ICLR 2024 | ProteinWorkshop: 评估蛋白质结构宇宙中的表示学习方法

获取详情及资源:
0 摘要
该研究介绍了ProteinWorkshop,这是一个面向蛋白质结构表示学习的综合性基准套件,重点关注几何图神经网络的应用。该基准覆盖大规模预训练以及多种下游任务,同时结合实验解析结构与预测结构,用于系统性评估所学习到的结构表示质量,以及其在捕获功能相关性并服务下游任务方面的有效性。结果表明,基于AlphaFold结构并结合辅助任务的大规模预训练,能够稳定提升旋转不变与等变图神经网络的性能,且表达能力更强的等变模型从预训练中获得的收益更为显著。该工作旨在为机器学习与计算生物学社区建立统一的评测基础,以促进蛋白质结构表示学习方法的严格比较与持续发展。其开源代码库通过提供高效的结构化数据加载器,支持包括AlphaFoldDB与ESM Atlas在内的大规模蛋白质结构数据库,并提供从整个PDB构建新任务的工具,从而显著降低了处理大规模蛋白质结构数据集的使用门槛。
1 引言
现代蛋白质结构预测方法的发展,极大推动了结构数据可用性的爆发式增长。尽管大量基于序列的功能注释可以直接映射到三维结构上,但结构数量的迅速扩张也导致了结构数据与有意义的结构注释之间的差距不断扩大。近年来的研究尝试从海量结构数据中提取生物学信息,一类方法通过识别具有代表性的结构来承载功能注释,另一类方法则通过将结构表示为更为简洁紧凑的形式,例如结构字母表或图嵌入,以降低大规模分析的计算成本。这些工作显著提升了结构数据在规模化处理和分析中的可行性,但如何借助深度学习方法进一步揭示蛋白质序列、结构与功能之间的内在联系,仍有待系统性的验证。
在蛋白质结构建模领域,多种深度学习方法已被提出,其中几何图神经网络逐渐成为学习生物大分子结构表示的主流架构。这类方法通常可以从两个维度进行划分,一是输入结构的特征化方式及其结构粒度,例如仅使用Cα原子、主链或全原子表示,二是是否显式引入物理对称性和归纳偏置,即采用不变表示或等变表示。尽管相关方法不断涌现,目前仍缺乏一个稳健且标准化的评测体系,以更细致且更贴近下游应用的方式跟踪不同方法的发展进展。
针对上述问题,该工作构建了一个统一且严格的评估框架,用于系统评测蛋白质结构编码器。所提供的预训练语料覆盖了已知的结构折叠空间,评测任务则从不同结构粒度层面考察模型学习信息性表示的能力。以往研究多集中于学习整体层面的结构表示,并主要通过功能分类或折叠分类任务进行评估,而对局部层面,即节点级结构表示的研究相对较少。然而,高质量的节点级表示对于多种结构注释任务至关重要,例如结合位点或相互作用位点预测,同时也是结构条件分子设计方法中的重要条件信号。深入理解这一精细尺度上的结构-功能关系,有助于揭示支撑特定功能的结构模体,从而推动蛋白质设计领域的发展。
具体而言,该工作系统整理了大量基于结构的预训练与微调数据集,重点关注能够为预测结构提供结构注释的任务,并构建了一个高度模块化的基准体系,便于研究者在不同任务、模型与预训练设置下快速评估蛋白质表示学习方法。同时,研究在多种结构粒度层面上系统评测了几何图神经网络,涵盖从通用模型到蛋白质特异性架构的多个类别,并首次将高阶等变图神经网络引入蛋白质结构表示学习的系统评估中。此外,该工作在目前已知范围内,使用了包含227万条非冗余结构的最大规模AlphaFoldDB蛋白质结构语料进行预训练与评测。**实验结果表明,基于序列与结构去噪的辅助任务以及结构去噪预训练,能够稳定提升几何图神经网络的性能。**值得注意的是,在引入结构特征后,基于序列预训练的ESM-2-650M模型在家族与超家族折叠预测以及基因本体预测任务上的表现,可与当前最先进的图神经网络方法相媲美。
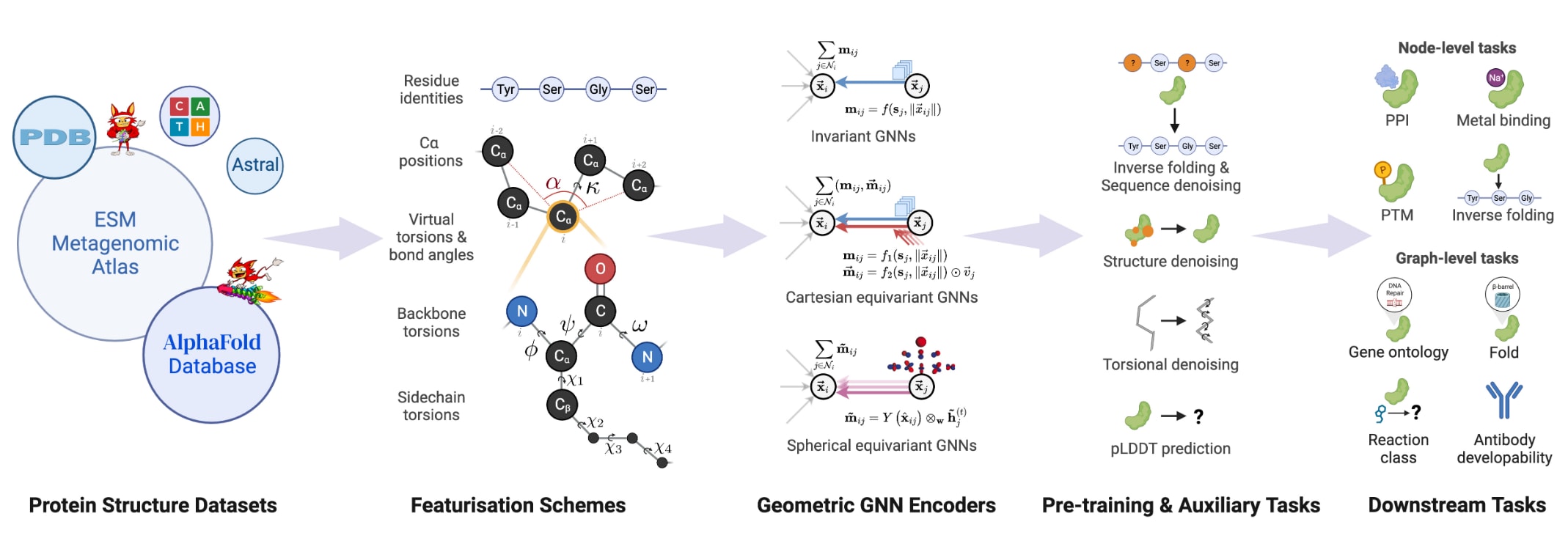
图1:ProteinWorkshop总体示意图。 该框架是一个综合性的基准套件,用于在大规模蛋白质结构数据上系统评估几何图神经网络的预训练策略及其结构表示学习能力。
2 ProteinWorkshop
ProteinWorkshop的总体目标在于尽可能全面地覆盖蛋白质结构表示学习方法的设计空间。为实现这一目标,该基准在设计上具有高度模块化特性,能够在广泛的有监督与无监督任务设置下,灵活评估不同结构编码器、蛋白质特征化方案以及辅助任务的组合。完整的用户手册见附录D,其中对各个组成模块给出了详细的列表与说明。
2.1 特征化方案
蛋白质结构通常被表示为几何图结构。由于全原子表示会引入大量节点,计算代价极高,研究中往往采用较为粗粒度的Cα原子图表示。然而,这种表示方式不可避免地会造成信息损失,例如主链的空间取向以及侧链结构等细节仅被隐式编码。鉴于全原子节点表示在计算上的高昂成本,该基准主要聚焦于基于Cα的图表示,并通过不同的特征化策略引入更高层次的结构信息。同时,基准也提供了相应工具,支持用户使用主链或全原子图进行实验。不同特征化方案的具体细节见附录D.4及表5。
2.2 预训练任务
该基准集成了一套较为全面的预训练任务,整体上可分为掩码属性预测、基于去噪的方法以及基于对比学习的方法。这些任务既可作为独立的预训练目标,也可作为下游有监督任务中的辅助任务使用。
序列去噪方面,基准提供了两种基于序列扰动过程的设置。给定一个氨基酸序列,通过在其中一定比例的位置引入扰动生成受损序列。第一种方式是将部分残基随机替换为其他氨基酸,模型需要恢复原始未受损序列。第二种方式为掩码残基预测,即将部分残基替换为掩码标记,并要求模型预测对应的真实残基。
结构去噪任务包括坐标去噪和扭转角去噪两类。在坐标去噪任务中,从正态或均匀分布中采样噪声,并按噪声因子进行缩放后施加到原子坐标上,从而破坏包括主链和侧链扭转角在内的结构特征。模型需要预测每个节点对应的噪声,或直接恢复原始坐标。在扭转角去噪任务中,噪声被施加到主链扭转角上,随后利用pNeRF方法并结合未受损的键长和键角重新计算笛卡尔坐标,模型同样需要预测残基层面的角度噪声或原始二面角。
序列-结构协同去噪是一种多任务形式,同时对序列和结构进行去噪,并为不同模态分别设置输出头。
在掩码属性预测方面,该基准将逆折叠任务作为预训练目标之一,并引入了距离、角度和二面角的掩码属性预测任务,以及主链二面角预测任务。
此外,还引入了pLDDT预测任务。结构预测模型通常为每个残基提供pLDDT置信度评分,该指标与无序区域具有较好的相关性。在此,该任务被表述为节点级回归问题,要求模型在预测结构上输出归一化后的残基级pLDDT值。
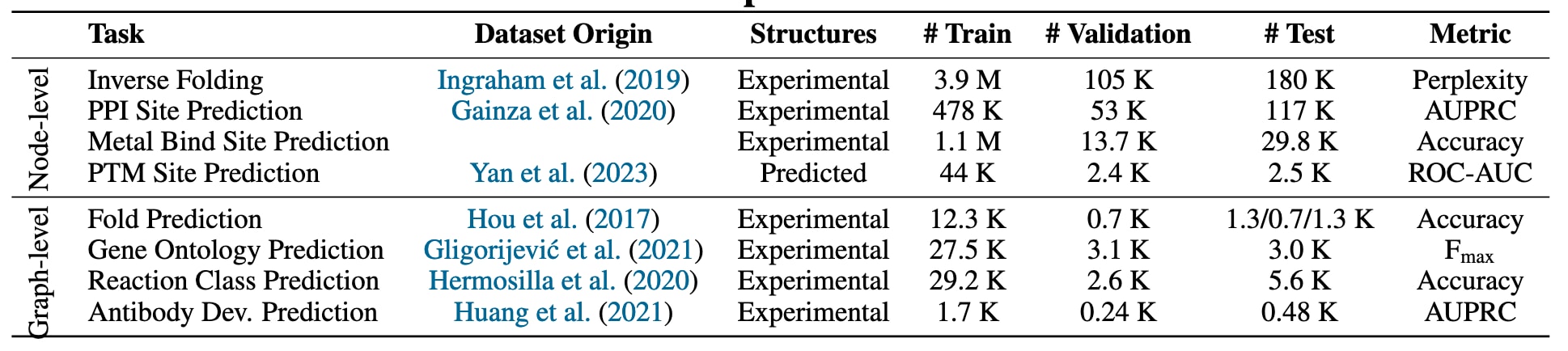
表1|有监督任务及其对应数据集的总体概览。
2.3 下游任务
该工作从已有文献和基准中整理了多种基于结构和基于序列的数据集,其概要列于表1。这些任务不仅用于评估模型在整体结构层面的表示能力,同时也用于考察模型学习信息性局部表示的能力,以支持残基层面的预测与注释任务。
在条件允许且考虑结构过时性的前提下,原始结构数据尽量直接从PDB或其他结构数据库中获取。这样做的原因在于,社区中部分已处理的数据集仅保留了Cα原子坐标而丢弃了完整原子信息,不适合开展更深入的实验分析。通过保留完整结构,用户可以根据需要自行设计预处理流程,例如去质子化或修复实验结构中常见的缺失区域等操作。
2.3.1 节点级任务
逆折叠任务是蛋白质从头设计流程中的关键组成部分。许多蛋白质设计生成模型通常只输出主链结构,随后需要为其设计对应的氨基酸序列。因此,逆折叠被定义为一个节点级分类任务,模型需要为每个残基预测其对应的氨基酸类型,标签空间大小等于氨基酸词表规模,在标准设置下为20种天然氨基酸。该任务具有较强的通用性,可应用于基准中的任意数据集,并且在相关研究中通常基于CATH数据集进行评估。
蛋白质-蛋白质相互作用位点预测在构建精细化相互作用网络和对接工具中具有重要意义,同时也为蛋白质工程与药物发现中的靶点识别提供生物学背景支持。该任务被表述为节点级二分类问题,目标是判断某一残基是否参与蛋白质-蛋白质相互作用界面。这里采用的是从PDB中整理得到的实验结构数据集,并沿用其原始的数据划分方式,但在标注策略上进行了调整,将界面定义为基于原子间距离小于3.5Å的接触,而非溶剂排除标准,且该阈值允许用户自行设定。该数据集通过一系列预处理步骤构建,包括筛选包含特定配体的结构、基于30%序列一致性的聚类以及随机下采样,最终包含1459个结构样本,并按72%、8%和20%的比例划分为训练集、验证集和测试集。此外,从结构中提取半径为12Å的局部片段,当片段中心点与对应配体原子的距离小于3Å时,该片段被标注为结合口袋的一部分。
金属结合位点预测关注蛋白质中与过渡金属离子发生配位的残基,这类相互作用在蛋白质功能中具有重要作用。该任务同样被定义为节点级二分类问题,每个残基被标注为是否位于用户指定金属离子或配体杂原子3.5Å范围内。基准提供了从PDB中自动构建该类数据集的工具,在运行时动态计算残基级的结合位点标注。尽管该任务支持在PDB的任意子集及不同配体上运行,基准中默认提供了一个锌结合位点数据集。该数据集通过对PDB进行30%序列一致性聚类以去除冗余后构建,筛选条件包括蛋白质长度小于3000个残基、结构中至少包含一个锌原子、分辨率优于2.5Å、并且不含核酸分子。若多个结构同时满足条件,则选用分辨率最高者。最终得到的训练集、验证集和测试集分别包含2085、26和59个样本,并保证验证集和测试集中的蛋白质与训练集之间不存在任何部分重叠。
翻译后修饰位点预测对于理解蛋白质调控机制以及设计靶向治疗策略具有重要意义。该任务被建模为多标签分类问题,每个残基对应一个取值范围为1到13的标签,用于区分不同类型氨基酸上的修饰形式,例如发生在丝氨酸、苏氨酸或酪氨酸上的磷酸化修饰,以及发生在天冬酰胺上的N-连接糖基化。该基准采用了一个包含48811条AlphaFold2预测结构的数据集,其中每条结构均包含构建残基级修饰位点标签所需的翻译后修饰元数据。数据集依据50%序列一致性和80%覆盖率进行划分,最终得到训练集43907个样本、验证集2393个样本以及测试集2511个样本。
2.3.2 图级任务
折叠类型预测任务主要用于检验模型区分不同蛋白质结构折叠的能力,可视为衡量结构表示质量的一项基础测试。直观而言,若模型在折叠分类任务上的表现较差,往往意味着其学习到的结构表示较为有限或质量不足。该任务被定义为多分类图级分类问题,每个蛋白质结构对应一个折叠类别标签,类别总数为1195。所采用的数据集最初基于SCOP 1.75构建,并根据拓扑相似性划分为三种测试设置:在Fold设置中,训练阶段不包含来自同一超家族的蛋白质;在Superfamily设置中,训练集中不包含来自同一家族的蛋白质;在Family设置中,同一家族的蛋白质可以同时出现在训练集中。
基因本体预测任务旨在以GO术语的形式对蛋白质功能进行注释,在蛋白质分析与工程中具有重要应用价值,例如用于聚类功能相关的结构,或引导蛋白质生成方法设计具备特定功能的新蛋白质。该任务被建模为多标签分类问题,需要为每个结构预测其对应的GO注释。GO注释涵盖三大本体分支,包括生物过程、细胞组分和分子功能。该基准采用了从PDB中整理得到的实验结构数据集,并保留了基于多重序列相似性阈值的原始划分方式,其中主要使用30%序列相似性作为划分标准。
反应类型预测任务依据酶所催化反应的酶学委员会编号进行分类,该编号包含四个层级。能够预测反应类型的模型有助于阐明新设计蛋白质的潜在功能。该任务同样属于多分类图级分类问题,每个蛋白质被映射到384种反应类别之一,标签由EC编号的四个层级共同确定。所使用的数据集最初从PDB中构建,并基于50%的序列相似性阈值进行数据划分。
抗体可开发性预测关注治疗性抗体在实际研发过程中是否具备良好的理化性质,这类性质与其靶点结合亲和力和特异性同样关键。该任务被表述为二分类图级分类问题,用于判断给定抗体是否具备开发潜力。数据集来源于SabDab数据库,包含2426个同时具有序列和PDB结构信息的抗体样本,每个样本均包含重链和轻链,且结构分辨率优于3Å。标签依据抗体可开发性指数进行二值化得到,该指数综合考虑了抗体的疏水性与静电相互作用。从基准评测的角度看,该任务具有一定特殊性,因为它能够在特定免疫球蛋白折叠类型上考察模型性能,从而评估通用结构编码器在折叠特异性任务中的适用性。
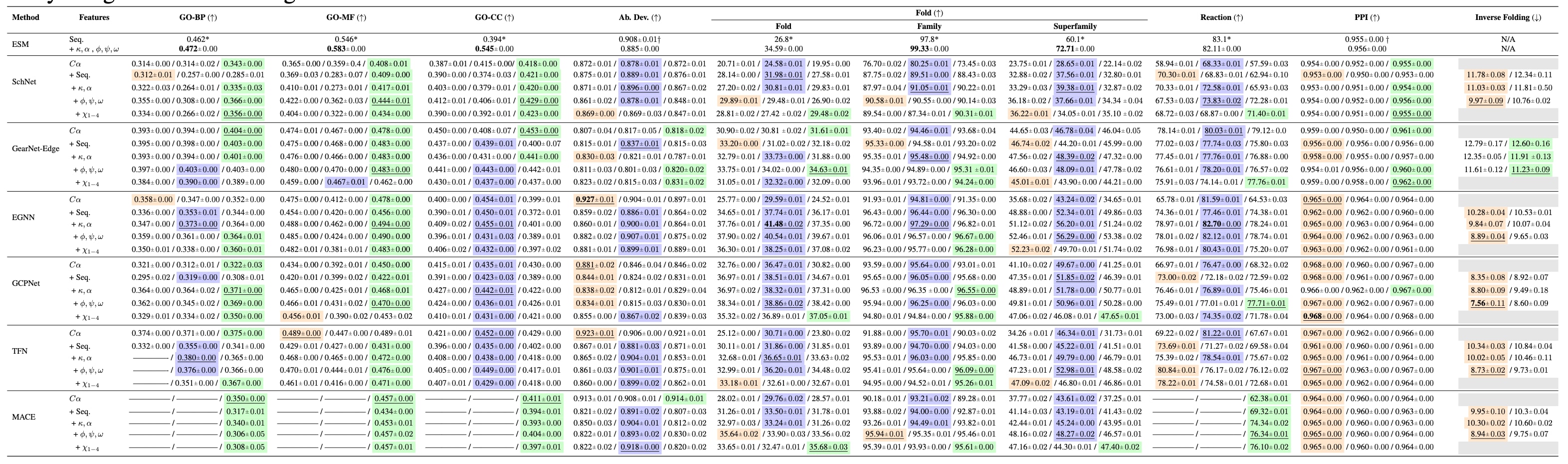
表2|未进行预训练条件下的基线评测结果。 每种模型与特征化方案组合的结果按以下顺序给出:不使用辅助任务 / 加入序列去噪 / 加入结构去噪。彩色标注表示在给定模型与特征化方案下表现最优的辅助任务,带下划线的结果表示该模型对应的最佳特征化方案,加粗结果表示在该任务上整体性能最优的模型。灰色单元格表示无效的任务设置组合,例如将逆折叠任务与序列去噪作为辅助任务同时使用,而“——”表示对应实验未能收敛。[*]表示来自已有工作的结果,即不包含结构特征的ESM-MSA-1b模型,[†]表示在新任务上评测的ESM-2-650M结果。主要结论包括以下几点:第一,相较于不使用辅助任务的情况,引入辅助任务在不同模型和主任务上均能稳定提升性能。第二,总体而言,等变图神经网络优于不变模型,但蛋白质特异性架构在各类任务中始终保持较强性能。第三,基于Cα原子、虚拟角度和主链扭转角的特征化方案整体表现最佳。第四,在引入结构特征后,ESM-2-650M在家族与超家族折叠分类以及基因本体预测任务上的表现相较于图神经网络具有较强竞争力。为便于阅读和分析,附录A中进一步提供了一系列刻画各任务结果的分析图。
2.4 预训练数据集
该基准包含多个规模较大的实验结构与预测结构数据集,可用于模型预训练或推理阶段。同时,基准提供了直接从PDB中配置有监督任务与数据划分的工具,并针对大规模预测结构语料构建了存储高效的数据加载器,支持AlphaFoldDB和ESM Atlas等资源。这些设计显著降低了使用大规模结构数据集开展研究的门槛。
2.4.1 实验结构
PDB部分提供了直接从蛋白质数据银行构建数据集的工具。除整体使用全部结构外,用户还可以基于结构相似性、序列相似性或时间策略对数据进行筛选和划分。支持的过滤条件包括蛋白质长度、链数、分辨率、提交日期、特定配体的存在与否以及结构解析方法等。
CATH数据集则来源于CATH 4.2中40%序列一致性的非冗余链集合,规模相对较小,作为补充性的预训练数据集使用。
ASTRAL数据集提供蛋白质结构域级别的结构信息。结构域通常能够在脱离整体蛋白质的情况下保持其结构和功能完整性,往往承担高度特异的功能,可视为蛋白质结构的基本构建单元。
2.4.2 预测结构
AlphaFoldDB代表性结构数据集包含约227万条代表性结构,这些结构通过FoldSeek对AlphaFold数据库中约2.14亿条预测结构进行大规模结构相似性聚类后获得。此外,基准还提供了其中一部分未被注释的代表性结构子集,即所谓的暗蛋白组,约占全部代表性结构的31%。
ESM Atlas与ESM High Quality数据集则是由ESMFold生成的预测结构压缩集合。ESM Atlas包含MGnify 2023版本中全部7.72亿条预测结构,而ESM High Quality则是在此基础上筛选得到的高置信度结构子集,其筛选标准基于平均pLDDT评分。
3 方法与实验设置
概述 为了展示该基准的实用性,研究系统考察了蛋白质结构表示方式、模型架构选择以及预训练或辅助任务的不同组合,并分析它们在多种任务上的预测性能表现。所选任务聚焦于具有实际应用价值的结构注释问题,以便从整体和局部两个层面评估模型的表示能力。为此,研究选取了一系列先进的蛋白质结构编码器以及通用的几何图神经网络架构,这些模型在消息传递阶数和张量阶数等设计维度上覆盖了几何GNN的主要设计空间。同时,还评估了多种结构表示形式,这些表示在不同程度上捕获了蛋白质结构的完整细节。
模型架构 该基准提供了多种旋转不变与旋转等变模型的统一实现。评测对象包括四种通用模型,分别为SchNet、EGNN、TFN和MACE,以及两种蛋白质特异性架构GCPNet和GearNet。此外,还将几何图神经网络与结合结构特征的序列预训练语言模型ESM进行对比。这里选择参数规模为650M的ESM-2模型,是因为在这一规模下,ESM已表现出显著的结构相关能力。
特征化方案 研究共考虑了五种特征化方案,通过逐步引入更多结构信息来增强模型输入,包括序列位置信息、基于Cα轨迹的虚拟二面角和键角、主链扭转角以及侧链扭转角等。各类特征化方案的具体定义见附录表5。
预训练数据集 所有预训练实验均基于AlphaFoldDB进行。该数据集包含227万条非冗余蛋白质结构,在保持可操作规模的同时,其结构多样性在现有基于结构的预训练语料中处于领先水平。原则上,在AlphaFoldDB上完成预训练的模型,由于已经接触过相同或相似的蛋白质折叠类型,应当能够很好地泛化到当前已知及预测得到的天然蛋白质结构空间。为支持对AlphaFoldDB和ESM Atlas等大规模数据集的高效处理,研究基于FoldComp开发了存储高效的数据加载器,相关细节见附录D.6。
预训练与辅助任务 在实验中,研究重点关注基于去噪的预训练与辅助任务,因为相较于对比学习或掩码属性预测任务,这类方法的系统研究仍相对较少。共考察了五种预训练任务,包括结构去噪、序列去噪、扭转角去噪、逆折叠以及pLDDT预测。其中,结构去噪和序列去噪同时作为下游实验中的辅助任务使用。此外,还评估了逆折叠预训练任务,并在CATH数据集上进行微调,用于下游逆折叠任务的性能评测。
加噪策略 在结构去噪任务中,从高斯分布中独立采样噪声并按σ=0.1进行缩放,用于扰动输入的坐标或二面角。几何标量和向量特征均基于加噪后的结构计算得到。对于序列去噪,采用突变策略,在每条蛋白质序列中随机扰动25%的残基。当序列去噪作为辅助任务时,其损失项权重设为λ=0.1。
训练设置 为了公平比较大规模数据集与模型,实验中尽量统一采用六层网络结构,每层包含512个隐藏通道。对于等变GNN模型,为适配单张80GB显存的NVIDIA A100 GPU,适当减少了网络层数和通道数。在下游任务中,最大训练轮数设为150,使用Adam优化器、批大小为32,并采用ReduceLROnPlateau学习率调度策略,验证指标的耐心值为5,衰减系数为0.6。模型训练至收敛,并在验证集指标上进行早停,耐心值为10。预训练阶段训练10个epoch,采用线性预热结合余弦退火策略。所有结果均在三种随机种子下重复三次,并报告标准差。
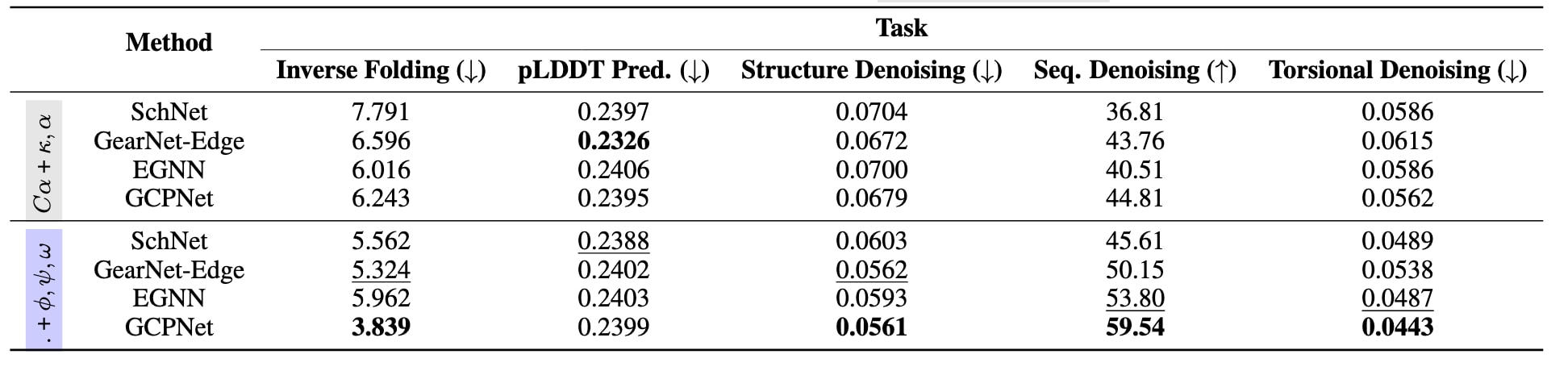
表3|在AlphaFoldDB上进行预训练任务的验证集性能结果。 评测指标包括:逆折叠任务使用困惑度;pLDDT预测、结构去噪和扭转角去噪任务使用均方根误差;序列去噪任务使用准确率。加粗结果表示最优性能,带下划线的结果表示次优性能。主要结论在于,相较于仅使用序列上的虚拟角度特征,在特征化中引入主链结构信息,即加入扭转角φ、ψ和ω,通常能够显著提升预训练阶段的性能表现。
4 结果与讨论
4.1 辅助去噪任务能够稳定提升基线性能
在表2中,首先分析了在不进行预训练的情况下,模型架构选择与去噪辅助任务的组合所带来的影响,主要结论包括以下几个方面。首先,在10项任务中,EGNN和GCPNet等等变模型在其中5项任务上取得了最佳性能,表明等变模型在多种场景下具有优势。值得注意的是,引入结构特征后的ESM-2-650M在家族与超家族折叠预测以及基因本体预测任务上的表现,已可与最先进的蛋白质特异性GNN方法相当。其次,在输入表示方面,结合Cα原子、虚拟角度以及主链扭转角的特征化方式,在60种模型与任务组合中的22种情况下取得了整体最优结果。这说明,仅依赖主链特征让模型隐式学习侧链取向和柔性,可能有助于避免对晶体结构伪影的过拟合。最后,无论是序列去噪还是结构去噪,在大多数情况下都能显著提升模型性能,在60种组合中有50种优于不使用辅助任务的设置。尤其是结构去噪,在GO预测和反应类型预测任务中,显著提升了MACE模型训练的稳定性。
4.2 引入更多结构细节可提升预训练效果
在表3中,研究进一步考察了结构预训练的效果。结果表明,在预训练阶段,引入包含二面角在内的更丰富结构信息,通常比单纯更换模型架构更有助于提升验证集指标。此外,不同预训练任务对不同类型模型的促进作用存在差异。逆折叠、结构去噪、序列去噪和扭转角去噪对等变模型的提升更为显著,而pLDDT预测则更有利于不变模型。这说明,不同预训练任务在不同模型类别之间具有选择性收益。受限于计算成本,目前尚未能对球等变GNN模型进行大规模预训练。
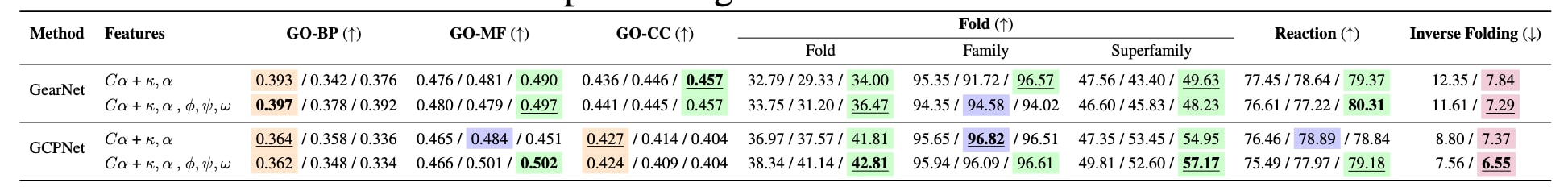
4.3 预训练与更精细的结构表示有助于下游任务表现
基于预训练阶段的观察,表4进一步分析了在下游任务上的微调结果。总体来看,等变GNN在多数情况下优于不变模型,且在结合结构相关预训练任务后收益最大,尤其是在输入特征中提供更丰富结构细节时更为明显。同时,无论是不变模型还是等变模型,相较于仅使用Cα原子表示,引入更多结构信息通常都能提升下游任务性能。其中,结构去噪预训练在两类模型上均表现出稳定的正向效果。
表4|预训练模型的基准评测结果。 每种模型与特征化方案组合的结果按以下顺序给出:不进行预训练 / 序列去噪预训练 / 结构去噪预训练。其中,在CATH数据集上的逆折叠任务采用在AlphaFoldDB上进行逆折叠预训练的模型。主要结论是,等变的GCPNet模型从预训练以及引入最大程度结构细节的特征化方案中获得的收益最为显著。
5 结论
该工作致力于构建一个面向蛋白质结构表示学习的综合性多任务基准。ProteinWorkshop通过统一的框架整合了大规模预训练语料、特征化方案、几何图神经网络模型以及多类评测任务,用于系统评估不同蛋白质结构编码方法的有效性。研究结果表明,结构预训练以及序列和结构去噪等辅助任务,能够在多种下游任务上显著提升模型性能,同时在特征化过程中引入更丰富的结构细节也有助于进一步提高表现。该基准在设计上具有良好的扩展性,便于纳入新的任务和数据集,并已向更广泛的研究社区开放。