Nat. Comput. Sci. 2025 | PhoreGen: 面向药效团的三维分子生成助力高效特征定制型药物发现
获取详情及资源:

0 摘要
分子生成是一项前沿技术,具备彻底变革智能药物发现流程的潜力。然而,当前报道的基于配体或结构的分子生成方法在实际药物研发中仍难以落地。为此,该研究提出了一种显式药效团导向的三维分子生成方法,命名为 PhoreGen。PhoreGen 在扩散-去噪过程中引入了异步扰动与更新机制,分别作用于原子和化学键信息,并融合了配体–药效团映射的先验知识,通过消息传递机制实现精确控制。评估结果表明,PhoreGen 能高效生成与给定药效团高度匹配的 3D 分子,同时保持良好的化学合理性、多样性、药物相似性及结合亲和力,且尤为重要的是,可高频率地产生具有目标特征的定制化分子。借助 PhoreGen,研究成功设计出一系列新型双环硼酸酯类抑制剂,靶向进化型金属和丝氨酸 β-内酰胺酶,有效增强了美罗培南对临床分离超级细菌的作用。此外,还发现了针对金属依赖性烟酰胺酶的新型抑制剂,后者为农药开发中的新兴靶点。该研究展示了一种显式约束的分子生成范式,并验证了其在特征定制化药物发现中的巨大应用潜力。
1 引言
本研究针对早期药物发现阶段“从庞大化学空间中筛选候选分子”的核心难题,提出并验证了一个全新路径:基于药效团的三维分子生成方法 PhoreGen,以应对现有方法在特征定制药物设计上的局限。传统分子生成方法主要分为基于配体的(LBMG)和基于结构的(SBMG)两类。LBMG 方法虽起步早、效率高,但由于缺乏靶标结构信息,难以生成靶标特异性分子。SBMG 方法则通过引入蛋白–配体互补原理提升生成效果,特别是扩散模型近年来的兴起,使 SBMG 在靶点适配性上取得显著进展。然而,SBMG 方法在处理共价药物、金属结合分子等复杂特征设计时表现仍有限,难以在实际应用中落地。
药效团作为作用机制的抽象表达,天然具备描述空间相互作用与配体–靶标互补性的能力,具备驱动三维结构生成的潜力。虽然已有部分工作尝试将药效团引入分子生成过程,但仍缺乏能够直接基于药效团模型生成三维分子的技术路径。
PhoreGen 正是填补这一空白的尝试。该模型构建了一个融合化学知识与空间信息的条件扩散框架,引入异质几何图以表征配体–药效团结构,并通过方向匹配的消息传递机制,引导生成过程与药效团约束精准对齐。其独特的“先键后原子”去噪策略有效避免了结构失真问题,同时通过辅助模块实现对分子尺度的合理控制。
在训练阶段,PhoreGen 使用了超百万对高质量配体–药效团数据对,涵盖三维配体、蛋白复合物及对接结构,特别加强了共价、金属配位等低频关键药效团特征的学习能力,以实现特征定制的分子设计。
全面评估结果显示,PhoreGen 能稳定生成与药效团高度匹配、构象合理、具备成药性和结合潜力的三维结构分子,尤其在金属结合与共价抑制剂等方向展现出独特优势。目前,PhoreGen 已被应用于新型 MBL/SBL 双重抑制剂的设计,显著提升了美罗培南对耐药菌的疗效,同时也发现了针对烟酰胺酶的潜在农药分子。
综上所述,PhoreGen 提供了一种具备显式结构引导、特征定制能力与高生成质量的三维分子生成新范式,为智能化药物设计拓展了新的可能,展示了其在从靶点机制到分子设计的全流程创新潜力。

XXXXXX
图 1 | PhoreGen 的整体架构 a, 展示了 PhoreGen 的扩散与生成过程。前向扩散从真实分子
XXXXXX
2 结果
2.1 基于药效团导向的三维分子生成框架
PhoreGen 是一个基于药效团约束的条件扩散模型,旨在生成与药效团特征高度匹配的完整三维分子结构(见 Fig. 1a)。其核心思路是在扩散过程中,通过药效团特征所编码的相互作用模式与空间限制进行引导,从随机噪声中逐步去噪,最终生成符合结构合理性的三维分子。
PhoreGen 遵循一个典型的扩散过程
模型结构上,PhoreGen 基于 E(3)-等变图神经网络 进行构建(Fig. 1b)。它以扩散中任一时间步
值得注意的是,药效团上下文
此外,为控制生成分子的规模,模型引入了分子大小预测模块(Fig. 1d),根据药效团的几何与形状特征推断生成所需的原子数。在生成过程中,模型首先根据训练好的该模块确定原子数量范围,以兼顾配体多样性与分子尺度控制。接着从标准高斯分布
为了让 PhoreGen 能够充分学习三维配体结构与药效团之间的本质关联,作者设计了两阶段训练方案,并构建了三个定制数据集:
- LigPhore:包含 2,350,797 对来自真实三维配体的配体-药效团对;
- CpxPhore:包含 12,581 对来自实验蛋白-配体复合物结构的数据;
- DockPhore:包含 76,203 对来自分子对接模拟的配体-药效团配对;
共考虑了 12 类药效团特征(其中包括 4 类共价特征,详见补充表 2 与 3)。
第一阶段使用 LigPhore 进行预训练,使模型能够在广阔的化学和药效团空间中学习到具有普适性的结构映射规律。第二阶段的微调则使用 CpxPhore 与 DockPhore,进一步增强模型应对配体-药效团不完美匹配情形与**诱导契合效应(induced-fit effects)**的能力。
此外,扩散过程中还引入了异步噪声策略(asynchronous noise schedule),即对化学键施加比原子更大的噪声扰动,从而更好地模拟真实分子的扰动方式。
消融实验表明,若去除预训练阶段或微调阶段,会显著降低模型在三维构象有效性、药效团对齐度与药物相似性等方面的表现(见补充表 4)。而在扩散去噪流程中剔除键信息,同样会导致生成分子在化学合理性与能量稳定性方面表现不佳。
考虑到药效团模型本身存在一定的配体映射容差,作者还对输入中的药效团特征中心与方向施加额外扰动,以增强数据多样性并缓解模型面临的过强约束,最终提升了模型在更广泛任务中的泛化能力。
XXXXX
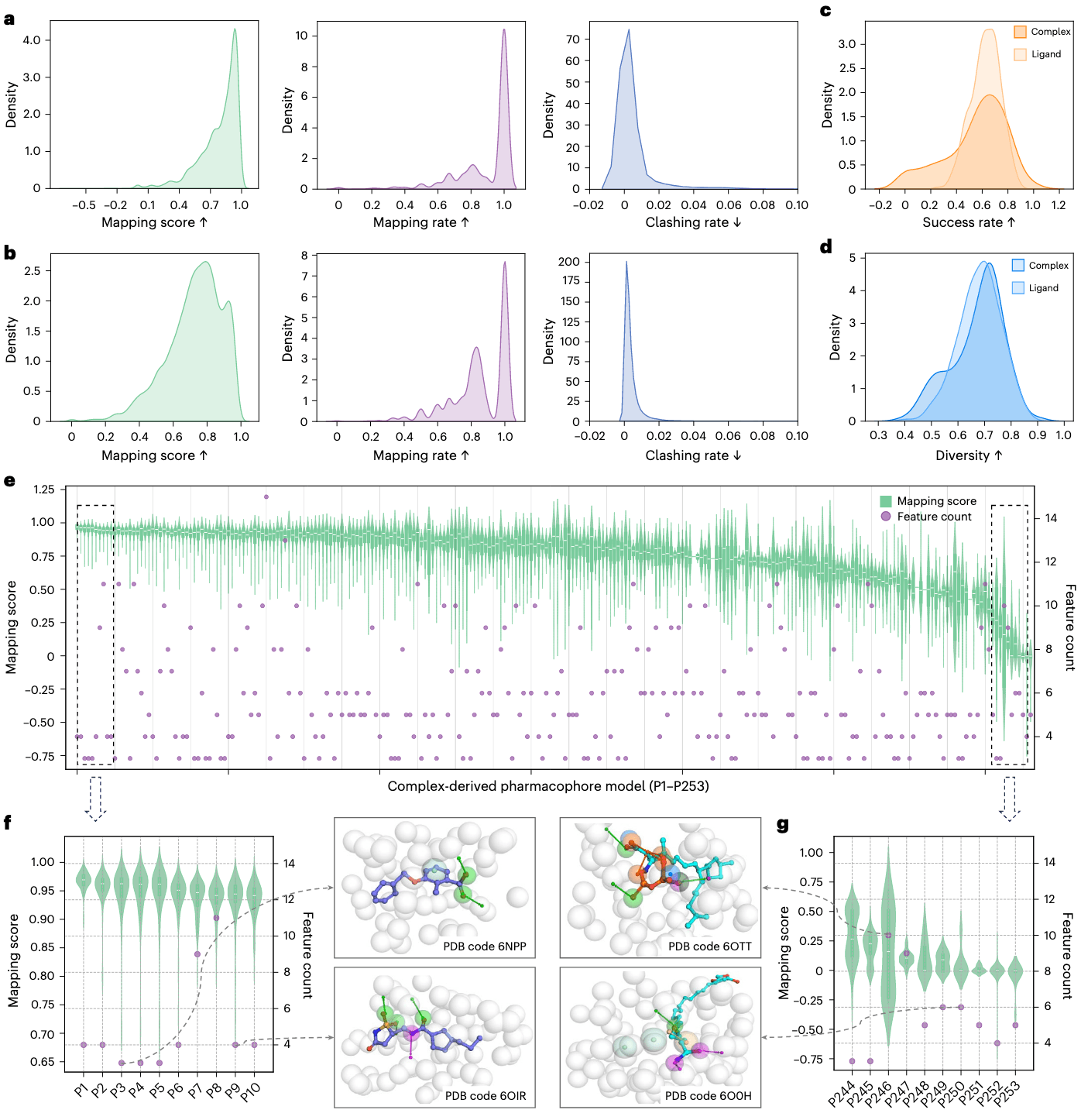
图 2 | PhoreGen 能够生成与复合物和配体来源药效团模型高度匹配的三维分子结构:a、b 展示了模型在处理复合物来源(a)与配体来源(b)药效团时生成分子的映射分数、特征匹配率和排斥球冲突率;c 为通过 PoseBusters 评估的构象真实性成功率分布;d 显示了两类药效团模型对应的生成分子的化学多样性分布,体现了模型的结构探索能力;e 分析了映射分数与药效团特征数量之间的关系;f、g 分别展示了 10 个复合物来源药效团模型中映射分数最高(f)与最低(g)的一组生成分子(示例编号包括 100、46、26 等),图中原子以板岩蓝或青色表示碳,蓝色表示氮,红色表示氧,金黄褐色表示硫,图示采用箱线图,箱体表示四分位范围,中线为中位数,须线为 ±1.5 倍 IQR,箱外为离群值。
XXXXX
2.2 生成与药效团高度匹配的三维分子结构
本节评估了 PhoreGen 在生成与给定药效团模型高度匹配的三维分子方面的能力。实验选用了 269 个复合物来源和 300 个配体来源的药效团模型,这些模型均未出现在训练数据中。评估采用三项指标:映射分数(mapping score)、特征匹配率(feature mapping rate)与排斥球冲突率(exclusion-sphere clashing rate)(详见“评估指标”章节)。
针对复合物来源的药效团模型,PhoreGen 生成的三维分子表现出良好的对齐能力,平均映射分数为 0.784,平均特征匹配率达 0.894,排斥球冲突率极低,仅为 0.006(见 Fig. 2a)。在配体来源模型上,虽然整体表现略低,但依然表现良好,平均映射分数为 0.725,匹配率为 0.847(见 Fig. 2b)。
构象合理性方面,通过 PoseBusters 评估,PhoreGen 在复合物来源与配体来源模型上分别达到了 0.552 与 0.626 的成功率(见 Fig. 2c)。同时,生成分子在化学结构上表现出显著的多样性,化学多样性得分分别为 0.673(复合物来源) 与 0.682(配体来源),体现了模型在保持构象合理性前提下具备探索广阔化学空间的能力(见 Fig. 2d)。
XXXXX
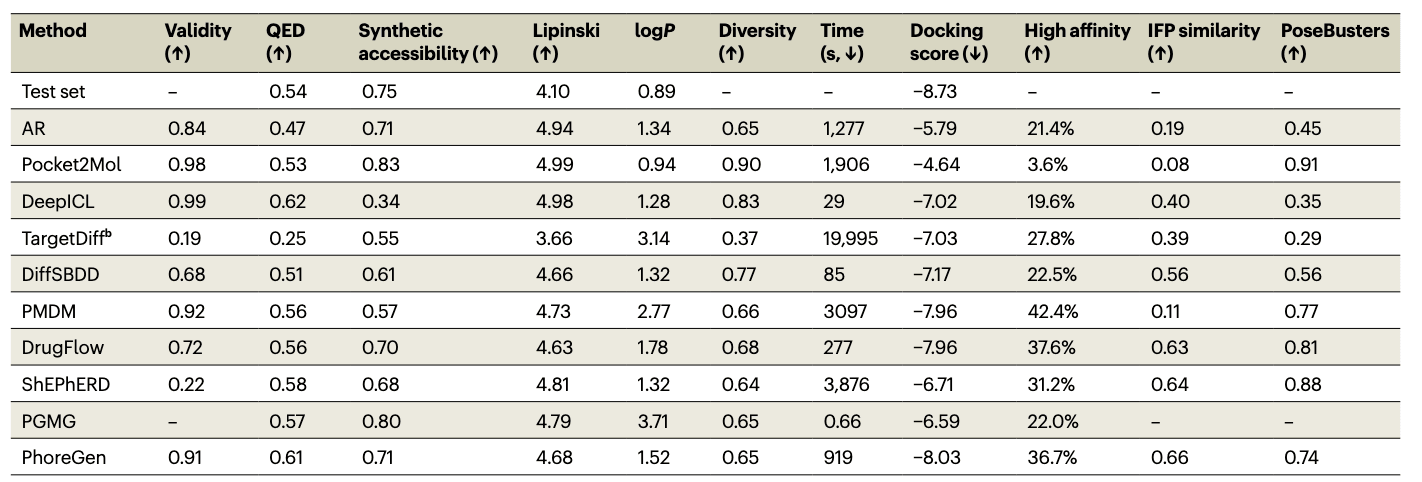
表 1|PhoreGen 与基线模型在各项指标下的性能对比
XXXXX
进一步分析显示,生成分子的质量(以映射分数与 PoseBusters 构象合理性为衡量)与药效团模型的若干关键因素密切相关,包括特征数量、特征组成以及排斥球的数量。图 2e 展示了生成分子的映射分数与复合物来源药效团模型中药效团特征数量之间的关系。
当药效团模型在特征数量、组成与药物-靶点常见相互作用之间达到良好平衡时(参见扩展数据 Fig. 1 与补充表 5–12),PhoreGen 通常可生成具有良好对齐度、合理化学结构与能量特征的分子(见 Fig. 2f)。相比之下,对于那些特征分布密集、同质化或空间分散的模型,PhoreGen 所生成的三维分子数量减少,结构多样性降低,质量亦不理想(见 Fig. 2g 与补充图 1),这类模型往往对应的是特定骨架结构、化学空间受限的分子类型。通过优化药效团模型设计或引入定制化训练集,有望提升这类模型的生成质量。
相似的趋势也出现在配体来源模型中,生成分子的质量同样与药效团模型的结构特征高度相关(见补充图 2 与补充表 13–21)。
与其他基于扩散的分子生成模型依赖经验分布来决定分子大小不同,PhoreGen 利用神经网络直接根据药效团特征与排斥球信息预测分子规模(见 Fig. 1d)。进一步分析表明,药效团特征数量、排斥球数量与 cavity size(即药效团特征与排斥球间的平均距离)与生成分子的原子数与化学多样性密切相关。
对于复合物来源模型,随着药效团特征数量与排斥球数量的增加,生成分子的原子数亦同步增加(见扩展数据 Fig. 2a,b),配体来源模型也呈现同样趋势(见扩展数据 Fig. 2c,d)。在 cavity size 上,生成分子的原子数与 cavity size 之间呈现显著正相关(
另一方面,药效团特征与排斥球数量的增加则会使生成分子的多样性下降(见补充图 3),其中,药效团特征数量对多样性的影响最为显著,超过排斥球数量与 cavity size(见补充图 4),这凸显了药效团特征在化学空间映射中的核心地位。
总体而言,在药效团特征、排斥球设置与生成分子多样性之间保持适当平衡,是提升模型实际应用成功率的关键策略。
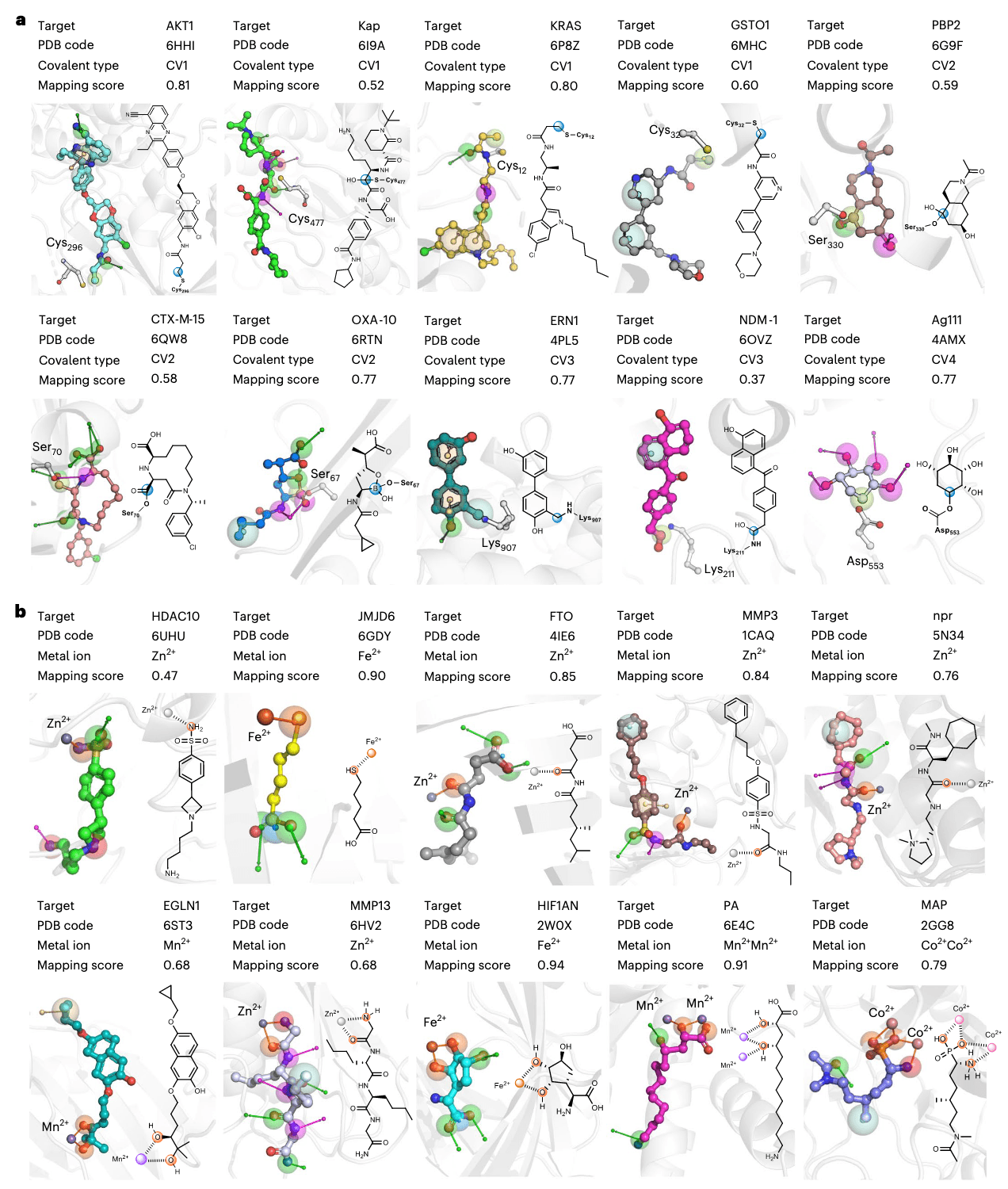
XXXXXX
图 3|PhoreGen 在共价药物与金属酶药物设计中展现出显著潜力:a 展示了针对十个具有共价反应位点的靶点所对应药效团模型生成的代表性三维分子,表明 PhoreGen 能够成功生成含有合理共价基序的分子,图中以蓝色球体标注共价原子;b 展示了针对十个金属酶靶点药效团模型所生成的代表性三维分子,体现了 PhoreGen 具备生成金属结合基团的能力,金属结合原子以橙色球体表示。图中各原子颜色编码为:硼(粉色)、氮(蓝色)、氧(红色)、磷(深橙色)、硫(金黄褐色)、氯(浅绿色),其余颜色均表示碳原子。
XXXXXX
2.3 从头药物设计(De novo drug design)
本节对 PhoreGen 在从头药物设计任务中的表现进行了评估,并与九种基线方法进行了对比,其中包括七种基于三维结构的分子生成模型(AR、Pocket2Mol、DeepICL、TargetDiff、DiffSBDD、PMDM 和 DrugFlow),以及考虑分子形状、电势表面和药效团信息的 ShePhERD 模型与基于药效团的 SMILES 生成模型 PGMG。
作者选取了 10 个未出现在训练集中的蛋白靶点,这些靶点与训练集在序列相似性(平均为 37%)和三维药效团相似性上都较低(见补充图 5),但具备明确的蛋白–配体相互作用模式(见补充图 6)。针对每个靶点,使用不同模型生成 100 个分子,并评估了采样有效率、药物相似性评分(QED)、合成可及性、对接评分(docking score)、相互作用指纹(IFP)相似性以及构象质量等指标(详见“评估指标”章节)。
PhoreGen 在这十个靶点上的平均采样有效率为 91%,显著优于扩散类模型(如 TargetDiff 和 ShEPhERD)(见表 1)。在 QED、合成可及性、Lipinski 法则与 logP 等方面,PhoreGen 表现与基线方法相当,表明其能够生成具有良好药物相似性与合成可行性的分子。
结构多样性方面,PhoreGen 与 AR、PMDM、DrugFlow 和 ShEPhERD 表现相当,略低于 Pocket2Mol、DeepICL 与 DiffSBDD,部分原因在于其采用了更严格的药效团约束(见表 1)。值得注意的是,PhoreGen 能够有效避免生成不良结构,如分子量过低或尺寸与靶点适配性差的分子,这一点是许多基于结构的生成方法难以克服的问题(见补充图 7)。这一优势得益于其所引入的分子规模预测模块(见 Fig. 1d)。
在计算效率方面,PhoreGen 平均仅需 919 秒即可为十个靶点各生成 100 个有效分子,具备可接受的运行效率(见表 1)。
作者进一步使用 AutoDock Vina 1.2.2 对所有模型生成的分子与各靶点进行对接分析,以确保公平比较。结果显示,PhoreGen 在对接评分上表现最佳,平均对接评分达到 −8.03 kcal/mol,优于所有基线方法(见表 1 和补充图 8),表明其更有可能生成高结合亲和力的候选分子。
通过对相互作用指纹(IFPs)的分析,PhoreGen 生成的分子与靶点的平均 IFP 相似性为 0.66,显著高于其他模型,如 AR(0.19)、Pocket2Mol(0.08)、DeepICL(0.40)、TargetDiff(0.39)、PMDM(0.11)、DiffSBDD(0.56)(见表 1 和补充图 9)。虽然 ShEPhERD 模型(使用与 PhoreGen 不同定义的方向性药效团)也能结合分子形状与静电特性,取得了相近的 IFP 相似性(0.63),但整体仍略逊一筹。
这些结果结合对高评分分子的分析(见补充表 22),说明 PhoreGen 所生成的分子更贴近常见的靶点相互作用模式,具有更高的活性潜力。另外,IFP 相似性与化学多样性的分析表明,高 IFP 相似性并未影响分子的结构多样性(见补充图 10)。
通过 PoseBusters 工具对构象质量的评估显示,PhoreGen 的整体构象成功率为 0.74,显著优于 AR(0.45)、DeepICL(0.35)、TargetDiff(0.29)和 DiffSBDD(0.56)等三维生成模型(见表 1)。虽然 Pocket2Mol 在构象上成功率最高(0.91),但其生成了大量体积偏小的分子(见补充图 7),这类分子生物活性有限,对接亲和力与 IFP 相似性均较低(见表 1 与补充图 8、9)。PMDM、DrugFlow 和 ShEPhERD 分别达到了 0.77、0.81 与 0.88 的构象成功率。
进一步分析生成分子的环结构分布发现,PhoreGen 在低频环结构(如三元环 3.3%、四元环 1.4%、八元环 0.3%、九元环 0.1% 以及多环结构 4.9%)方面出现频率较低(见补充表 23),表明其在控制环结构化学与能量合理性方面表现良好。
此外,在 CrossDocked2020 测试集(包含 100 个靶点)上的扩展测试也验证了 PhoreGen 的稳定性,尤其在结合亲和力与 IFP 相似性方面表现出色,竞争力优于多数基线方法(见补充表 24)。不过,对于某些具有非典型或复杂药效团模型的靶点,PhoreGen 的表现稍显逊色,这类靶点通常需要更加结构特异性的生成分子。
作者注意到 CrossDocked2020 测试集在化学空间、药效团空间及配体–药效团分布上与训练集存在差异(见补充图 11),暗示未来通过引入更多结构多样化的配体–药效团对用于训练,有望进一步提升模型泛化能力与实际适应性。

XXXXX
图 4|PhoreGen 在新型 MBL/SBL 双重抑制剂发现中的应用 a 显示了与药效团模型高度匹配的生成三维构象。考虑到化学合成的可行性,最终选择了化合物 CB1 进行合成,该分子与生成分子编号 157(见补充图 13–15)具有相同的硼酸酯核心骨架和等排体侧链。b 展示了 CB1 与 CB2 对临床相关 SBL 酶(KPC-2 和 OXA-48)及 MBL 酶(NDM-1、VIM-2 和 IMP-1)的 IC₅₀ 曲线。数据以平均值 ± 标准误(s.e.m.)表示,来自 3 组生物复重复实验(n = 3),误差条代表 s.e.m.。c 为 OXA-48 与 CB1 的复合物晶体结构视图(PDB 编号:9KSA),显示抑制剂与催化残基 Ser70 形成共价键,并与 Arg250 形成离子相互作用,同时与多个残基形成氢键,包括:Ser70:
XXXXX
2.4 特征定制的药物设计
在以靶点为中心的药物发现中,设计具有特定药效团特征的分子(如锚定位点、共价基团、金属配位结构等)往往至关重要。本节评估了 PhoreGen 在特征定制型药物发现中的适应能力,重点聚焦于共价药物与金属酶靶点药物的设计。
在共价药物设计方面,研究者根据参与共价反应的化学基团类型与靶点残基(主要参考 CovalentInDB 2.0 数据库,见补充表 3),定义了 4 种共价特征。用于训练的数据包含 674,507 对配体来源的“反应前”配体–药效团对 与 635 对复合物来源的“反应后”配体–药效团对。在评估阶段,作者针对 10 个包含共价反应位点的靶点(未出现在训练集中),每个靶点生成 100 个分子。
结果显示,PhoreGen 所生成分子中有 平均 76% 含有合理的共价基序,平均映射分数为 0.64(见补充表 25 和 26)。值得强调的是,大多数生成的共价基序呈现“反应后”状态(见 Fig. 3a),说明在训练中引入复合物来源配对数据可有效引导模型学习反应后构象,从而提升模型对真实化学过程的建模能力。
在金属酶药物设计方面,训练数据包含 1,659,215 对配体来源 和 1,686 对复合物来源 的配体–药效团对,均含有金属配位特征。在测试阶段,PhoreGen 针对 10 个金属酶靶点(训练集中未见)进行分子生成,结果显示 超过 95% 的生成分子包含合理的金属配位结构,且具有较高的药效团匹配分数(见补充表 27 和 28)。
不仅如此,PhoreGen 还能为结构复杂的金属酶靶点(如 PA 与 MAP,见 Fig. 3b)生成化学合理且多样化的金属结合基序,即便这些酶含有多配位位点或多个金属离子。相比之下,现有的基于结构的分子生成(SBMG)方法在这些目标上难以有效生成包含共价或金属结合特征的分子。
以上结果凸显了 PhoreGen 在共价药物与金属酶药物设计中的应用潜力,展现了其作为面向药效团的三维分子生成方法在特征定制型药物发现任务中的独特优势。
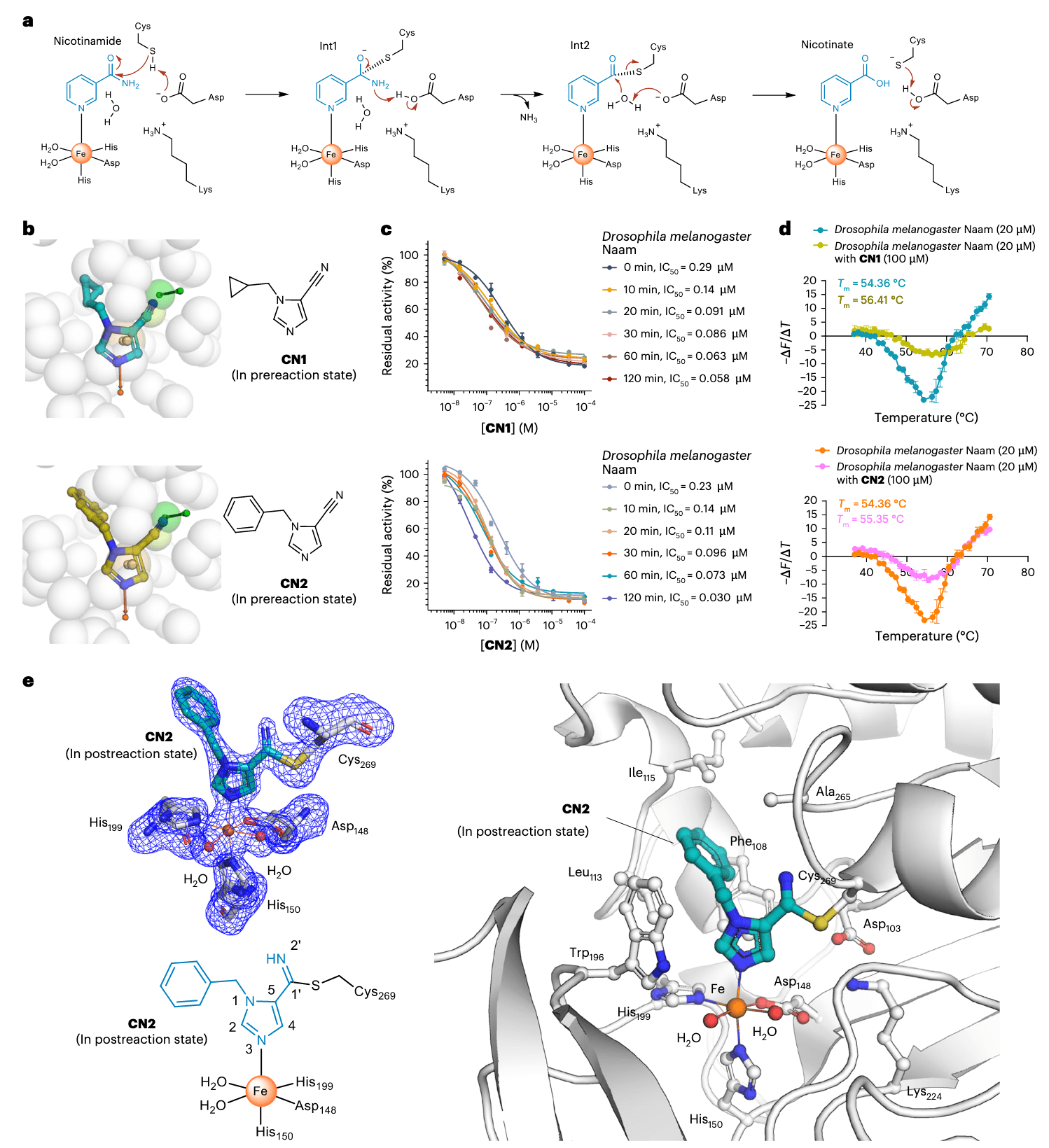
XXXXX
图 5|PhoreGen 的应用促成了 Naam 共价抑制剂的发现 a 展示了 Naam 催化烟酰胺(nicotinamide)转化为烟酸(nicotinate)的化学反应机制,此机制构成了药效团模型构建与分子生成的理论基础。其中 Int1 与 Int2 表示催化路径中的两个中间体。b 在 PhoreGen 所生成的 300 个候选分子中(见补充图 17–19),筛选出两种具代表性的化合物 CN1 与 CN2 用于合成,它们的 腈基结构(cyano group)可作为共价反应位点,与催化性半胱氨酸形成共价键。c CN1 与 CN2 对果蝇(Drosophila melanogaster)Naam(15 nM)的 IC₅₀ 曲线表明,两种化合物均表现出时间依赖性抑制作用,推测是由于其腈基与活性位点 Cys 残基发生了共价反应。数据为 3 次生物重复实验的平均值 ± 标准误(s.e.m.),误差条表示 s.e.m.。d 展示了果蝇 Naam 蛋白(20 μM)在存在或不存在 CN1(100 μM)或 CN2(100 μM)条件下的解链曲线(解链一阶导数),结果表明两种化合物均能结合并热力学稳定酶结构。数据同样为 n = 3 的平均值 ± s.e.m.。e 展示了 CN2–果蝇 Naam 复合物的晶体结构图(PDB 编号:9U8M),显示 CN2 与 Fe²⁺ 形成配位键(N3–Fe²⁺ 距离为 2.1 Å),并与 Cys269 形成共价键(C1′–S_Cys269 距离为 1.8 Å)。此时 CN2 呈现“反应后”状态,表明其已被 Cys269 亲核攻击。蓝色网格为 mFo–DFc 电子密度图(OMIT map,3σ 等高线),显示 CN2 与其配位残基均被可靠建模。CN2 的预测结合构象与晶体结构的均方根偏差(r.m.s.d.)仅为 0.21 Å,验证了模型预测的准确性。图中原子颜色编码为:氮原子(蓝色)、氧原子(红色)、硫原子(金黄褐色),其余颜色表示碳原子。
XXXXX
2.5 MBL/SBL 双靶点抑制剂的设计
对 β-内酰胺类抗生素的耐药性正日益加剧,其核心机制之一是进化后的金属 β-内酰胺酶(MBLs)与丝氨酸 β-内酰胺酶(SBLs)的广泛传播,这些酶能够高效水解 β-内酰胺骨架结构。目前,仅有极少数具备 MBL/SBL 双抑制活性的化合物处于临床试验阶段,其中 QPX7728 表现出对多种 MBL/SBL 同工酶在纳摩尔级别下的强效广谱抑制能力。
研究中,作者基于 QPX7728 与 MBL/SBL 酶复合物的晶体结构(见补充图 12)构建药效团模型,利用 PhoreGen 进行分子生成。在 300 个生成分子中(见补充图 13–15),最终筛选出两个新型双环硼酸酯化合物 CB1 和 CB2 进行合成(见 Fig. 4a)。
实验结果显示,这两种化合物对临床相关的 SBL 与 MBL 酶均表现出广谱抑制活性(见 Fig. 4b)。其中:
- CB1 对 SBL 靶点 KPC-2 与 OXA-48 的 IC₅₀ 分别为 0.031 μM 与 0.16 μM
- CB2 对 KPC-2 与 OXA-48 的 IC₅₀ 更优,分别为 0.11 μM 与 0.013 μM
此外,两者对 MBL 靶点 NDM-1 与 VIM-2 表现出强效抑制,对 IMP-1 也具有中等活性。
在结构层面,OXA-48:CB1 复合物晶体结构(PDB 编号:9KSA) 显示 CB1 可与催化位点的 Ser70 形成共价键,同时与 Arg250 形成离子作用,并与 Ser70、Ser118、Thr209、Tyr211 和 Arg250 形成多个氢键(见 Fig. 4c),该结合模式与 QPX7728 保持一致(见补充图 16),也与模型预测的结合构象高度一致。
细胞水平的抗菌活性评估显示,CB1 与 CB2 均可增强美罗培南(Meropenem)对耐药革兰阴性菌的杀菌效果,最高可将最低抑菌浓度(MIC)降低达 512 倍(见 Fig. 4d)。其中,CB2 尤具潜力,代表了抗碳青霉烯类耐药的新型先导结构。
2.6 共价 Naam 抑制剂的设计
Nicotinamidase(Naam)是一种依赖 Fe²⁺ 的酶,能够催化烟酰胺转化为烟酸,并已被鉴定为神经毒性杀虫剂的潜在靶点。目前,针对害虫 Naam 的小分子抑制剂几乎尚未有报道。
基于 Naam 的催化机制(见 Fig. 5a),研究者构建了一个药效团模型,包含四种关键特征:共价键特征、金属配位特征、芳环结构与氢键受体(见 Fig. 5b)。在使用 PhoreGen 生成的 300 个分子中(见补充图 17–19),作者筛选出两种 1H-imidazole-5-carbonitrile 类化合物 CN1 与 CN2(见 Fig. 5b),它们对果蝇(Drosophila melanogaster)Naam 显示出 时间依赖性的抑制活性,在孵育 120 分钟后,IC₅₀ 分别为 0.058 μM 与 0.030 μM(见 Fig. 5c)。
这两种化合物还能热力学稳定 Naam 蛋白结构,其中 CN1 的 ΔTm 为 2.05°C,CN2 为 0.99°C(见 Fig. 5d)。通过 液相色谱-串联质谱(LC–MS/MS)分析确认,CN1 与 CN2 均可与果蝇 Naam 的催化残基 Cys269 形成共价键(见扩展数据 Fig. 4)。
进一步通过 X 射线晶体学解析 CN2–果蝇 Naam 复合物的晶体结构(PDB 编号:9U8M,见补充表 29)表明,CN2 通过典型的八面体配位几何结构与 Fe²⁺ 形成配位键,并与 Cys269 形成共价键(见 Fig. 5e),与 LC–MS/MS 结果相符。预测结合模式与共晶结构的叠合结果显示出高度一致性,均方根偏差(r.m.s.d.)仅为 0.21 Å。
此外,CN1 与 CN2 还对害虫 Myzus persicae 和 Bemisia tabaci 的 Naam 酶表现出时间依赖性的抑制作用,并能热稳定这些酶(见扩展数据 Fig. 5)。
这些 共价型 Naam 抑制剂 展现出用于 新型杀虫剂开发的潜力,特别是在针对害虫中尚未充分开发的靶点上。
3 讨论
基于特定作用机制(mode of action)进行化学实体设计,是现代药物发现中的关键策略之一。许多获 FDA 批准的小分子药物,往往是天然产物、内源性配体或已有药物的衍生物或模拟物。然而,目前在特征定制型药物发现方面仍缺乏智能化的建模方法。
本研究系统探索了 纯药效团驱动分子生成方法(pure PBMG)的潜力,并开发出 PhoreGen 这一实用工具,迈出了通往智能药物设计的重要一步。实验结果表明,PhoreGen 综合了配体驱动(LBMG)与结构驱动(SBMG)方法的优势,适用于源自配体、蛋白质、复合物或催化机制等多种来源的药效团模型。
PhoreGen 所生成的分子在生物活性概率上具有明显优势,这在重对接分析与具体案例研究中得到了体现。虽然 ShEPhERD 模型同样取得了一定成果,依赖其引入的方向性药效团特征和额外的分子形状与电势学习,但类似 PharmacoBridge 等方法在缺乏共价、金属结合与排斥球等特征支持下,其适用性仍然受限。相比之下,PhoreGen 能高效生成具备共价结构、金属配位团或特征性官能团(如硼酸酯)的分子,体现出其在特征定制化设计中的独特潜力。
药效团模型本身的灵活性,尤其是使用集成药效团模型(ensemble pharmacophores),能进一步提升生成分子的质量、多样性与成功率。与固定子结构或骨架约束相比,PBMG 提供了更高的设计自由度,可支持更丰富的化学骨架与功能定制,这使得 PhoreGen 相较于 SBMG 和 LBMG 方法更具实用性与生成优势。
PhoreGen 通过对药效团特征的嵌入与扰动,有效处理了配体与药效团之间的映射偏差。模型将“配体–药效团映射规律”嵌入到消息传递机制中,能够精准捕捉药效团与三维分子构象之间的关键关系,从而提高采样效率。同时,模型还融合了化学键等结构知识,在原子与键的联合生成过程中显著提升了化学合理性。
此外,PhoreGen 利用药效团特征与排斥球所蕴含的空间信息来控制分子大小,降低生成失配尺寸化合物的可能性。这一性能的实现还得益于其高质量的配体–药效团对数据集与层次化训练策略。通过进一步扩展训练数据,PhoreGen 有望应用于更复杂的方向,如金属药物、放射性药物或富含特征官能团的分子(如糖类似物)设计。
进一步地,借助基于受体的药效团建模方法,还可实现**“无参考”药物生成(reference-free generation)**,即直接从靶点结构中提取药效团特征,从而支持无需已知配体的药物设计。
作为一个融合知识驱动与数据驱动机制的代表性模型,PhoreGen 为人工智能辅助药物发现奠定了坚实基础。在本研究中,PhoreGen 成功发现了新型双环硼酸酯(尤其是 CB2),作为 MBL–SBL 双重抑制剂,为应对碳青霉烯类耐药性提供了全新起点;同时也识别出针对 Naam 的共价抑制剂,为新型杀虫剂开发提供了可行性方案。
这些案例表明,PhoreGen 无论针对具有明确药效团特征的“经典靶点”,还是受催化机制或特殊结构引导的“新靶点”,均展现出良好的应用效果。
当然,PhoreGen 仍存在一些局限性,包括:在特征妥协匹配与精确化学构象之间的平衡尚不完美;在处理非典型药效团模型时生成合理化合物存在挑战;未显式考虑合成可行性问题等。
总体而言,该研究为智能药物发现提供了一种全新的有价值模式与多个成功案例,展现了知识驱动药物生成方法在未来药物开发中的广阔前景。