映射到口袋残基,并保留与口袋重叠度最高的结构域作为口袋结构域。系统被标注为全配体、人工产物或离子,并附有详细的配体定义和属性。该研究对分子结构进行了全面处理和清理,修正了键、价态和手性等问题,确保绝大多数数据能够被深度学习模型常用的预处理和特征提取流程顺利处理,对无法处理的部分进行了明确标注。
2.2 蛋白-配体系统相似性
为了对该研究整理的数据集进行聚类和划分,采用MMSeqs和Foldseek计算蛋白质和结合口袋的相似性,结合唯一的PLIP相互作用对蛋白-配体相互作用(PLI)进行相似性评估,并使用ECFP4指纹计算配体相似性。对所有系统中的PDB链进行MMSeqs和Foldseek搜索(E值<0.01,最低序列等价性为0.2,最多返回5000条序列),获得比对结果和查询覆盖率。蛋白质相似性基于lDDT评分(仅限Foldseek)、序列等价性百分比、序列相似性及经过查询覆盖率调整的全局评分计算。结合逐残基的比对信息,进一步计算结合口袋级别的相似性得分。PLIP相互作用通过相互作用类型及其特定属性去重,并结合结合口袋信息,使用加权的Jaccard相似性指标计算蛋白-配体相互作用层面的相似性。具体细节见附录B。
针对每种相似性指标及设定的阈值(50、70、95、100),构建系统节点图,图中若两个系统的相似性得分超过阈值,则在它们之间建立一条边。所有相似性值均被调整到0-100的范围内。利用NetworKit识别图中的强连通分量、弱连通分量及基于Parallel Louvain方法的社区结构,并为每个聚类中的系统分配相应的聚类标识,为系统添加基于相似性的附加注释。
2.3 训练-验证-测试集划分
该研究使用算法1将数据集划分为原始训练集和测试集,以确保满足训练集规模、测试集质量、测试集多样性以及低信息泄露的要求。需要注意的是,不同预测任务对这些要求的侧重点有所不同。例如,对于刚性对接方法,如果训练集和测试集中存在相似蛋白,但结合口袋的位置、构象或配体相互作用足够不同,这通常不会被视为信息泄露,这样可以模拟将不同配体对接到已知药物靶点的常见场景。然而,对于共折叠任务,这类训练-测试对可能会高估模型表现,因为预测的蛋白结构在常用准确度指标中的贡献可能超过配体构象。同样地,对于基于结合口袋条件的配体生成任务,配体结构和口袋相互作用的相似性对信息泄露的影响通常比受体序列相似性更大。

图A1展示了利用PLINDER相似性注释,测试集和验证集可以进一步细分为特定任务的子集,使得该资源适合对基于新蛋白质、配体、结合口袋及蛋白-配体相互作用的多种任务方法进行严格评估,同时也包含了它们的交集,形成通用的测试集。
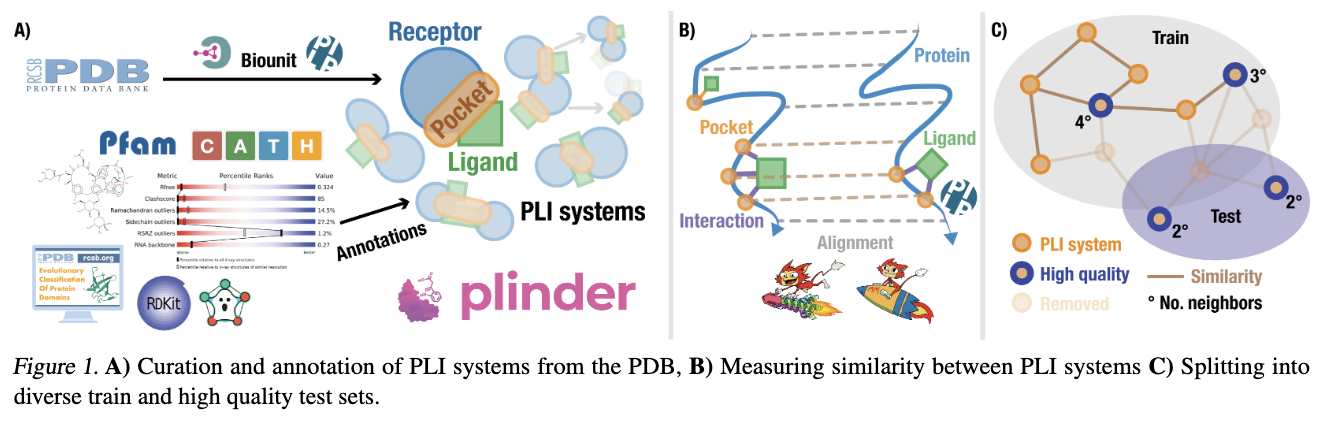
该研究的划分算法由一组图G配置,每个图基于特定相似性指标、阈值以及邻居深度D定义,邻居深度D确定两个系统之间最短路径的最大长度,超过该长度则视为非泄漏。函数pass_quality()判断系统的实验质量是否足够高(见表A2),以决定其是否可归入测试集。最小邻居数m用于避免测试集中出现孤立或连接稀疏、可能不现实的系统;最大邻居数M限制一个测试系统从原始训练集中剔除的系统数量,以保持训练集规模的合理性。测试集冗余性通过限制每个聚类成分中最多保留n个代表系统来控制。
除了泄漏数量外,该研究还优先选择属于同系匹配分子系列(MMS,详见附录A.3)中的测试代表系统,以及关联的apo结构和预测结构数量。对于属于同系MMS的测试系统,满足质量标准的整个系列成员都会被纳入测试集,同时对应的泄漏系统会从原始训练集中移除。获得的原始训练集进一步按照指定指标和阈值基于聚类成分进行90%训练、10%验证的随机划分(本文所用划分基于结合口袋≥50的弱连通分量)。该算法在所有PLINDER全配体系统上应用了表A10中列出的四种配置。该研究选择了最适合盲对接的配置1,重新训练DiffDock模型,以研究信息泄露对模型性能的影响。
2.3 泄漏系统比例
对于给定的训练-测试划分、相似性指标及阈值,该研究统计测试集中至少与训练集存在一条相似性不低于阈值的连接(即边)的系统比例(同样方法也用于验证集与测试集及训练集与PoseBusters的比较)。由于DiffDock专注于单配体对接,该研究聚焦于仅含单一配体的全配体PLI系统,并针对每个唯一的PDB ID与CCD代码组合选取一个系统,得到包含106,745个系统和35,255个唯一配体SMILES的非冗余数据集,命名为PLINDER-NR,用于DiffDock训练。针对PLINDER-NR应用划分配置1(使用结合口袋lddt > 50的图G)得到的划分称为PLINDER-PL50。此外,该研究还基于时间(PLINDER-TIME)和ECOD拓扑结构(PLINDER-ECOD)对PLINDER-NR进行了划分,以便与PLINDER-PL50进行对比(详见附录D)。为评估DiffDock在PoseBusters数据集上的表现,PoseBusters中的PDB ID在三种划分的训练、验证和测试集中均被剔除。DiffDock模型按照附录E所述在这三种划分上重新训练。
3 结果
3.1 数据集数量
截至本文撰写时,PLINDER包含1,344,214个蛋白-配体相互作用(PLI)系统,来源于162,978个PDB条目,其中449,383个为全配体(holo)系统,剩余包括573,169个常见实验伪影系统、318,060个离子系统,以及3,602个包含超过五条蛋白链和/或配体链的系统(系统分类详见附录A.1)。在全配体系统中,26%的系统含有多个配体,25%的系统含有多个相互作用的蛋白链,且通过X射线衍射法确定的系统中有34%符合附表A2中的高质量X射线标准。由于数据整理流程覆盖整个PDB,该研究同时标识出564,240条PDB链为未结合态(apo),因此PLINDER还自动生成了一个无检测到配体相互作用(除伪影或离子外)的apo链数据集。
表1展示了PLINDER与常用的PDBBind和DockGen数据集的比较。PLINDER中的每个系统均包含超过500条详细注释(类别见附录A)。在449,383个全配体系统中,共615,932个配体覆盖了46,988个独特的CCD代码,其中233,760个(37%)符合Lipinski的五项规则(Ro5),146,444个(23%)含有共价键连接,122,741个(19%)为辅因子,105,836个(17%)为寡糖、寡核苷酸或寡肽,55,987个(9%)为片段。另有15,383个系统属于2,117个同系匹配分子系列(MMS),每个系列至少包含三个具有共同核心结构的配体。

3.2 数据集划分
如表2所示,PLINDER-PL50划分在训练集与测试集之间表现出最低的信息泄漏水平,相较于PLINDER-TIME和PLINDER-ECOD更为优越。用于划分的结合口袋lddt图在阈值超过50%时,成功消除了训练集与测试集间的连接,同时有效剔除了基于相互作用、结合口袋位置和蛋白序列相似性的多数连接。尽管两集中存在相似配体,但它们结合于不同的蛋白结合口袋,这为刚性对接任务和评估方法提供了合理的测试案例。

PLINDER-ECOD划分尽管比PLINDER-TIME的泄漏更少,但暴露出结构域注释不完整的问题。由于选择了无ECOD注释的系统作为测试集,这些系统中许多实际上与训练集存在相同的结构域和结合口袋。类似的DockGen划分显示,细致且完整的ECOD结构域分配确实能减少泄漏,但这限制了可分配数据量,并且依赖人工整理。
PLINDER庞大的数据规模保证了无论采用何种划分策略,数据集都具备高度多样性。PLINDER-PL50测试集中所有系统均满足设计的高质量标准,而PLINDER-ECOD和PLINDER-TIME仅分别有21%和19%的系统符合高质量标准,确保了PLINDER-PL50的真实可靠性。
表2还比较了不同划分下训练集、验证集和测试集的系统数量,展示了该研究创建PLINDER-PL50划分的能力:该测试集拥有比常用PoseBusters测试集多十倍的复合物数量,同时与对应训练集之间的信息泄漏依然保持在低水平。
3.3 DiffDock在不同数据集划分上的表现
该研究按照附录E的说明,使用NVIDIA BioNeMo在PLINDER-PL50、PLINDER-ECOD和PLINDER-TIME三个划分上重新训练了DiffDock模型,并在对应的测试集以及标准化的PoseBusters基准集上评估其性能。所有评估均使用单个A100 GPU进行推理。推理流程包括在蛋白质上生成配体构象,尝试从真实测试集中识别蛋白结合口袋及配体构象。生成的构象由预训练的置信度模型排序(如Corso等人2023年所述),并计算每个构象的RMSD值。
由于该研究训练了新的评分模型但使用了已有的置信度模型,Top-1构象的选择可能存在偏差。因此,该研究生成10个构象,并报告Top-1和Top-10中RMSD小于2埃的成功率。Top-1构象基于未优化的置信度模型选择,而Top-10构象则考虑所有生成构象的整体可能性,从而减少置信度模型带来的影响。这种差异强调了优化置信度模型对于准确报告结果的重要性。
图2和附录表A12展示了基于PLINDER-TIME、PLINDER-ECOD和PLINDER-PL50训练集重新训练的DiffDock模型的准确率表现。在PoseBusters基准集上,DiffDock基于PDBBind-DiffDock划分训练的Top-1基线性能为38%。仅通过增加训练数据量(未调整架构或处理泄漏问题),Top-1和Top-10的准确率分别提升至47.8%和58.2%,凸显了训练集规模和多样性对深度学习模型准确度的关键影响。然而,采用更严格划分策略(如PLINDER-PL50和PLINDER-ECOD),测试集与训练集之间信息泄漏减少,模型准确率相应下降至15%-18%(Top-10为21%-26%)。
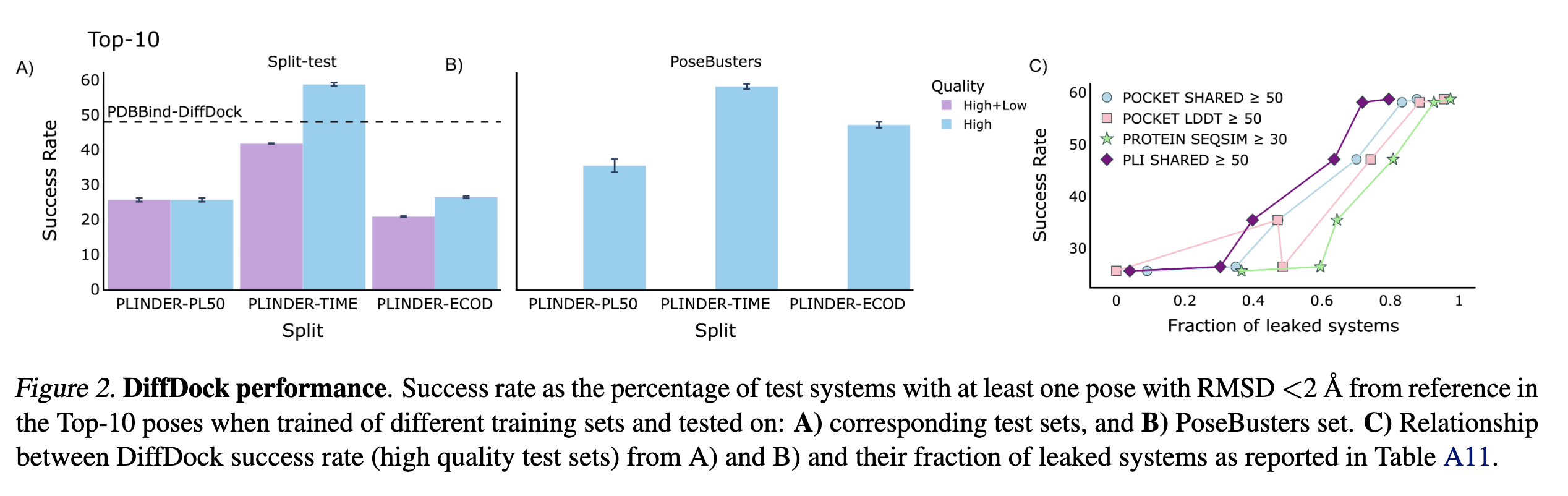
该研究推测测试集质量对性能测量影响显著。正如图2A和附录表A12所示,当向测试集中加入低质量系统时,DiffDock在PLINDER-TIME和PLINDER-ECOD上的表现分别从45.2%降至29.2%,以及从19%降至14.7%。图2C显示,不同相似性指标对应的测试集泄漏比例与DiffDock性能呈线性关系:大多数指标下,泄漏越高,模型在高质量测试集上的表观准确率(Top-10构象成功率)越高。该研究在附录F中对此关系进行了深入探讨。这些定量证据验证了长期以来关于不同相似性指标在减少信息泄漏中重要性的直觉。
4 数据集和代码的可用性及更新
该研究的自动化整理流程支持可重复的周期性更新。除了提供系统注释外,该研究还为每个系统增量更新蛋白质相似性和配体相似性数据集,支持对比查询,并基于新相似性数据直接分配聚类。每半年将新系统整合进现有相似性数据集,重新进行聚类,并发布新版本。数据集以CC-BY 4.0许可证公开发布,托管于Google Cloud Storage。数据集的架构会随着更新不断调整,草案文档也可供参考。
PLINDER源码采用Apache 2.0许可证,用户可从 https://github.com/plinder-org/plinder 下载。附录C介绍了支持快速高效数据整理和图查询的工程设计与软件选型。除数据整理外,该研究还提供评估蛋白-配体复合物预测的软件,支持任意大小复合物,并提供多种配体(RMSD、lDDT-PLI、PoseBusters)、结合口袋(lDDT-LP)及蛋白质(lDDT、多聚体评分)层面的准确性指标。未来,该研究计划发布支持高效数据加载和多样性采样的软件工具,并提供含分层验证集和测试集的PLINDER-2024数据划分版本,以方便机器学习社区广泛使用。
5 当前限制与未来方向
随着全球科研持续产出新实验数据,本工作也在不断推进,力求为蛋白-配体相互作用预测领域提供最全面的资源。该研究正致力于增加更多注释,如经典对接评分、交叉对接评分、测量和预测的结合亲和力、隐蔽结合口袋及多配体标签,以便优先级排序和分层测试集,同时支持辅助任务标签。该研究探索利用无配体实验结构和预测方法进行数据增强,以提升覆盖的相互作用、结合口袋及蛋白折叠多样性,目前已有的相互作用和泄漏检测算法可直接应用于这些扩展数据。
当前版本每个系统仅关联一个AlphaFold预测模型,未来将利用UniProt中的冗余信息,链接多个AlphaFold数据库中的预测结构。鉴于PLIP在某些情况下未能识别高度相似的近乎相同结合口袋,该研究也将尝试其他蛋白-配体相互作用分析工具以优化PLI系统整理。当前对高质量测试系统的筛选偏好较小分子,可能导致仅有低质量结构的蛋白质或配体类别代表性不足,未来计划探索基于原子级权重的准确性指标。
此外,目前仅利用X射线结构的质量信息,电子显微镜结构的验证报告已包含逐残基Q分数,未来可将其纳入质量评判标准。为公平评估方法进展,该研究计划建立PLINDER数据集上的排行榜,涵盖基于完整PLINDER划分训练和测试的主流模型,包括本研究DiffDock训练子集之外的多样性采样、冗余及增强系统。该研究还将覆盖更贴近实际的评估场景,如交叉对接或在apo及预测受体结构中预测构象,结合口袋将按构象变化程度进行分层。
6 结论
该研究推出了PLINDER,一个规模庞大、内容全面且自动化的蛋白-配体相互作用数据集资源。该研究展示了基于蛋白-配体复合物的可扩展相似性度量方法及优先保证测试集质量与低信息泄漏的划分算法的价值。通过在不同划分数据集上重新训练DiffDock模型,该研究证明了所提出的方法和结果不仅为数据集构建提供了坚实基础,也实现了对数据质量和信息泄漏的定量评估与有效控制。
7 参考
[1] Durairaj, Janani, Yusuf Adeshina, Zhonglin Cao, et al. “PLINDER: The Protein-Ligand Interactions Dataset and Evaluation Resource.” bioRxiv, ahead of print, July 19, 2024. https://doi.org/10.1101/2024.07.17.603955.