ICLR 2025 | DPLM-2: 多模态扩散蛋白语言模型
蛋白质的功能由其氨基酸序列与三维结构共同决定,二者密切关联且相辅相成。现有蛋白质生成与设计方法多聚焦于单一模态(序列或结构),难以同时捕获序列与结构的复杂内在联系,限制了联合理解与生成能力。针对这一挑战,有研究提出 DPLM-2 —— 一种基于离散扩散机制的多模态蛋白语言模型,能够同步建模并生成蛋白质序列与对应结构。DPLM-2 采用创新的无查表结构量化方法,将连续的三维结构转化为离散标记,结合高效的预热策略融合进化序列信息,提升多模态学习效果。实验表明,DPLM-2 可无缝生成高度兼容的蛋白质序列与结构,且在折叠、逆向折叠及基序骨架设计等多样条件生成任务中表现优异,显著推动多模态蛋白质生成模型的发展与应用。

获取详情及资源:
0 摘要
蛋白质作为生命体内关键的大分子,其氨基酸序列决定了三维结构,进而影响功能。蛋白质生成建模因此需要多模态方法,能够同时建模、理解与生成序列与结构。但现有方法多为分模态单独建模,难以捕获序列与结构间复杂关联,导致联合理解与生成性能受限。本文提出 DPLM-2,基于离散扩散的蛋白语言模型 DPLM 扩展而来,支持序列与结构的联合建模。为使语言模型学习结构信息,采用无查表量化(lookup-free quantization, LFQ)将三维坐标转换为离散标记。通过结合实验及高质量合成结构数据训练,DPLM-2 学习序列与结构的联合分布及边缘和条件分布。引入高效预热策略,融合进化序列数据与基于预训练序列语言模型的结构归纳偏置。实验证明,DPLM-2 可同时生成高度匹配的氨基酸序列与三维结构,无需两阶段生成流程,并在多种条件生成任务中表现优异,包括蛋白折叠、逆向折叠及多模态基序骨架搭建,同时提供结构感知的预测任务表征。
1 引言
蛋白质功能由其氨基酸序列和三维结构共同决定。近年来,扩散模型在基于结构的蛋白质生成中表现卓越,大规模蛋白语言模型则成为序列基础的蛋白质表征与生成核心。DPLM 作为离散扩散的蛋白语言模型,已在序列生成与理解领域取得最先进性能。
许多工程应用(如基序骨架搭建、抗体设计)要求序列与结构的联合确定,然而现有多采用单一模态生成模型,另一模态依赖独立模型处理,限制了联合理解与生成能力。多模态蛋白生成模型的需求因此凸显。
近期如 Multiflow 采用流匹配融合序列与结构,但在序列兼容性及折叠任务表现不足,归因于缺少基于进化序列的归纳偏置。鉴于基于序列的 DPLM 在生成与理解上的优势,本文以 DPLM 为基础,面向多模态联合学习,提出 DPLM-2。
关键技术与贡献
- 结构离散化:开发无查表量化结构分词器,将三维坐标转为离散令牌,解决语言模型处理连续结构数据的难题。
- 高效预热:利用序列预训练模型的结构归纳偏置,设计暖启动训练策略,促进多模态学习。
- 自混合训练:针对离散扩散中的暴露偏差,设计自混合训练策略,提升生成质量与多样性。
DPLM-2 支持无条件的结构与序列联合生成,生成蛋白质在二级结构统计上更符合天然蛋白特性,性能优于纯结构生成模型。其多模态特性使其能完成多样的条件生成任务,包括序列条件下的折叠、结构条件下的逆向折叠以及多模态基序条件下的骨架搭建。此外,DPLM-2 学习的结构感知表征提升了多种预测任务的表现。
同期工作比较
在开发 DPLM-2 过程中,注意到近期提出的多模态生成蛋白语言模型 ESM3(Hayes et al., 2024),该模型也通过生成式掩码语言模型联合建模结构与序列的离散化表示。尽管两者目标相似,DPLM-2 在以下几个关键方面有所区别:
-
多模态蛋白质生成
DPLM-2 设计上平等对待结构与序列模态,强调同步联合生成兼容的蛋白质序列与结构;而 ESM3 以序列为主导,训练中对其他模态进行随机丢弃,生成过程按模态顺序级联进行。
-
数据与计算效率
ESM3 从零开始进行多模态预训练,依赖海量合成数据,模型规模从 14 亿至 980 亿不等,且受限于严格许可及训练基础设施,难以供社区复现和定制。相比之下,DPLM-2 仅依托较小规模的数据集(PDB + SwissProt),基于开源的预训练序列语言模型 DPLM(规模从 1.5 亿到 30 亿不等),充分利用其进化知识和强大的序列理解及生成能力。
此外,DPLM-2 承诺开源模型及训练推理代码,推动多模态蛋白生成语言模型在社区的普及与应用。综上,DPLM-2 为社区贡献了独特且具实用价值的多模态蛋白生成基础模型。
2 预备知识
2.1 蛋白质生成式建模
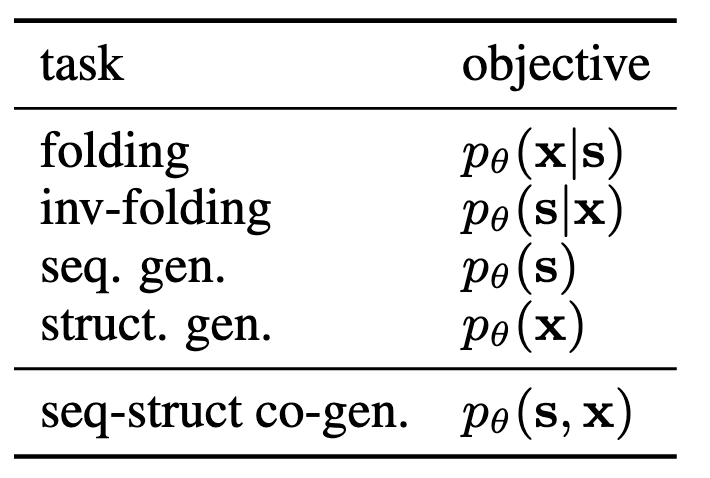
生成蛋白质建模的目标是通过学习概率模型
概率模型
因此,大多数蛋白质任务可以看作是在这两种模态之间指定输入条件和输出目标(见表 1),包括:
- 基于序列条件的结构预测(折叠任务,参考 Jumper et al., 2021;Lin et al., 2022;Huguet et al., 2024);
- 基于结构条件的序列生成(逆向折叠或固定骨架设计,参考 Dauparas et al., 2022;Hsu et al., 2022;Zheng et al., 2023b);
- 序列的学习或生成(参考 Rives et al., 2019;Nijkamp et al., 2022;Alamdari et al., 2023;Wang et al., 2024);
- 结构生成(参考 Yim et al., 2023;Watson et al., 2023;Ingraham et al., 2023);
- 序列与结构的联合生成(参考 Jin et al., 2021;Shi et al., 2022;Campbell et al., 2024)。
2.2 扩散蛋白语言模型(DPLM)
语言模型(LM),通常由 Transformer(Vaswani et al., 2017)参数化,因其可扩展性和优异的表达能力已成为多个领域的事实标准(OpenAI, 2023)。其中,蛋白质语言模型在蛋白质序列学习(Rives et al., 2019;Lin et al., 2022)和生成(Nijkamp et al., 2022;Alamdari et al., 2023)方面,已成为重要的人工智能基础。
扩散蛋白语言模型(DPLM,Wang et al., 2024)基于吸收式离散扩散框架(Austin et al., 2021;Zheng et al., 2023a),表现出在蛋白质序列生成与表示学习上的卓越性能。设
该过程将数据
模型学习逆向去噪过程
其中,
这里
在推断时,DPLM 可通过离散扩散的逆向迭代去噪过程生成氨基酸序列:
具体而言,在时间步
对于序列的表示学习,直接将序列输入 DPLM 即可获得有效嵌入,用于下游预测任务。
这种框架支持多种条件生成应用,允许对单一模态或混合模态进行部分条件生成,例如基序骨架搭建和抗体设计等。
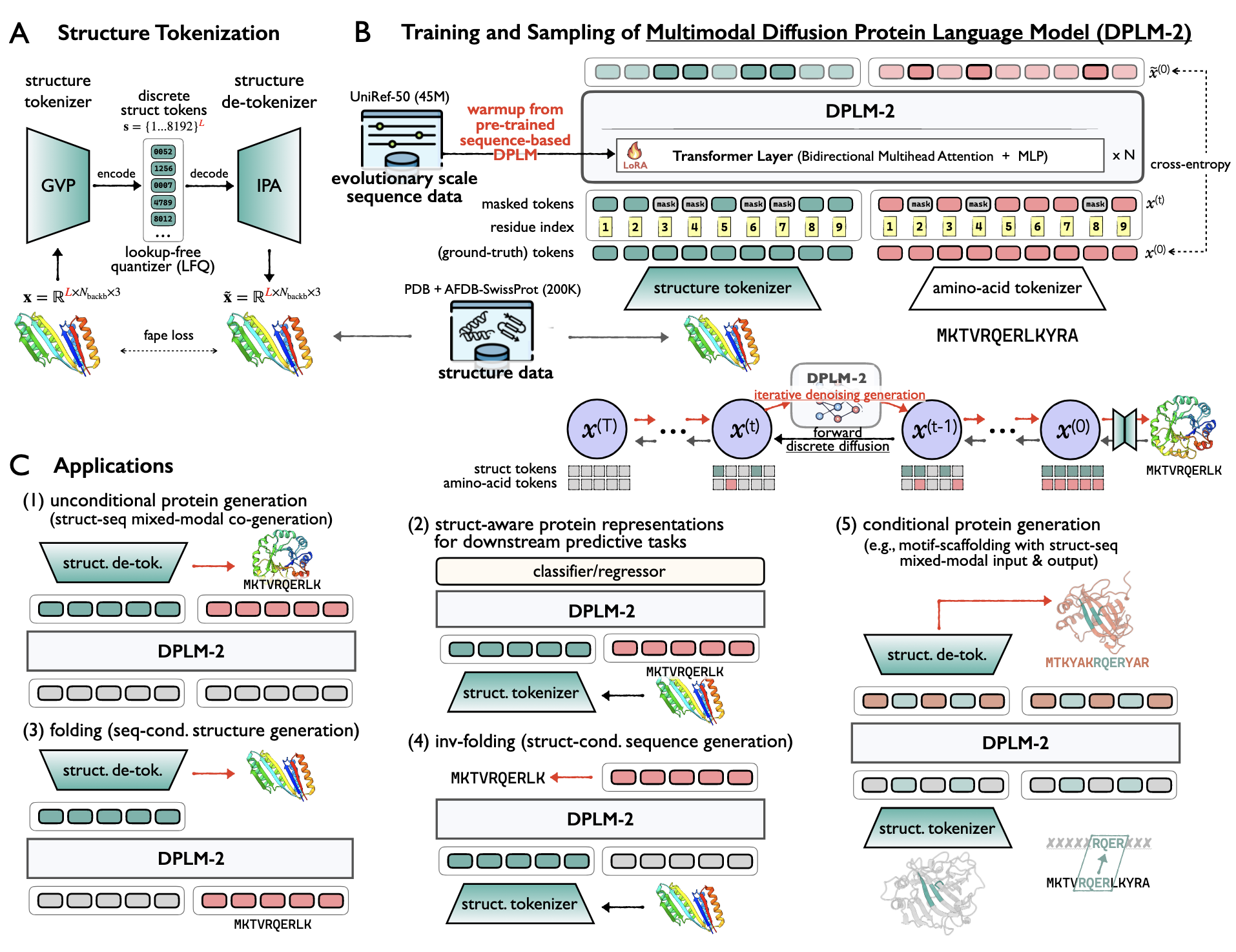
3 DPLM-2:多模态扩散蛋白语言模型
3.1 总览
图 1 展示了 DPLM-2 的整体架构。DPLM-2 基于最先进的序列生成蛋白语言模型 DPLM(Wang et al., 2024),采用离散扩散概率框架,同时建模蛋白质序列及对应结构。
为了让语言模型能够学习结构信息,设计了一种基于分词器的蛋白结构表示,将蛋白质主链三维坐标
训练数据包含约 2 万条来自蛋白质数据库(PDB)的高质量聚类实验结构及 20 万条来自 AFDB SwissProt 分割集的预测结构,长度均小于 512。训练过程中,DPLM-2 需在从完全噪声到完全干净的不同噪声水平下对输入序列去噪。多模态训练目标基于式(1)定义为:
其中通过假设条件独立,
为了增强模型对结构与序列的区分能力,分别为两种模态设计了独立的噪声调度器
此外,针对离散扩散中的暴露偏差问题(Ranzato 等,2016;Bengio 等,2015),提出了受计划采样启发的自混合训练策略,显著提升生成质量和多样性(详见附录 A.1)。
3.2 基于预训练序列模型的高效预热
蛋白序列编码了关键的进化信息,反映了残基对的协同进化,这些协同变异往往对应三维空间中的相互作用,有助于蛋白折叠预测(Melnyk 等,2022b)。Lin 等人(2022)证明,基于大规模进化数据训练的蛋白语言模型隐式捕获了该信息,促进结构预测。
基于这一联系,DPLM-2 采用基于序列的预训练 DPLM 进行高效预热,充分利用现有的进化知识,提升蛋白质结构建模能力。由于结构数据远少于序列数据库(20 万 vs. 4500 万),预热允许有效微调,避免从零开始训练。为保持序列知识不被遗忘并限制参数偏离,采用 LoRA(Hu 等,2021)技术控制微调幅度。此策略不仅降低训练成本,还有效转移了宝贵的进化信息。
3.3 结构分词学习
多模态蛋白语言模型的核心难点在于使语言模型学习结构信息,这既复杂又尚未充分解决。将连续数据模态离散化为紧凑、有意义的表示(Van Den Oord 等,2017)已在图像合成等领域广泛应用,有助于有效压缩和高效生成,尤其适合基于序列的 Transformer 模型。近期研究尝试将此方法应用于蛋白结构坐标(Van Kempen 等,2024;Liu 等,2023;Gao 等,2024;Lu 等,2024),促进语言模型学习局部结构元素的组成规律。
然而,如何设计高效的结构分词器仍是活跃研究课题。典型的 VQVAE(Van Den Oord 等,2017)框架下的结构分词过程可概括如下:
公式及说明如下:
-
**结构编码器(encoder)**将主链三维坐标
编码为不变特征 -
**量化器(quantizer)**将编码特征
转换为长度为 的离散标记序列 其中 是有限大小的码本。 -
**结构解码器(decoder)**从离散标记
重建三维坐标
具体量化过程采用 Lookup-Free Quantizer (LFQ),其潜在空间被分解为一组单维二值变量的笛卡尔积:
给定编码特征
对应的结构标记索引为:
该 LFQ 结构分词器基于重构损失、承诺损失及熵正则化训练,重构损失采用 FAPE(frame aligned point error)。
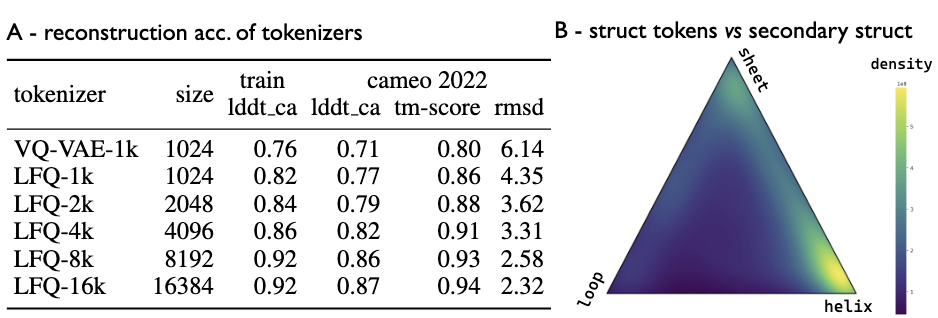
评估结果显示,LFQ 在重构准确度上显著优于传统 VQ-VAE,且训练时间大幅缩短(8 张 A100 GPU 上 2 天对比 15 天)。码本大小增加可提升重构效果,其中码本大小为 8192 时实现最佳压缩与重构平衡。
同时,结构标记与蛋白质二级结构高度相关,很多标记聚集在α-螺旋和β-折叠的顶点区域,一些标记位于区域之间,说明结构标记有效捕捉了主链局部环境的细粒度结构元素。
4 实验评估
本节评估了 DPLM-2 在多种生成和理解任务中的表现,涵盖无条件蛋白生成(包括结构、序列及结构-序列联合生成,见 §4.1)以及多种条件生成任务,如折叠 (§4.2)、逆向折叠 (§4.3)、基序骨架搭建 (§4.4),以及一系列蛋白质预测任务 (§4.5)。
4.1 无条件蛋白生成
无条件蛋白生成旨在同时生成蛋白质的三维结构和氨基酸序列。传统方法通常采用级联策略,先生成结构再预测序列,或反之。本文重点评估 DPLM-2 同时生成结构和序列的能力,并比较级联与同时生成在无条件结构生成、序列生成和结构-序列联合生成三方面的表现。
评价指标遵循 Multiflow 设定,涵盖生成蛋白的质量、新颖性和多样性。质量通过设计性(设计的结构能否对应有效序列)和折叠性(序列能否折叠成合理结构)衡量。设计性使用 ESMFold 对生成序列进行折叠预测,再通过 co-generated 结构计算 sc-TMscore 和 sc-RMSD 评估相似度。折叠性以 ESMFold 的 pLDDT 评分(>70 视为合理)进行评估。新颖性以生成结构与 PDB 已知结构的 TMscore(pdb-TM,越低表示新颖性越高)衡量。多样性通过样本间的 TMscore(inner-TM,越低越多样)及 FoldSeek 聚类数目(归一化)评估。
4.1.1 DPLM-2 实现高质量、多样且新颖的蛋白序列与结构生成
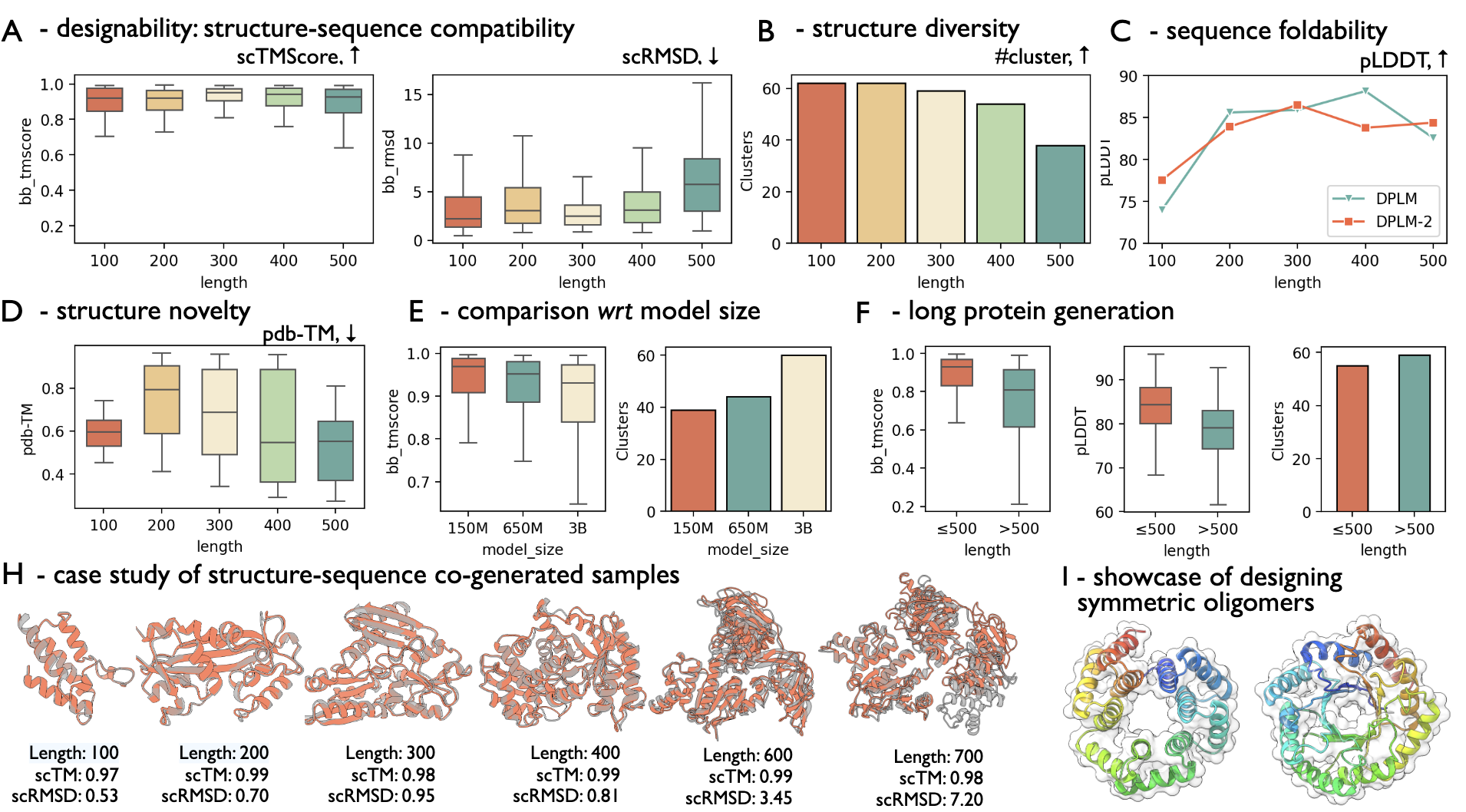
表 2 和图 3 汇报了 DPLM-2 无条件蛋白生成的主要结果,重点如下:
- DPLM-2 能够生成多样且结构合理的蛋白,实现结构与序列的联合生成。针对长度为 100、200、300、400 和 500 的蛋白,各采样 100 个样本。图 3A/B 显示 DPLM-2 生成的序列和结构在不同长度下均具备较高的设计性(大部分 sc-TM 超过 0.9),且聚类结构多样。图 3D 显示,蛋白的新颖性(pdb-TM)随着蛋白长度增加而提升。DPLM-2 支持同时生成两种模态,也支持单模态逐步生成。表 2 显示联合生成的 sc-TM 最高,证明联合建模确实有益于蛋白生成。
- DPLM-2 在联合生成及单独生成结构或序列任务中均表现出与强基线竞争的性能。表 2 显示 DPLM-2 的 sc-TM 接近 PDB 中天然结构的质量。相比之下,ESM3-Open(序列优先生成)在无条件生成任务上表现欠佳。与 Multiflow 相比,DPLM-2 达到相当的联合生成质量。值得注意的是,Multiflow 直接用带有天然序列的结构训练时,序列生成表现较差,导致联合生成性能显著下降,除非借助外部逆向折叠模型(ProteinMPNN)进行知识蒸馏。我们用自有数据重新训练 Multiflow,其联合生成性能依然不佳,远落后于 DPLM-2,说明 DPLM-2 能够直接且高效地从复杂的结构-序列联合分布学习。此外,DPLM-2 在单一模态生成时也能匹配该任务的最佳模型,进一步验证其多模态生成能力。
- DPLM-2 可生成超出训练长度范围的长链蛋白。虽然训练时序列长度截断为 512,图 3F 评估了长度为 600、700、800、900、1000 的蛋白。结果显示,对于超过最大训练长度的蛋白,DPLM-2 生成序列的 pLDDT 分数与 DPLM 相近,表明 DPLM-2 保留了序列预训练中继承的生成能力,实现了长度外推。
- 案例展示。图 3H 展示了 DPLM-2 生成的最大 700 残基蛋白样本,图 3I 演示了通过强制重复结构与序列模式,DPLM-2 可被操控设计对称多聚体。
![表 2:无条件蛋白生成的基准比较,涵盖结构-序列联合生成、仅骨架结构生成和仅序列生成。对每种方法,在长度为 [100, 200, 300, 400, 500] 的蛋白质上生成 100 个样本。* 表示我们重新训练的 Multiflow 变体,分别使用了未经过 ProteinMPNN 蒸馏的原始 PDB 数据和与 DPLM-2 相同的训练数据(即 PDB + SwissProt)。](https://cdn.molastra.com/weixin/2025/08/a4758e0a6d17e6051a3ffdf6c8d9cbb4.png)
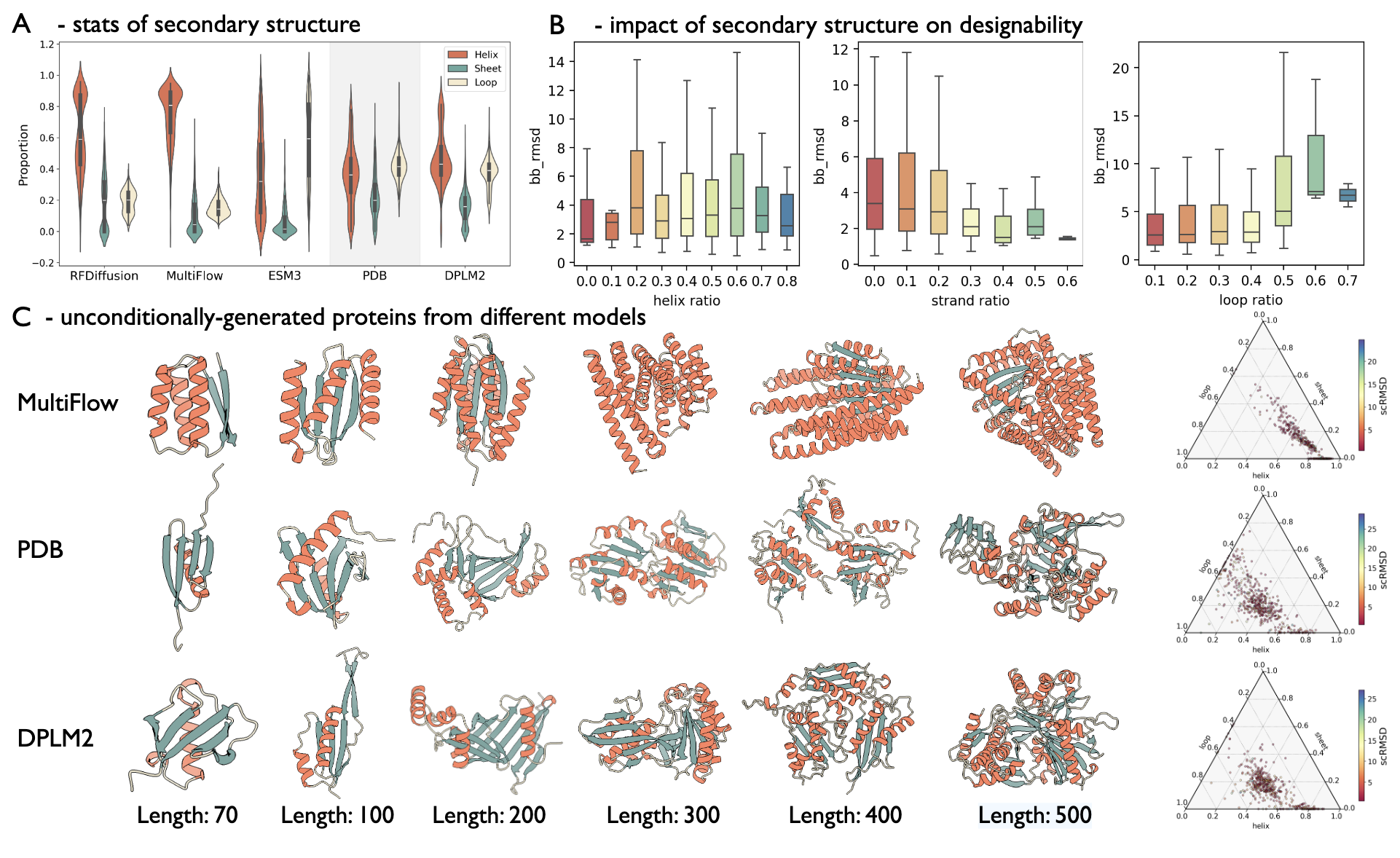
4.1.2 DPLM-2 生成的蛋白质更接近天然蛋白质
为了进一步分析不同模型的性质,研究人员比较了它们生成蛋白质的二级结构分布与 PDB 天然蛋白的相似度。结果显示,DPLM-2 生成的蛋白质在二级结构上与天然蛋白最为接近。图 4A 显示,结构生成模型如 RFDiffusion 和 MultiFlow 倾向于生成更多的 α 螺旋、较少的 β 折叠和环状结构,而蛋白语言模型如 ESM3 和 DPLM-2 则没有明显的 α 螺旋偏好,但 ESM3 倾向于生成更多的环状结构。在各方法中,DPLM-2 产生的二级结构比例与 PDB 蛋白质最为接近。图 4C 中,MultiFlow 生成的蛋白质含有大量螺旋,且随着序列长度增加趋向球状,表现为理想化的二级结构,而 DPLM-2 生成的蛋白质结构更均衡,螺旋较少,β 折叠和环状结构较多。Simplex 图显示 MultiFlow 的蛋白质聚集于螺旋丰富区域,而 DPLM-2 的蛋白质分布更广泛,类似天然蛋白,但其较少生成主要由 β 折叠和环状结构组成的蛋白,这类蛋白在自然界存在。图 4B 显示环状结构比例对设计性影响显著,环越多,结构自一致性 RMSD 越高,因环结构高度柔性。因此,DPLM-2 常生成含长环的蛋白,其 scRMSD 较高,与表 2 的结果一致。
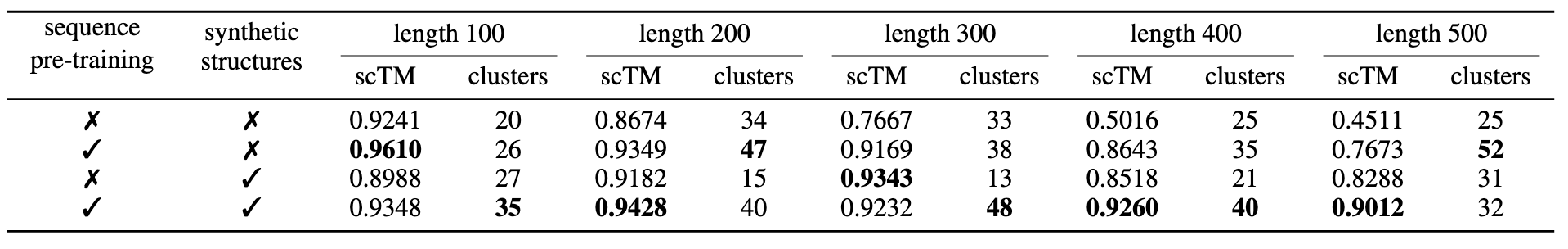
4.1.3 消融实验
训练 DPLM-2 时,先从预训练的序列模型 DPLM warm-up,利用已有进化信息,并扩增了约 20 万条 SwissProt 的高质量 AlphaFold 预测结构与 PDB 聚类结构。该节评估序列预训练及数据增强对无条件蛋白生成的影响。
4.2 正向折叠(基于序列的结构预测)
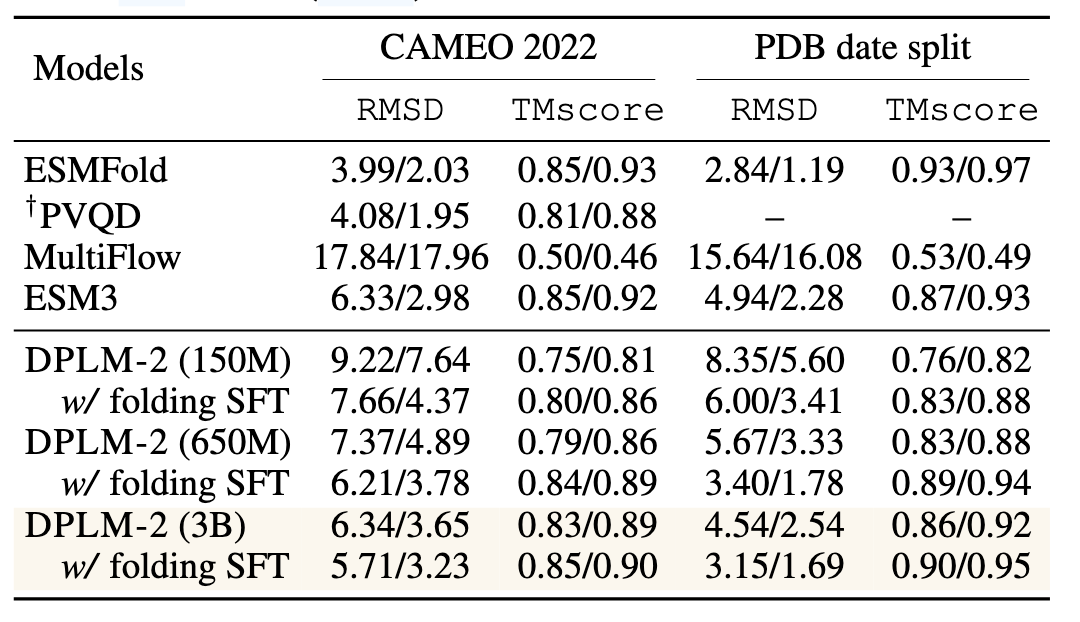
折叠任务旨在预测给定氨基酸序列对应的三维结构。作为多模态生成模型,DPLM-2 可自发地完成序列条件结构预测任务(见图 1C-3)。评估使用 CAMEO 2022 和 Multiflow 使用的 PDB 数据集,采用预测结构与真实结构的 RMSD 和 TMscore 进行评估。DPLM-2 采用 argmax 解码,采样 100 次。表 4 显示 DPLM-2 在零样本条件下已具备良好折叠能力,进一步通过监督微调(最大化
4.3 逆向折叠(基于结构的序列生成)
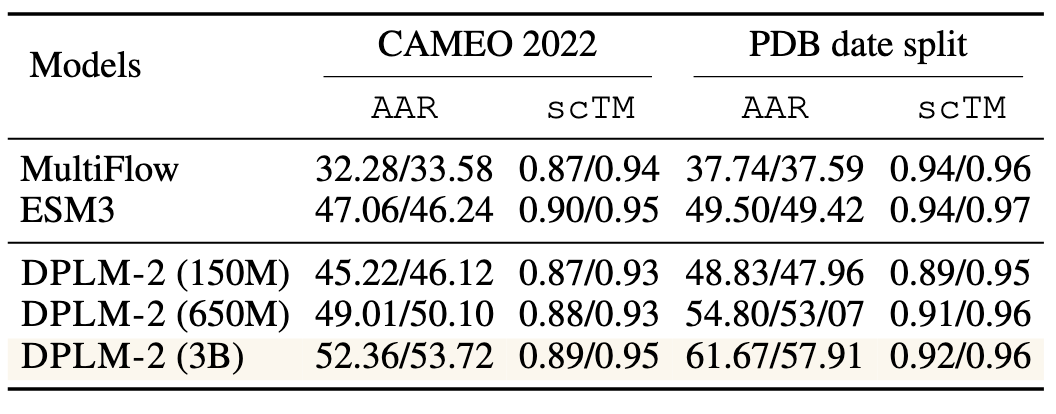
逆向折叠任务旨在寻找可折叠至给定骨架结构的氨基酸序列。序列评价采用氨基酸恢复率(AAR),结构评价通过生成序列预测结构与原始结构间的自一致性 TMscore(scTM)。DPLM-2 能生成合理序列折叠成给定结构。表 5 显示 DPLM-2 性能优于或与其他联合生成模型(MultiFlow、ESM3)持平。随着模型规模提升,序列恢复和结构一致性均得到提升,体现与折叠任务相同的规模效应。多模态训练有效对齐结构与序列,使得 DPLM-2 无需额外训练即可生成对应序列。
4.4 具有混合模态基序条件的骨架搭建
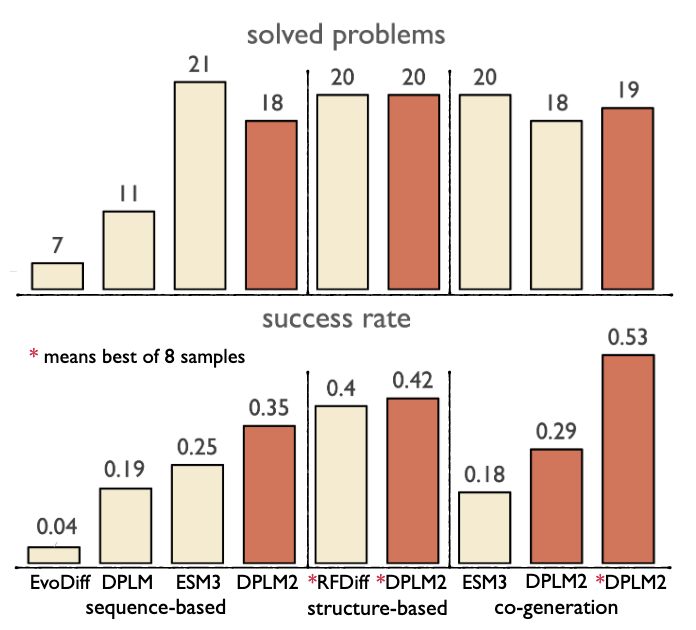
基序骨架搭建旨在生成合理骨架以保持基序结构,保证功能完整。参考 Yim 等(2024)的实验设计,涵盖 24 个基序搭建问题,每个采样 100 个骨架:首先确定骨架长度,其次基序片段保持不变,条件采样骨架。骨架长度从 Yim 等给定范围采样,多基序时保持基序顺序一致。以基序的结构和序列作为 DPLM-2 输入。作为多模态模型,DPLM-2 分别用基于序列、结构和联合生成方法评估。成功判定条件:(1)整体设计性,序列模型中 pLDDT > 70 或结构模型中 scTM > 0.8;(2)基序保持性,预测基序结构与真实基序的 RMSD < 1Å。图 5 显示 DPLM-2 可为功能基序生成合理骨架。在序列、结构和联合评估中,DPLM-2 在大多数情况下优于或持平对应方法,解决更多问题且成功率更高。相比仅序列方法,DPLM-2 表现更佳,因其支持基序结构输入,有助于基序结构和功能保持。值得注意的是,DPLM-2 在仅生成骨架结构时与 RFDiffusion 表现相当,同时在联合设计骨架序列和结构时表现优于 ESM3。尽管尚无实验验证,这表明多模态条件和生成有望带来更成功的条件蛋白设计。
4.5 蛋白质表示学习评估
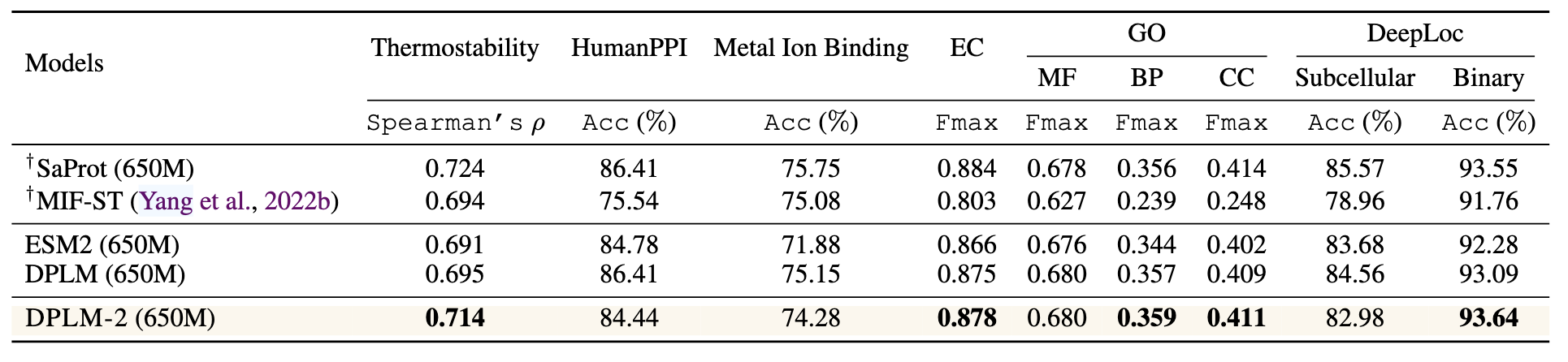
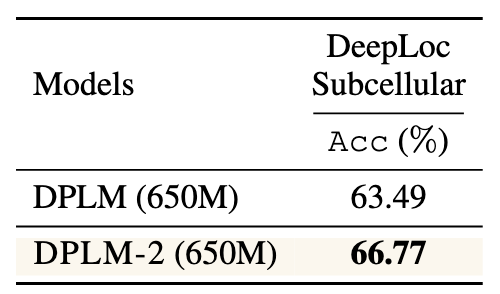
直接访问结构信息有助于下游蛋白预测任务。利用 SaProt 数据集,DPLM-2 输入蛋白结构 tokens 和序列,实现多模态表示学习。表 6 显示 DPLM-2 在部分任务中优于仅序列方法(ESM2、DPLM),说明其能利用蛋白结构生成包含多模态信息的更优表示,用于下游任务。然而,在大多数任务上 DPLM-2 落后于结构感知最先进蛋白语言模型 SaProt,且部分任务不及 DPLM。推测原因是 DPLM-2 结构训练数据量较小(仅 PDB 和 SwissProt),且与 DPLM 预训练用的 UniRef50 不同,导致灾难性遗忘和表示不佳。为验证此点,在 DeepLoc 亚细胞任务中,未进行大规模序列预训练的 DPLM-2 明显优于 DPLM。表 7 显示:(1)引入结构信息确实提升性能;(2)小规模数据导致灾难性遗忘,影响预训练优势。未来方向是利用更大规模的预测结构数据进一步提升预测性能。
5 讨论
本文介绍了 DPLM-2,一种多模态扩散蛋白语言模型,能够理解、生成并推理蛋白质的结构与序列,旨在成为蛋白质的多模态基础模型。尽管在蛋白质的联合生成、折叠、逆向折叠以及基于多模态输入输出的条件基序骨架构建等任务中表现出令人鼓舞的效果,但仍存在一些亟待解决的局限性:
(1) 结构数据:虽然结构信息有助于提升预测任务的性能,但受限于结构数据量的不足,DPLM-2 学习到的表示尚不够稳健。未来工作中还需考虑更长的蛋白链和多聚体的建模。
(2) 离散潜变量表示的权衡:将结构离散化为符号有助于构建多模态蛋白语言模型及联合生成,但可能导致精细的结构细节和控制能力的丢失,例如精确的原子位置和原子间距离。未来的研究应致力于将基于数据空间的结构生成模型的优势,与基于序列的多模态语言模型相结合,最大化二者的优点。