NMI 2025 | RXNGraphormer: 用于跨任务的反应性能预测与合成路径规划的统一预训练深度学习框架
随着人工智能在化学领域的持续推进,如何实现反应性能预测与合成路径规划的统一建模,已成为合成化学与计算科学交叉中的重要挑战。前者通常涉及回归任务,后者则依赖序列生成,两者在模型结构与训练策略上存在根本差异。为解决这一瓶颈,该研究提出了 RXNGraphormer —— 一个统一的预训练深度学习框架,能够同时执行反应性与选择性预测、逆合成分析以及正向产物预测任务。该框架融合图神经网络(GNN)与 Transformer 架构,并引入创新的 delta-mol 图结构以编码反应机制中的键变换信息。在超过 1300 万条真实与构造反应数据的预训练基础上,RXNGraphormer 在多个标准基准数据集和文献验证集上实现了当前最优性能(SOTA),并展现出卓越的泛化能力与反应类型识别能力。该研究不仅为建立更具通用性与解释性的化学 AI 模型提供了范式,也标志着结构–性能建模与合成设计任务在深度学习框架内的首次有效统一。
获取详情及资源:

0 摘要
人工智能正在重塑精准有机合成领域。以机器学习与深度学习为代表的数据驱动方法,在反应性能预测与合成路径规划方面展现出巨大潜力。然而,反应性能预测依赖数值回归,合成路径规划则基于序列生成,二者在方法论上的根本差异使得构建统一的深度学习架构面临重大挑战。该研究提出了 RXNGraphormer —— 一个通过统一预训练策略协同解决这两类任务的框架。该方法结合 图神经网络(GNN)用于分子内部模式识别,与 基于 Transformer 的模型用于分子间相互作用建模,并通过精心设计的训练策略,在 1300 万条反应数据上进行预训练。RXNGraphormer 在 八个反应性或选择性预测、正向合成或逆合成规划的标准基准数据集上均达到了当前最优性能(state-of-the-art),并在三个真实外部数据集上同样取得优异表现。尤为值得一提的是,模型生成的分子嵌入具有明确的化学意义,无需监督即可自动将不同类型反应聚类。这项工作打通了化学人工智能中反应性能预测与合成路径规划之间的关键壁垒,为反应预测与合成设计提供了一个强大而通用的工具。

1 引言
精准有机合成的四大基础支柱包括:反应性预测、选择性预测、逆合成分析,以及反应产物预测。其中,构建稳健的结构–性能关联模型是实现高准确性反应性与选择性预测的关键。逆合成任务旨在识别目标产物的可行前体,而反应产物预测则用于判断给定反应物能生成的最可能产物。
在早期的研究中,这两类任务往往依赖形式化的反应模板,这些模板通过子图模式匹配从专家系统(如 LHASA、Chematica)中提取。模板方法能够复用已有的反应规律,在模板匹配良好的情境下实现可靠预测,但一旦遇到超出模板库的新型反应,则预测能力明显受限。
近年来,以机器学习和深度学习为代表的数据驱动方法在反应预测中的广泛应用,已引发了该领域的重大变革。这些模型通过大量实验合成数据中挖掘出的统计规律,能有效预测多种转化反应中的关键因素,如反应性与选择性,显著提升了合成效率与质量。例如,已有研究将量子化学描述符与随机森林模型结合,或使用分子指纹作为特征输入,均实现了高精度预测。
除了基于描述符的传统方法,端到端的深度学习模型也展现出强大性能。例如,YieldBERT 利用预训练编码器从 SMILES 序列中提取分子表征,通过回归层实现产率预测;GraphRXN 则采用图神经网络(GNN)对每个分子编码,并融合所有分子嵌入来预测反应性能。
与此同时,深度学习方法也推动了计算辅助合成的快速发展,特别是在正向与逆合成路径规划方面。Segler、Jensen、Coley 等团队的工作推动了多种架构的发展,从基于模板的方法到去模板方法,不断拓展了合成规划的能力边界。
尽管深度学习在反应预测和合成设计上都表现出色,但由于回归预测与序列生成的计算方式差异显著,将这两个任务统一建模仍然极具挑战。目前,尽管已有研究尝试通过文本表示实现跨任务预测,但基于分子图的建模方法仍较为割裂 —— 许多工作各自独立地使用 GNNs 处理反应性能预测或合成规划,缺乏整合。
基于此,该研究提出:构建一个统一框架,结合分子图编码器与分子间交互编码器,同时搭配回归层与序列解码器,可望在一个系统内同时实现反应性能预测(涵盖反应性与选择性)与合成路径规划(包括逆合成与正向合成)。
为此,该研究提出了 RXNGraphormer —— 一个统一的反应预测框架,在多个方面实现了创新突破。该方法采用专用的反应编码机制,先通过图神经网络提取分子嵌入,再由 Transformer 编码各反应组分间的相互作用。模型在 1300 万条真实与构造反应数据上进行预训练,使其具备广泛适应性和表达能力。
此外,模型同时配备了 回归预测模块 与 序列生成模块,既可预测反应性能,也可输出 SMILES 格式的反应物与产物。在回归任务中,为提升性能,模型还引入了 “delta-mol” 图 —— 通过插值反应物与产物之间的键差,构建出一种包含反应机制信息的中间表示,无需借助任何量子化学计算,但却能更好捕捉反应本质。
实验证明,RXNGraphormer 在反应性、对映选择性(enantioselectivity)、区域选择性(regioselectivity)等多个任务上,均在 四个标准基准数据集与 三个真实文献数据集中取得了当前最优性能(SOTA)。同时,在多个合成路径规划任务(如 USPTO-50k、USPTO-full、USPTO-STEREO、USPTO-480k)中也保持了卓越表现。
更值得关注的是,该模型的预训练编码器具有出色的表示能力,能在无显式监督的条件下自动识别不同类型的反应,展现出对化学语义结构的深度理解。
这种多任务统一设计,使 RXNGraphormer 成为解决复杂反应预测问题的有力工具,兼具精度与效率,展示了跨任务深度学习在化学 AI 领域的巨大潜力。
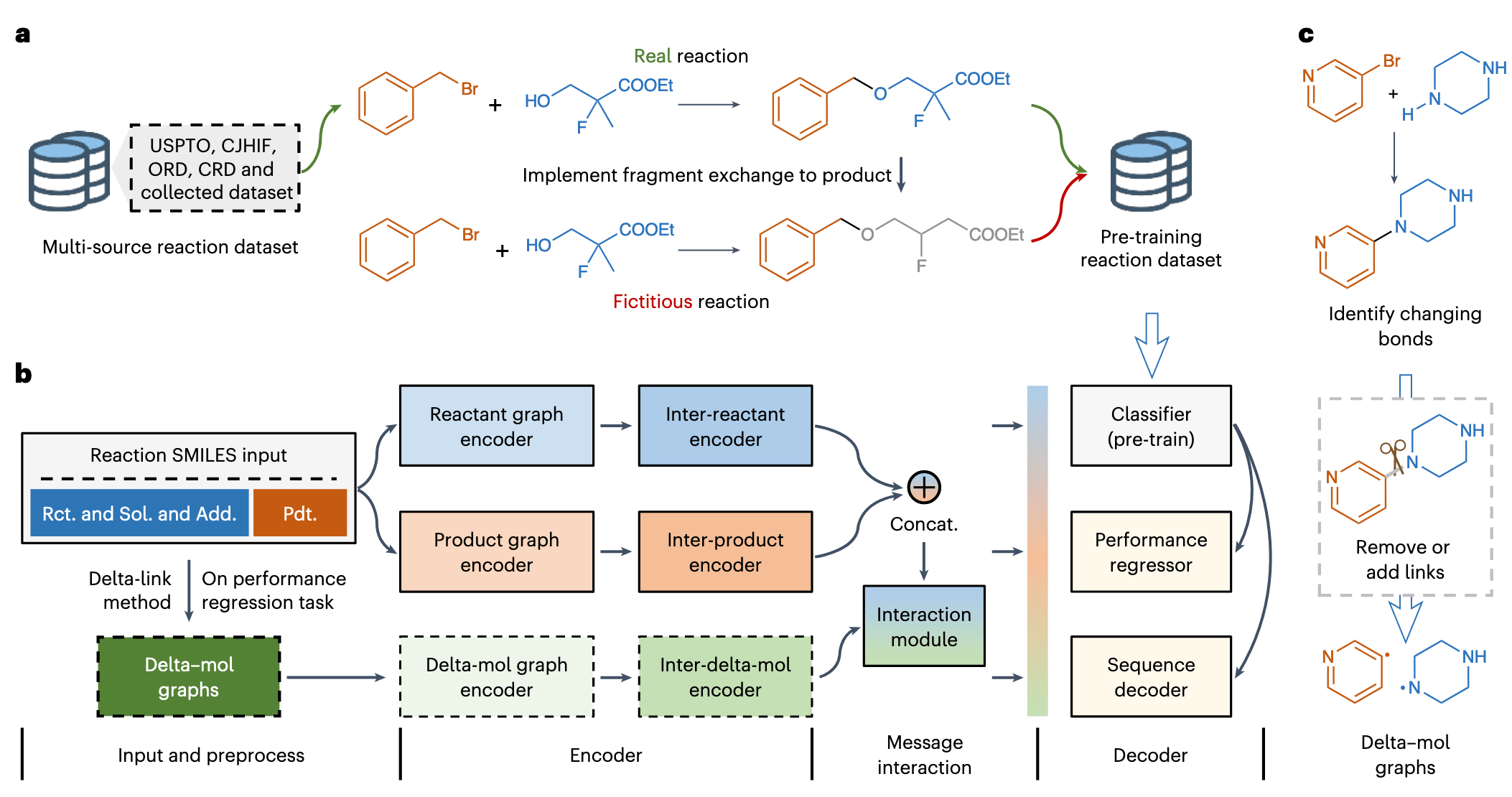
2 结果
2.1 RXNGraphormer 的设计
该研究认为,一个具有鲁棒性的预训练编码器能显著提升化学反应建模中跨任务预测的能力。为此,研究者构建了一个包含 超过 1300 万条化学反应记录的大规模数据集,并设计了基于对比学习的预训练策略。具体而言,该研究整合了多个开源数据库,并从世界知识产权组织(WIPO)数据库中精选出 逾 100 万条反应记录。数据集的详细信息见补充表 1。经过 SMILES 结构校验、标准化和去重处理后,最终得到约 680 万条高质量反应数据。
考虑到键变换是化学反应的基本过程,为了让模型在如此大规模数据中学习反应中的键变化模式,研究团队设计了一种 “片段置换算法”,用于对产物分子进行改造,生成具有错误键变化特征的负样本反应(见图 2a)。因此,预训练任务被设定为二分类对比学习问题,目标是区分真实反应样本与虚构负样本。
该片段置换算法被应用于超过 400 万个真实产物分子,生成数量相当的虚构反应样本(包含超过 500 万个算法生成的产物),使得整个数据集总量扩展至 1300 万条以上(图 2a)。关于片段置换算法的细节及虚假负样本的处理流程可参见方法部分。
本预训练任务旨在使模型学会区分真实与虚构反应,掌握有机转化过程中分子骨架保持与反应位点识别的规律,进而提升其对分子结构与反应特征的表达能力。值得一提的是,受 不变风险最小化(invariant risk minimization)理论启发,该研究选择生成与真实产物结构相似的虚构分子,而非更直接的方式(如引入反应物中未出现的原子等),以保证键变换规律成为预训练的主要学习目标。
基于上述预训练策略,研究团队构建了 RXNGraphormer 模型架构,支持分类、回归与序列生成等多种任务(图 2b)。模型采用经 Mole-BERT 改编的图神经网络(GNN)对反应中的各分子进行编码,捕捉其内在属性,生成类似自然语言处理中的“token embedding”的分子嵌入。随后,这些嵌入将被基于 Transformer 的交互编码器处理,用于建模分子之间的相互作用。
在分类预训练阶段,反应物与产物分别通过 GNN 与 Transformer 编码,并将两者的嵌入(包含交互信息)拼接为一个统一的反应表征向量,用于二分类任务中识别真实或虚构反应。此外,该架构可适应任意数量反应组分的建模,并使用 RDKit 对 SMILES 进行标准化,以降低分子排列顺序对模型性能的干扰。
为进一步提升模型在反应性与选择性预测任务中的表现,该研究不仅编码反应物与产物,还引入了描述键变化信息的 “delta-mol” 图结构。通过所提出的 “delta-link 方法”,比较反应物与产物之间的键变化,插值生成反应过程中的中间“帧” —— 即 delta-mol 图(图 2c)。这些图随后经过图编码器与交互编码器处理,将反应机制中的断键与成键过程显式纳入建模。
最终,delta-mol 嵌入与反应物/产物嵌入结合,经全连接交互模块整合后,形成完整的反应表示,并输入回归层以预测反应性能。
在逆合成任务中,模型仅使用产物侧的图编码器与交互编码器生成产物嵌入,随后通过 Transformer 解码器将其转化为对应反应物的 SMILES 序列(图 2b);而在正向合成任务中,则使用反应物侧嵌入预测产物的 SMILES 表达。
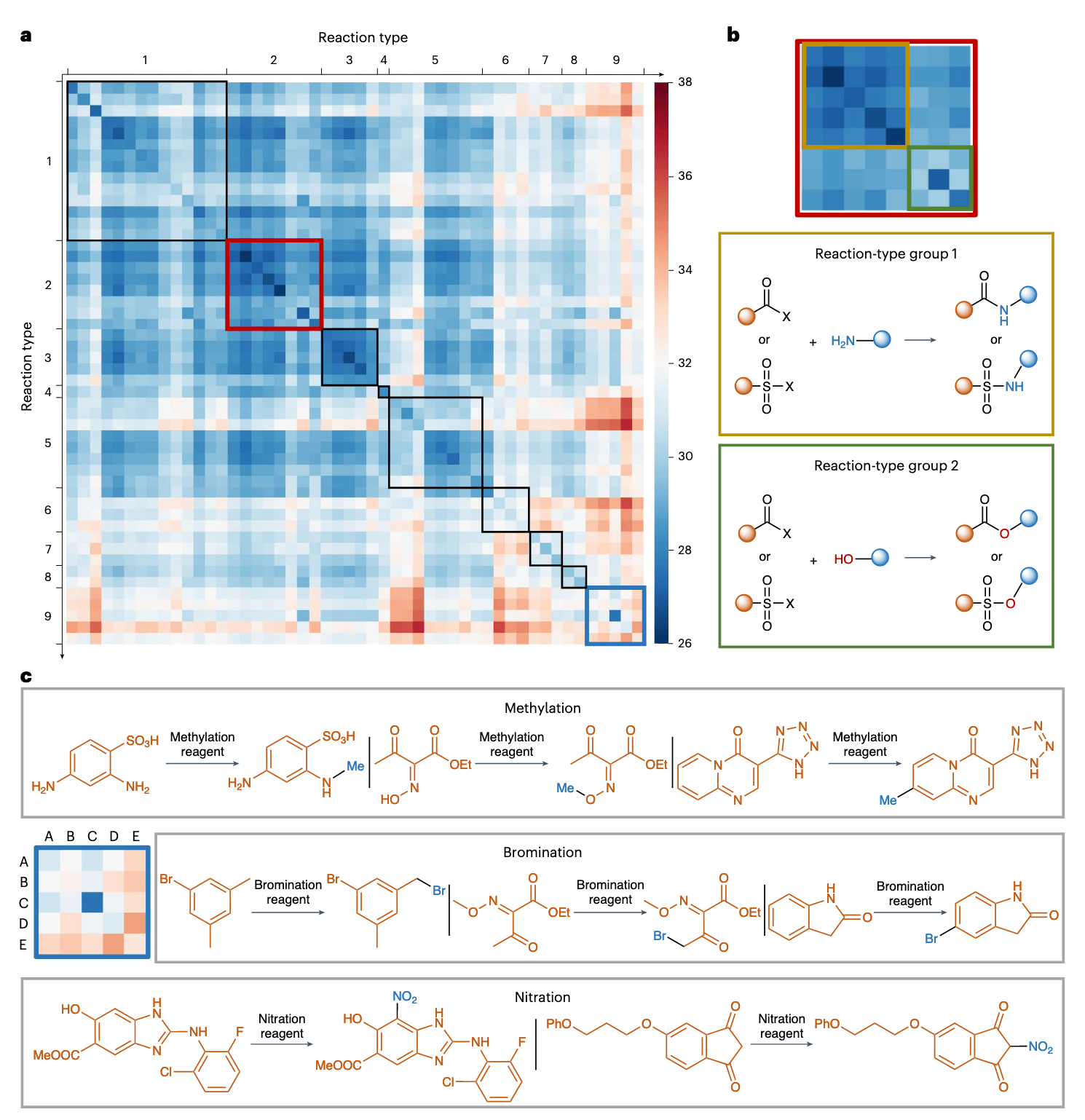
2.2 利用预训练嵌入区分反应类型
Schwaller 等人曾提出一种创新性的预训练任务,用于区分不同类型的化学反应。受此启发,该研究进一步探究所构建的预训练反应编码器是否也具备类似能力 —— 即是否能自动识别反应类型。由于化学家通常基于键变化对反应进行分类,而这一原则正与该研究的预训练目标键变换学习高度契合,因此具备理论上的可行性。
需要强调的是,该研究的预训练任务并未包含反应类型的任何标签信息,目标仅在于区分真实与虚构反应。因此,为验证模型能否内隐地区分不同反应类型,研究者未训练额外的分类模型,而是直接比较不同反应间嵌入的两两距离。
实验在 USPTO-50k 数据集上进行验证(该数据集中包含 50 类反应,每类 1000 条记录)。研究者提取了分类模型倒数第二层的嵌入表示,计算了不同反应类型之间的欧几里得距离,并据此构建了所有类型间的平均距离矩阵,最终可视化为热图(见图 3a)。关于这 50 类反应的具体信息与距离计算方法详见补充表 3。
在图 3a 中,热图中较冷(蓝色)代表模型潜在空间中反应类型之间距离较近,而较热(红色)则代表距离较远。对角线(从左上至右下)表示同类型反应之间的距离,因相似性高而呈现蓝色。图中以黑框标出的区域表示属于相似主类别的反应(例如 C‒C 键形成),呈现出明显的聚类相关性。
图 3b 展示了模型对不同反应间异同的分辨能力。例如热图左上角的反应类型组 1,代表一组高度相似的反应类型,而右下角的组 2也形成了另一独立聚类。深入分析发现,组 1 主要为酰胺和磺酰胺形成反应,组 2 则为酯和磺酸酯形成反应。这种聚类反映了其不同的反应性特征:羰基和磺酰基均具有类似的亲电性,因此其与不同亲核试剂(胺 vs 醇)反应时,分别形成酰胺/磺酰胺(组 1)和酯/磺酸酯(组 2),并在嵌入空间中表现出聚类分离,但两组之间的嵌入距离仍保持较小,体现出其官能团间电子特性的相似性。
值得注意的是,USPTO-50k 的分类体系存在主观性和粒度不均,导致热图对角线的“蓝度”并不均匀。特别是在对角线右下角区域出现明显异常偏热(红色),对应的反应主类别为官能团加成,其内部包含五个亚类(图 3c):溴化(A)、氯化(B)、Wohl–Ziegler 溴化(C)、硝化(D)与甲基化(E)。
其中,甲基化反应可分为 C‒N、C‒O 与 C‒C 键形成等多个子类,尤其 C‒C 与其他类型之间差异显著,因而在键转化角度上,甲基化与其他类型表现出内在差异性,导致其在嵌入空间中呈现出独立特征。
溴化类亚型包括苄位(如 Wohl–Ziegler)、α-羰基和芳香环溴化。虽然它们都涉及 C‒Br 键形成,但由于反应环境不同,导致亚类间仍存在一定差异。Wohl–Ziegler 属于苄位/烯丙位溴化,内部变异最小;而硝化反应在芳香环与羰基 α 位的反应位点之间也存在一定变异性。
这些分析结果表明,该研究提出的预训练策略能够有效引导模型学习键变化规律与反应电子效应特征,从而使模型具备无监督识别反应类型的能力。
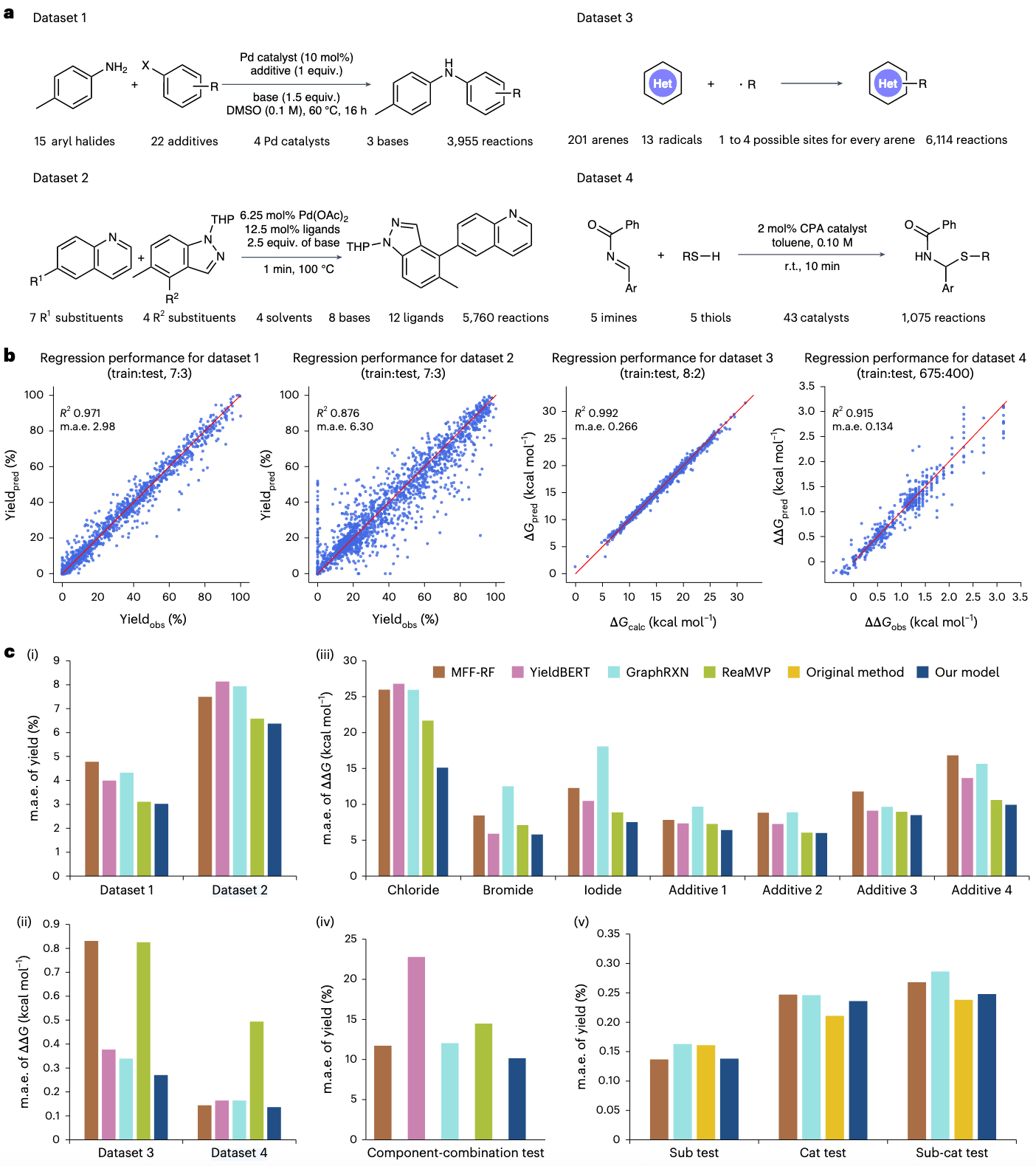
2.3 RXNGraphormer 的回归预测性能
该研究系统评估了 RXNGraphormer 在多个关键反应性能维度上的预测能力,包括反应性、区域选择性(regioselectivity)与对映选择性(enantioselectivity),并使用了一系列高质量、广泛认可的数据集进行测试。
在产率预测方面,分别使用了 Buchwald–Hartwig 反应数据集(图 4a 中的数据集 1)和 Suzuki–Miyaura 反应数据集(图 4a 中的数据集 2)。此外,还使用了自由基 C–H 官能化数据集(数据集 3,图 4a)与不对称硫醇加成数据集(数据集 4,图 4a),分别用于区域选择性和对映选择性的预测。除 C–H 官能化数据集来自高精度密度泛函理论(DFT)计算外,其余均来源于实验数据。
数据集按照原始文献或已有模型的比例进行随机划分为训练集与测试集,分别进行了 10 次预测实验。图 4b 展示了每个数据集的代表性测试结果。
整体来看,RXNGraphormer 在四个数据集上都表现出优异的预测能力。在产率预测(数据集 1 和 2)中,模型分别实现了
研究进一步将 RXNGraphormer 与多个代表性 SOTA 方法进行了比较,这些方法均无需额外的 DFT 计算,包括:
- 随机森林结合多种分子指纹特征(MFF + RF)
- 文本表示模型 YieldBERT
- 图神经网络模型 GraphRXN
- 结合二维与三维分子图的 ReaMVP
如图 4c(i)、(ii) 所示,在反应性、区域选择性与立体选择性的三个任务(数据集 2、3、4)上,RXNGraphormer 均明显优于其他模型,m.a.e. 分别为 6.37、0.270 kcal/mol 与 0.136 kcal/mol。在 Buchwald–Hartwig 反应的产率预测(数据集 1)中,RXNGraphormer 与专为产率任务优化的 ReaMVP 表现相当(m.a.e. = 3.02 vs 3.11)。
然而,虽然 ReaMVP 在产率预测上表现突出,但在两个选择性预测任务中,表现明显较弱。
为进一步验证模型的泛化能力,该研究在数据集 1 和 4 上执行了更具挑战性的样本外测试(OOS):
- 数据集 1 构建了基于卤代芳烃(氯、溴、碘)和添加剂的多个 OOS 测试集,并创建了一个涉及四个维度都未见过组分的组合测试集(图 4c(iii)、(iv))
- 数据集 4 按照文献 [10] 的方法划分为底物(sub)、催化剂(cat)和底物-催化剂联合(sub-cat)三个测试集
在数据集 1 的所有 OOS 测试中,RXNGraphormer 始终优于其他方法,即使在最难的组分组合测试集中,也维持了 m.a.e. = 10.12 的表现。
数据集 4 中,模型在 sub 测试集上与 MFF-RF 几乎持平(m.a.e. = 0.138 kcal/mol vs 0.137 kcal/mol),在 cat 与 sub-cat 测试集上表现更优。然而文献 [10] 中使用的平均空间占据(steric occupancy)描述符对未见催化剂结构的细微变化更敏感,因此在部分测试中性能优于 RXNGraphormer。
详细的模型对比指标见补充材料 3.2 节。这些结果充分证明了 RXNGraphormer 在建模结构–性能关系方面的强大泛化能力,尤其在面对新化合物或组合反应时的预测潜力。
此外,该研究还分析了模型潜在空间中反应性能标签的分布,详见补充材料 3.3 节。
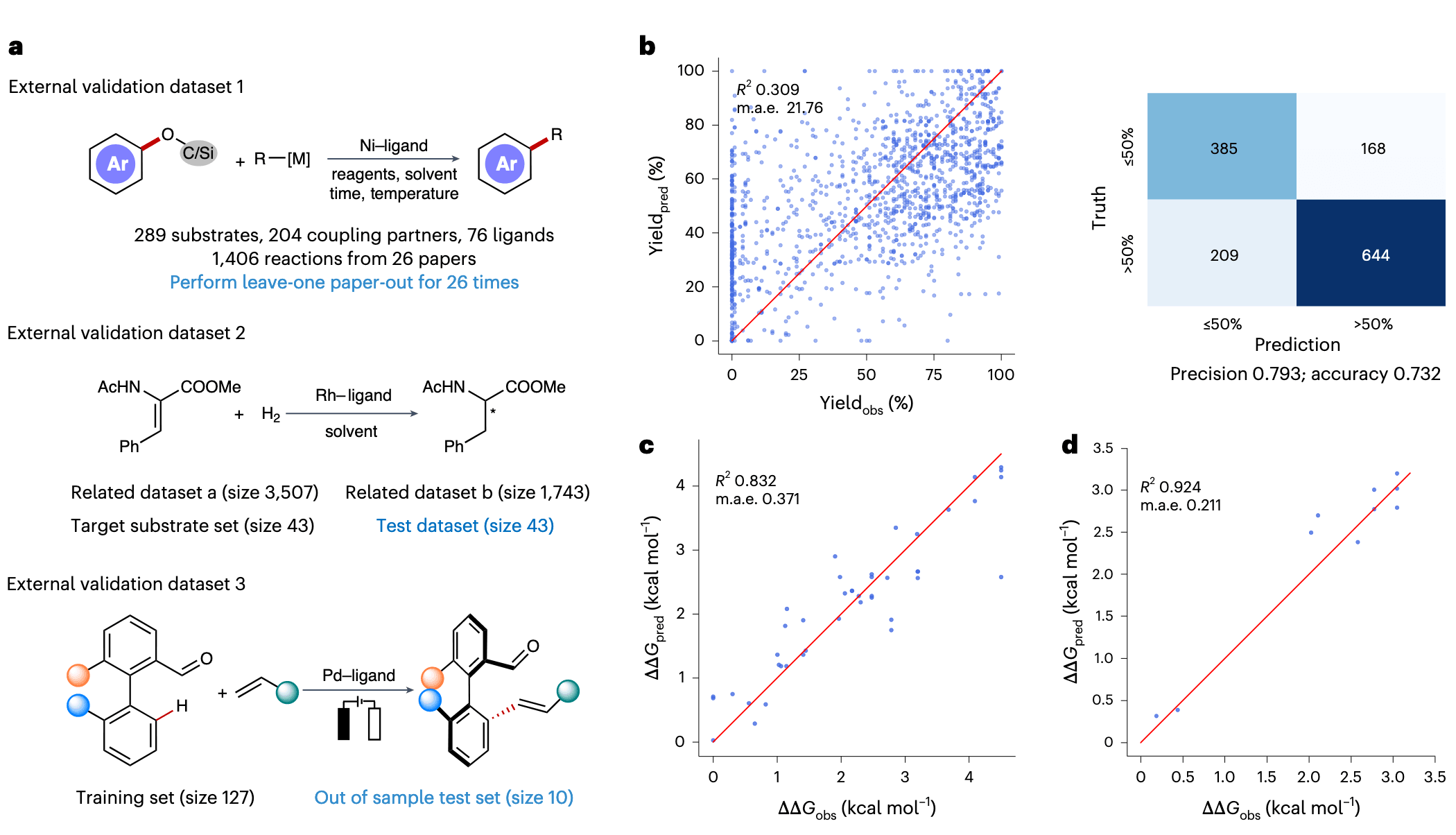
为验证模型在更贴近真实实验场景中的实用性,研究进一步在三个文献数据集中进行了测试(图 5a):
- 镍催化 C–O 偶联反应数据集
- 烯烃的不对称加氢反应数据集
- 钯电催化 C–H 活化反应数据集
在镍催化数据集上,采用与原文一致的 DOI 留出法进行 26 次验证,RXNGraphormer 直接产率预测的表现为:m.a.e. = 21.76,
在不对称加氢反应数据集中,按照原文对底物 (Z)-2-acetamido-3-phenylacrylate 的划分方式评估,RXNGraphormer 的预测结果为
在钯电催化数据集中,RXNGraphormer 对 10 个 OOS 测试样本的预测表现为:
这些结果说明:RXNGraphormer 不仅能在标准数据集上取得最优表现,也具备在真实应用环境中进行准确预测的能力。
2.4 RXNGraphormer 在合成路径规划中的表现
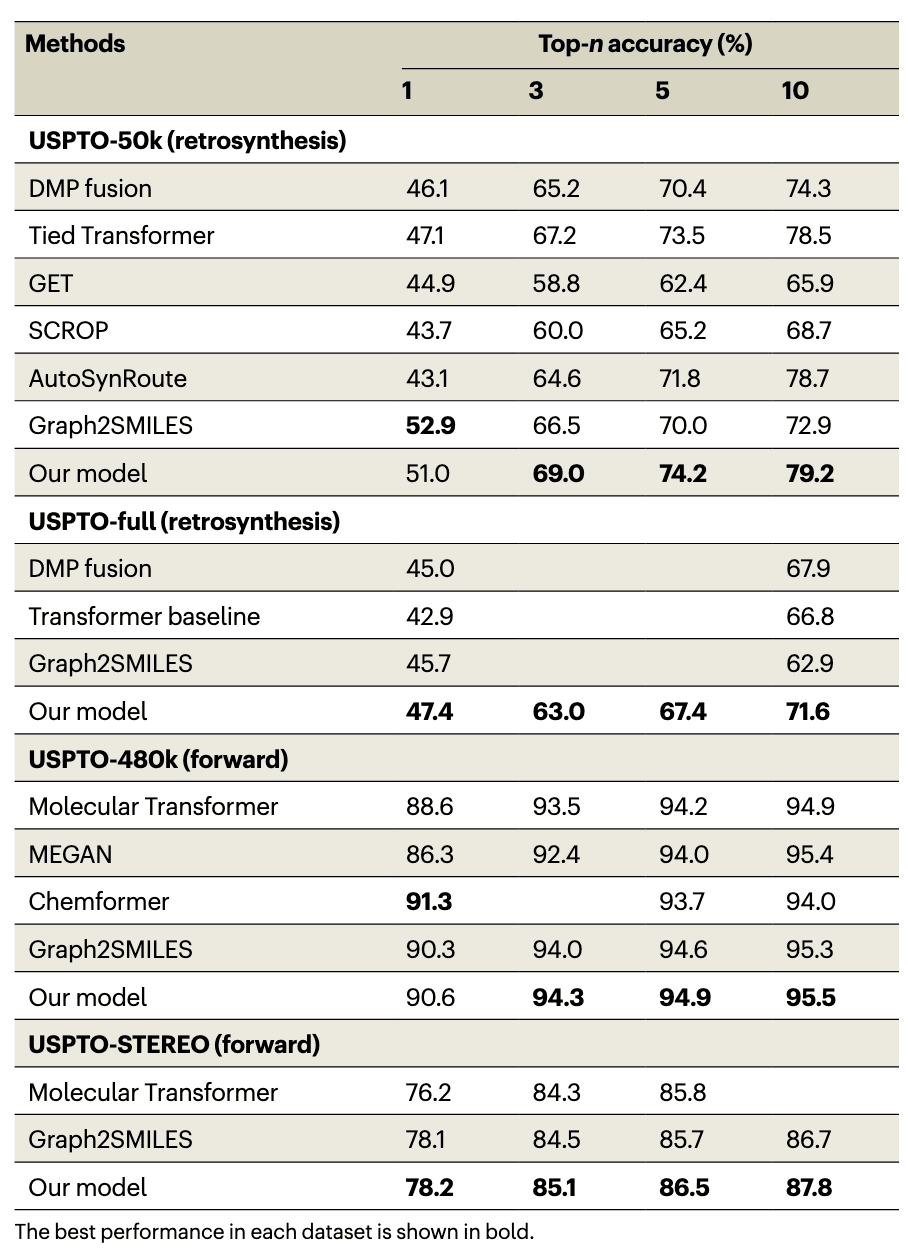
为评估 RXNGraphormer 在单步反应预测任务中的性能 —— 这是多步合成路径规划的基础模块 —— 该研究与多个 SOTA 模型进行了对比分析,并在四个来自 USPTO 数据库 的反应数据集上分别进行独立评估(见表 1)。
为保持与其他研究的一致性,这些数据集均按既有文献中使用的比例随机划分为训练集、验证集与测试集。其他模型的 top-n 准确率指标直接引用原始论文数据。
在逆合成任务中,为了确保公平比较,该研究排除了使用 模板策略、原子映射(atom mapping)或 SMILES 增强技术 的方法,因为这些技术会独立于模型架构提升性能,从而掩盖模型本身的能力【19】。
在 USPTO-50k 数据集上,该研究将 RXNGraphormer 与六个代表性模型进行了对比,包括:
- DMP fusion
- Tied Transformer
- GET
- SCROP
- AutoSynRoute
- Graph2SMILES
其中,Graph2SMILES 的 top-1 准确率为 52.9%,略高于 RXNGraphormer 的 51.0%。但在 top-3、top-5 和 top-10 准确率上,RXNGraphormer 全面超越其他模型,分别达到 69.0%、74.2%、79.2%。
在规模更大、噪声更多的 USPTO-full 数据集上,RXNGraphormer 在 top-1 到 top-10 的所有准确率指标上均超越了 DMP fusion、Transformer baseline【27】和 Graph2SMILES 等代表性模型,创下新的 SOTA 水平。未使用任何性能增强手段的前提下,top-1 准确率从 45.7% 提升至 47.4%,top-10 准确率从 67.9% 提升至 71.6%。
在正向合成任务中,研究对比了 RXNGraphormer 与 Molecular Transformer【20】、MEGAN【24】、Chemformer【26】与 Graph2SMILES,在 USPTO-480k 上的表现。RXNGraphormer 的 top-1 准确率为 90.6%,略低于 Chemformer 的 91.3%,但在 top-3、top-5 与 top-10 准确率上均优于所有模型。
在更具挑战性的 USPTO-STEREO 数据集上,该数据集包含复杂的立体化学信息,模型需处理更高维度的结构特征。RXNGraphormer 相比 Molecular Transformer 与 Graph2SMILES 取得了 SOTA 结果,在所有四个 top-n 准确率指标上均优胜,具体为:78.2%、85.1%、86.5%、87.8%。
这些结果表明,得益于统一的反应预测框架与创新的预训练–微调策略,RXNGraphormer 不仅在基于回归的反应性能预测中表现出色,也在大多数场景下超越了当前最先进的合成路径规划模型,无论是正向预测还是逆合成设计。
3 讨论
总而言之,该研究提出了 RXNGraphormer,一个统一的深度学习框架,能够同时实现 反应性预测、选择性预测、逆合成路径规划与正向产物生成 等关键任务。
为实现这一目标,研究构建了一个包含 超过 1300 万条反应数据 的预训练数据集,该数据集结合了精心整理的真实反应与通过 自定义“片段置换算法”合成生成的虚构反应,旨在训练模型有效区分真实与虚构反应。这一大规模预训练过程赋予 RXNGraphormer 丰富的分子与反应表示能力,随后通过微调被用于多个下游任务。
具体而言,微调后的 分子与反应编码器 被应用于反应性能预测任务,并结合 delta-link 方法 引入的 delta-mol 图结构,用于捕捉关键的反应机制中间态;而在逆合成与正向合成预测中,仅需使用预训练的分子编码器即可完成任务。
这一统一深度学习框架在多个反应数据集上经过严格验证,表现出强大的预测能力。在 Buchwald–Hartwig、Suzuki–Miyaura、自由基 C–H 官能化 与 不对称硫醇加成 数据集中,RXNGraphormer 无论是在随机划分的测试集还是未见分子的 OOS 测试集中,均在反应性与选择性预测方面超越其他模型,表现出更高的准确性。
此外,模型还在三个真实文献数据集上进行了外部验证,进一步证明其在更具实际挑战性的化学场景中依然具备出色的预测能力。
在四个 USPTO 数据集上,RXNGraphormer 在逆合成与正向产物预测任务中均展现出优异表现。尤其在包含近 100 万条反应的 USPTO-full 数据集中,RXNGraphormer 显著领先于所有现有方法,确立了其作为大规模反应预测高精度解决方案的地位。
除了卓越的预测性能之外,该研究精心设计的预训练策略还赋予模型识别不同反应类型的能力。由于该任务使模型能学习到化学键变化模式,而这正是大多数化学家进行反应分类的依据,因而预训练模型可在潜在空间中自然聚类出结构上差异显著的反应类型。
这一特性也使得 RXNGraphormer 不再是一个不可解释的“黑箱”,而是向“灰箱”模型迈进,使其更具可解释性。
通过建立一个贯穿多个关键任务的统一架构,RXNGraphormer 打通了反应性能预测与合成路径规划之间的关键断层,为数据驱动的合成转化设计提供了强大、灵活的解决方案。