arXiv 2025 | Protenix-Mini: 高效结构预测器——紧凑架构、少步扩散与可切换pLM
该研究提出 Protenix-Mini,通过紧凑架构与2 步 ODE 采样显著降低生物分子结构预测的计算成本,仅损失 1–5% 精度。作者发现 AF3 模型存在冗余模块,可裁剪 Pairformer 与 Diffusion Transformer 块,并用 ESM 蛋白质语言模型替代高开销的 MSA 模块。实验表明,Mini 在 RecentPDB 与 Posebusters 上表现接近完整模型,Tiny 成本降约 85%。ESM 版本在大部分任务中接近 MSA,但蛋白–蛋白界面预测显著下降。比较 EDM 与 Flow Matching,性能差异极小。针对少步 ODE 的塌陷与碰撞问题,调整步长策略可提升质量并保持多样性。未来将探索稀疏注意力、更优架构及结构增强型 pLM 预训练,实现长序列与多组分体系的高效预测。

获取详情及资源:
0 摘要
轻量化推理对于生物分子结构预测及其他下游任务至关重要,它能够在大规模应用中实现高效的实际部署与推理阶段的可扩展性。本研究针对模型效率与预测精度之间的平衡,提出了几项关键改进:首先,将多步的 AF3 采样器 替换为仅需少量步骤的 常微分方程(ODE)采样器,显著降低了推理时扩散模块的计算开销;其次,在开源 Protenix 框架中发现,一部分 pairformer/diffusion transformer 模块对最终结构预测并无显著贡献,因此可进行架构裁剪与轻量化重设计;最后,引入 ESM 模块 取代传统的 MSA 模块,从而减少多序列比对的预处理时间。
基于这些核心洞见,研究提出了 Protenix-Mini——一种紧凑且优化的高效蛋白质结构预测模型。该模型采用了更加高效的架构设计与两步ODE采样策略,在去除冗余Transformer组件并精简采样流程的同时,显著降低了模型复杂度,仅带来极小的精度损失。在基准数据集上的评估表明,与完整版本相比,其性能下降仅为 1–5%,但推理速度与资源利用率得到大幅提升。这使得 Protenix-Mini 特别适合于计算资源受限但仍需高精度结构预测的应用场景。
1 高效采样与轻量化设计的核心思路
- 少步ODE的高效采样
近年来,基于分数的生成模型研究表明,扩散过程可用常微分方程(ODE)进行近似,从而实现确定性采样,跳过传统随机微分方程(SDE)求解器中的随机性。分析发现,无论是使用 EDM 框架(如 AlphaFold3)还是 flow matching 框架训练的模型,都对采样步数的大幅削减表现出惊人的鲁棒性。仅使用2步ODE采样,依然能够生成结构精确的蛋白质构象,挑战了“高保真采样必须依赖几十次迭代”的传统认知。
- 冗余组件的压缩
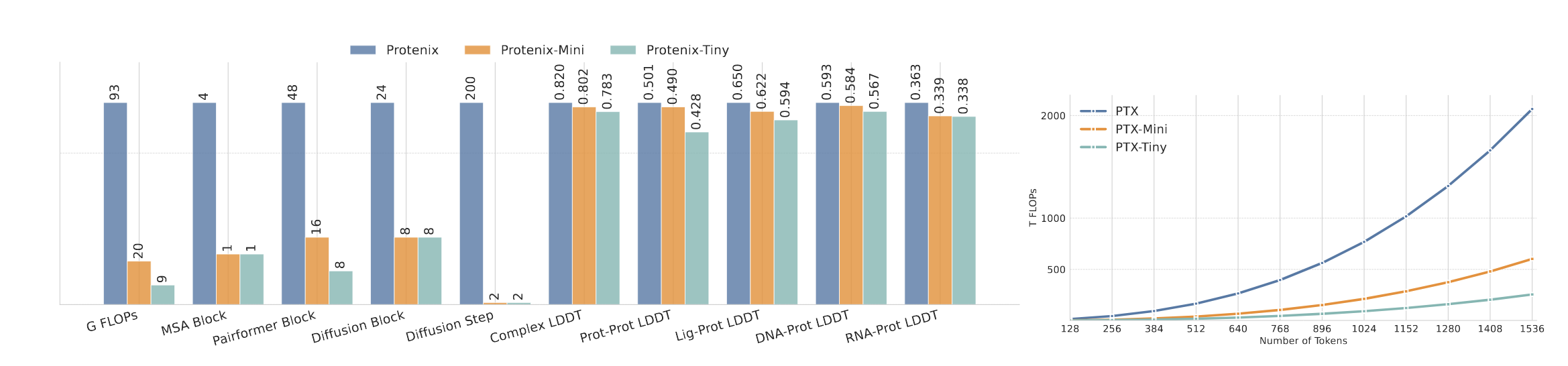
在对开源 Protenix 框架(基于扩散的结构预测器)分析时发现,其部分 pairformer/diffusion transformer 模块对最终预测结果几乎没有贡献。实验证明,在后训练阶段推理时直接移除这些模块,只会带来轻微的性能下降。通过识别并删除这些非关键组件,重新配置模型架构,可有效降低计算量。正如图1所示,在不同token数量和MSA规模下,FLOPs均有明显下降。
- 用pLM替代MSA模块
考虑到MSA搜索与计算模块的高计算成本,提出用**预训练蛋白质语言模型(pLM)**生成的嵌入替代MSA特征,以降低开销。具体实现中,使用 ESM2-3B 模型提取蛋白质表示,完全跳过耗时的MSA预处理与计算步骤。
基于以上洞见,提出了Protenix-Mini:在减少Transformer模块数量、采样步数(如1–2步)的同时,实现了对复杂生物分子结构的高效预测。实验结果表明,在 interface LDDT、complex LDDT、ligand RMSD 成功率 等指标上,性能下降仅为 1–5%,但推理速度与资源效率大幅提升。
这一成果证明了轻量化模型在生物分子结构预测中可与大型模型媲美。通过同时优化采样效率与架构开销,Protenix-Mini 向高保真结构预测的普及化迈出了一步。未来,作者计划结合架构设计、蒸馏、量化等方法进一步提升推理效率。
2 推理流程概述
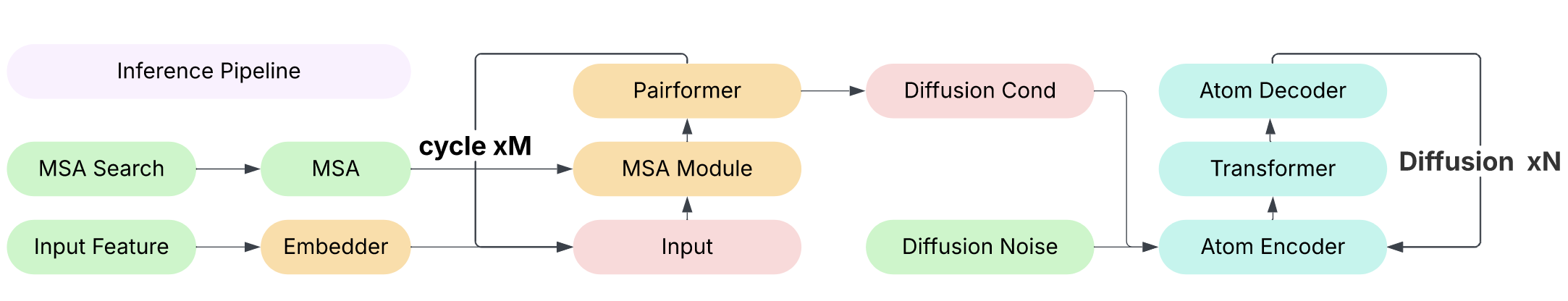
本节简要介绍了 AF3 风格结构预测模型(如 [1, 20, 21, 24])的架构与推理步骤,其整体流程(见图2)由**条件编码(Conditioning)与扩散生成(Diffusion)**两个核心阶段组成。
① 条件编码(Conditioning)
模型首先计算条件信号。设输入序列为
这两个表示将作为后续扩散模块的条件输入。
② 扩散生成(Diffusion)
扩散过程通过逐步去噪,将初始随机样本生成目标的三维原子结构。设
其中,正向过程通过逐步加噪扰动真实结构,反向过程则利用训练好的去噪器恢复原始信号。
在 AF3 中,这一过程由采样算法实现(见算法1),遵循 EDM 框架 [6]:在每一步迭代中,按照预设的时间表向当前样本

3 轻量化结构预测器
3.1 通过采样器配置实现少步扩散
在基于扩散的结构生成中,最大的性能瓶颈之一是长采样轨迹带来的高计算开销。AF3 风格模型默认使用多达 200 步的采样器 [1]。然而,这样的长采样计划在推理时是否真的必要?
令人意外的是,答案是否定的——只要采样算法配置得当,采样步数就可以大幅减少而不显著影响性能。研究发现,即使在无需重新训练的情况下,AF3 风格模型在采样步数大幅缩减时依然表现出极强的有效性。例如,200 步采样可以直接替换为仅 2 步 ODE 采样,仍能获得接近完整版本的预测效果;甚至在单步推理下,也能给出合理的结构预测,仅伴随轻微的性能下降。
这种结果颠覆了“高保真生成必须依赖长迭代链”的传统认知,为扩散模型的高效推理提供了直接可用的途径。
在直接将默认的 AF3 采样器 用于较少推理步数时,实验发现当步数低于 10 时,模型性能会急剧崩溃——生成的结构往往残缺或不合理。
进一步分析表明,这种退化并非源于模型本身的能力不足,而是由于默认采样算法并不适合少步推理。通过对采样过程的系统分析,研究找出了两项简单但关键的改动,使得少步推理成为可能:
- 切换为 ODE 采样器:将
,从而移除额外的噪声注入; - 设置步长比例
:确保与底层基于速度(velocity-based)的公式保持一致性。
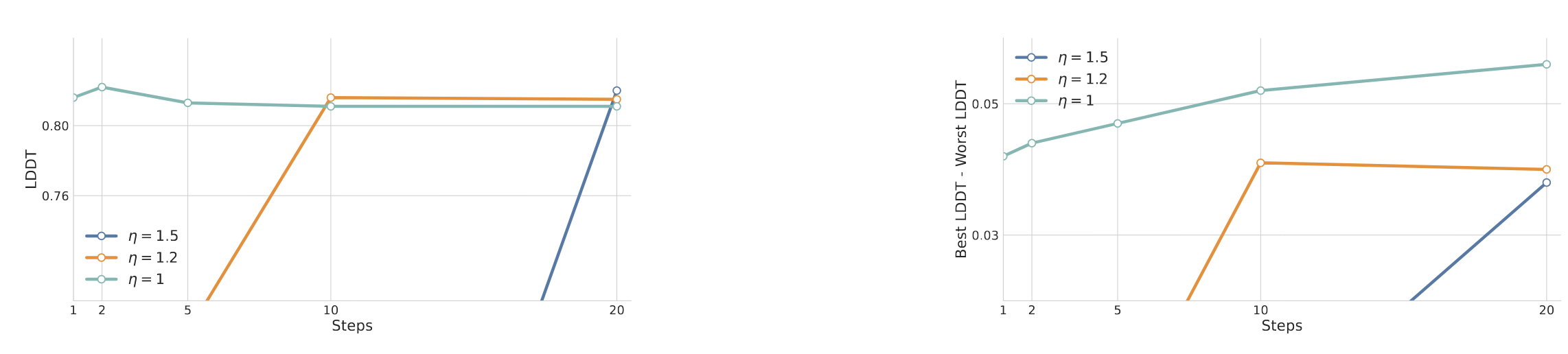
影响少步推理性能的核心因素之一是步长比例
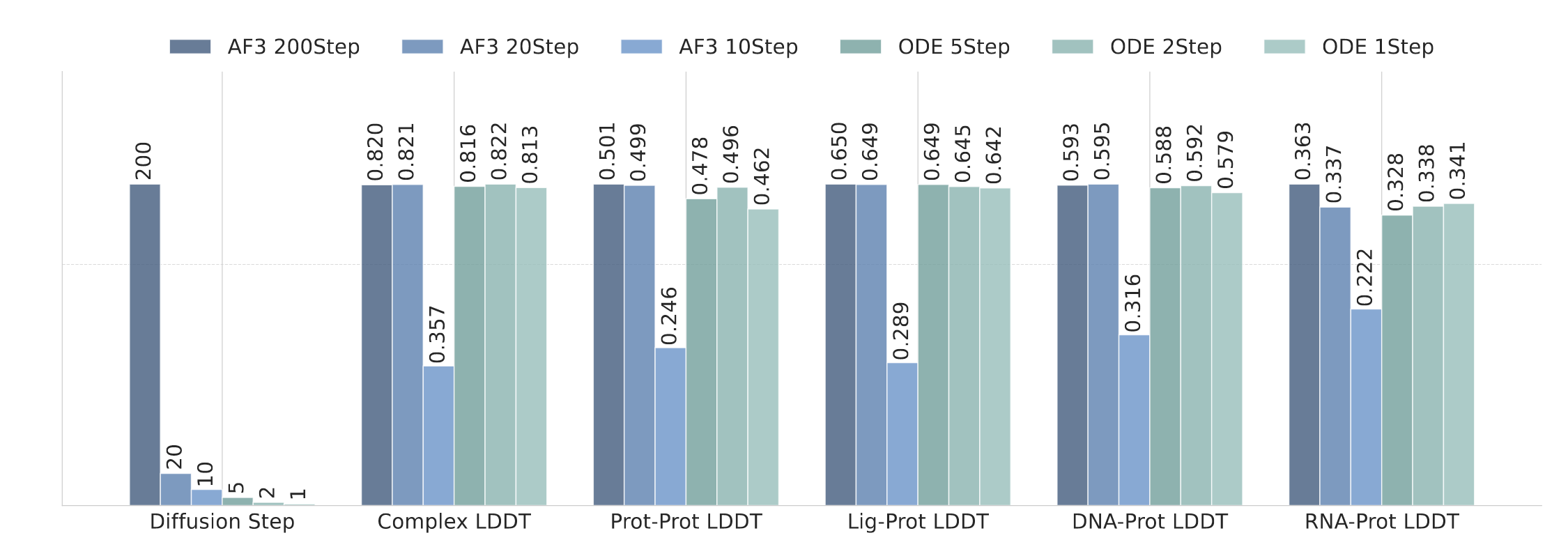
图4 对比了默认 AF3 采样器(
虽然少步推理在平均性能上表现优异,但仍会偶尔出现结构问题,例如物理合理性下降或原子碰撞等现象,具体示例见第4.2.3节。为了完整性,研究还在第4节评估了使用 flow matching 训练的模型,结果显示其在少步采样中同样具有较强的鲁棒性。
3.2 压缩 Protenix 模型中的冗余模块
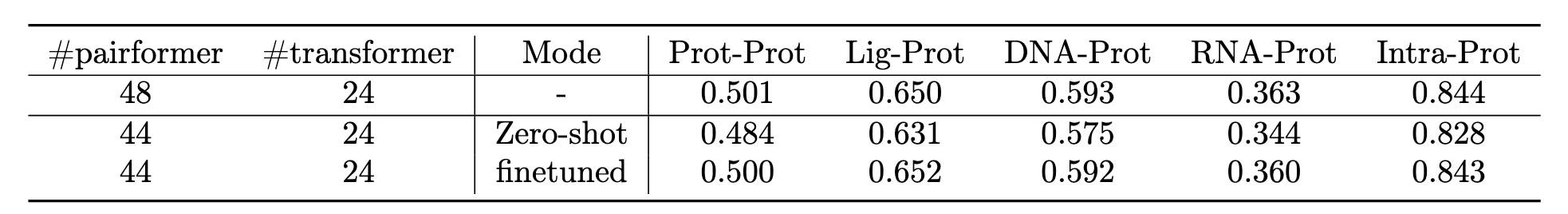
AF3 的架构包含 48 个 Pairformer 块(建模 token 两两间的距离关系)以及 24 个 Diffusion Transformer 块(生成三维坐标)。虽然这种深层网络在捕捉复杂结构依赖上表现出色,但其计算开销巨大,也引发了架构冗余的疑问——是否每一层都对最终预测有实质贡献。类似现象在先前研究中也有报道。
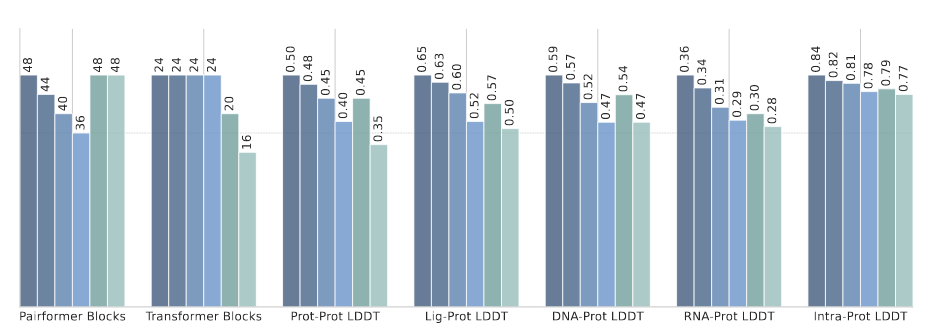
为此,作者进行了逐块消融实验,按顺序移除 Pairformer 网络的早期模块。结果显示,移除前 4 个 Pairformer 块在基准数据集上仅带来轻微精度下降(见图5);进一步如表1所示,在对剩余参数进行微调后,即使去掉若干块也不会显著影响性能。这一发现促使作者尝试更紧凑的架构配置。
基于此,提出了两种训练方案:
- 剪枝 + 微调:移除部分模块后微调剩余模型;
- 直接缩小架构并从零训练。
通过架构搜索,发现 Protenix-Mini 在 LDDT 分数与效率之间达成了较优平衡(见图1与表8)。此外,作者还进行了更激进的裁剪——移除最靠近输入的 前 8 个 Pairformer 块,在 800 次迭代微调后,complex LDDT 下降约 10%(从 0.80 降至 0.72),并由此得到更小的 Protenix-Tiny 模型。详细训练配置见表8。
3.3 基于 pLM 的 Protenix-Mini
由于 MSA 搜索与计算往往需要大量内存与并行计算资源,作者提出用**预训练蛋白质语言模型(pLM)**替代传统的 MSA 流程,以显著降低计算负担。
在架构上,首先将输入序列转化为嵌入表示,再通过线性变换层调整维度。在实现中,采用 ESM2-3B 模型生成上下文相关的序列嵌入,并将其添加到结构预测流水线中 input embedder 模块的
为促进 MSA 模型与 ESM 架构之间的知识迁移,作者设计了混合训练策略:在每次训练迭代中,以 50% 概率选择使用 MSA 模块或 ESM 特征,两条路径共享所有核心组件(如原子编码器、原子解码器、Transformer 块、Diffusion Transformer 块),保证特征表示一致性。该方法可视作一种隐式知识蒸馏,即 ESM 模块在无显式监督的情况下学习逼近 MSA 模块的输出分布,从而生成紧凑但信息丰富的嵌入。
在推理阶段,完全移除 MSA 模块,仅使用 ESM 模块 进行预测,从而实现高效的端到端结构生成。
4 实验结果
4.1 模型配置与训练细节
基于前文分析,作者训练了 Protenix-Mini 模型,配置为 16 个 Pairformer 块、8 个 Diffusion Transformer 块、1 个 MSA 模块块、1 个原子解码器块、1 个原子编码器块,其余训练与架构参数与开源 Protenix [20] 保持一致。模型分别在 EDM 和 flow matching 框架下训练,性能相近。
Protenix-Mini 的训练参数为 batch size 64、200K 次迭代、学习率
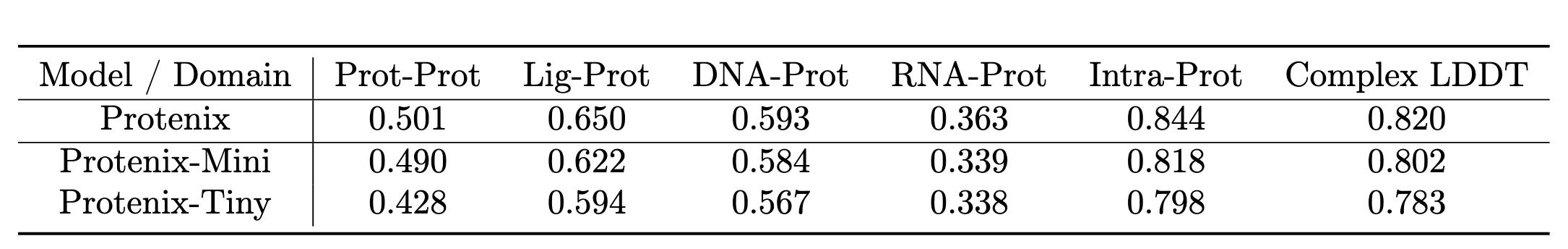
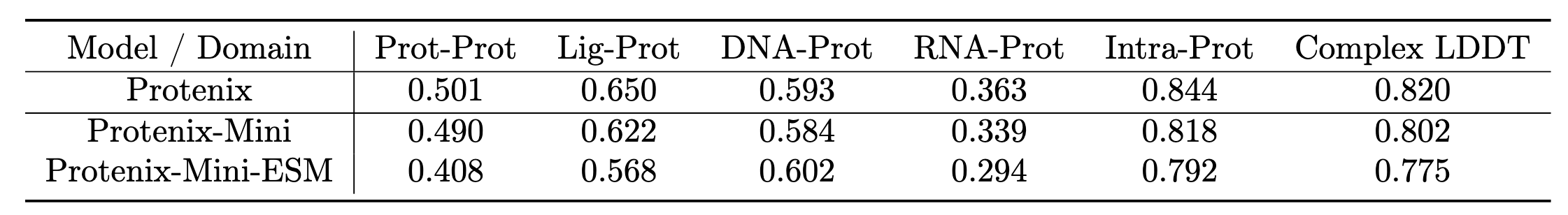
此外,将 Mini 模型加载权重后,将 MSA 置空,引入 ESM2 表示作为输入的一部分,得到 Protenix-Mini-ESM 模型,并额外训练 100K 次迭代。推理时,骨干网络循环 4 次,扩散模块采用 2 步 ODE 采样(Alg. 2)以节省计算。评估指标为 complex LDDT 与 interface LDDT,数据集为 token 数少于 768 的 RecentPDB 子集。

4.2 实验结果
在 RecentPDB(≤768) 与 Posebusters(≤768) 数据集上的表现如下:
- Mini 模型:在 complex 与 interface LDDT 上比标准 Protenix 略低;
- Tiny 模型:各类界面 LDDT 降低约 2–3%,但相比 Protenix 推理计算量减少约 85%(见图1);
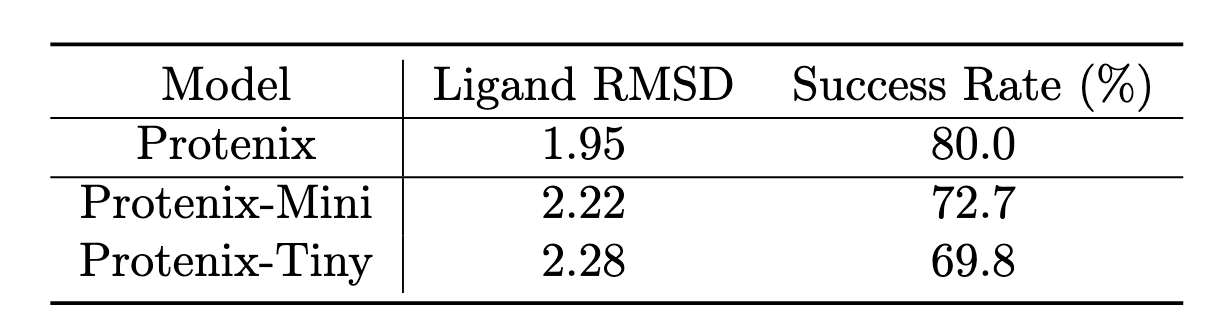
- Posebusters 数据集:以平均配体 RMSD 与 RMSD ≤ 2 的成功率为指标,Mini 与完整 Protenix 表现接近,Tiny 略差(见表5)。
4.2.1 ESM 模型性能
表3显示,Protenix-Mini-ESM 在多数测试指标上略低于 MSA 版本,例如 complex LDDT 从 0.80 降至 0.775;但在蛋白–蛋白界面上降幅显著,从约 0.50 降至 0.40,下降超 10%。这凸显了成对 MSA 特征在蛋白–蛋白界面预测中的关键作用。未来计划开发结构感知型蛋白语言模型以提升此类任务精度。
4.2.2 Flow Matching 模型性能
在 flow matching 框架下,将
并通过
计算附加损失(平滑 LDDT、键长损失等)。训练时
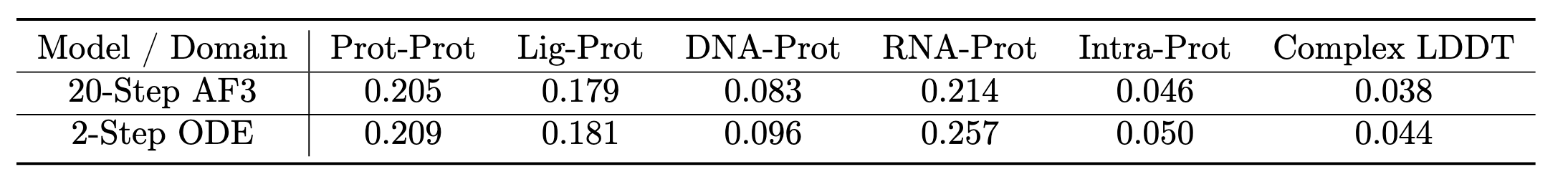
表4显示,Flow Matching 与 EDM 在不同交互类型上的性能差异极小(差值 -0.005 至 +0.009,Complex LDDT 差仅 0.013),说明二者对最终性能影响不显著。
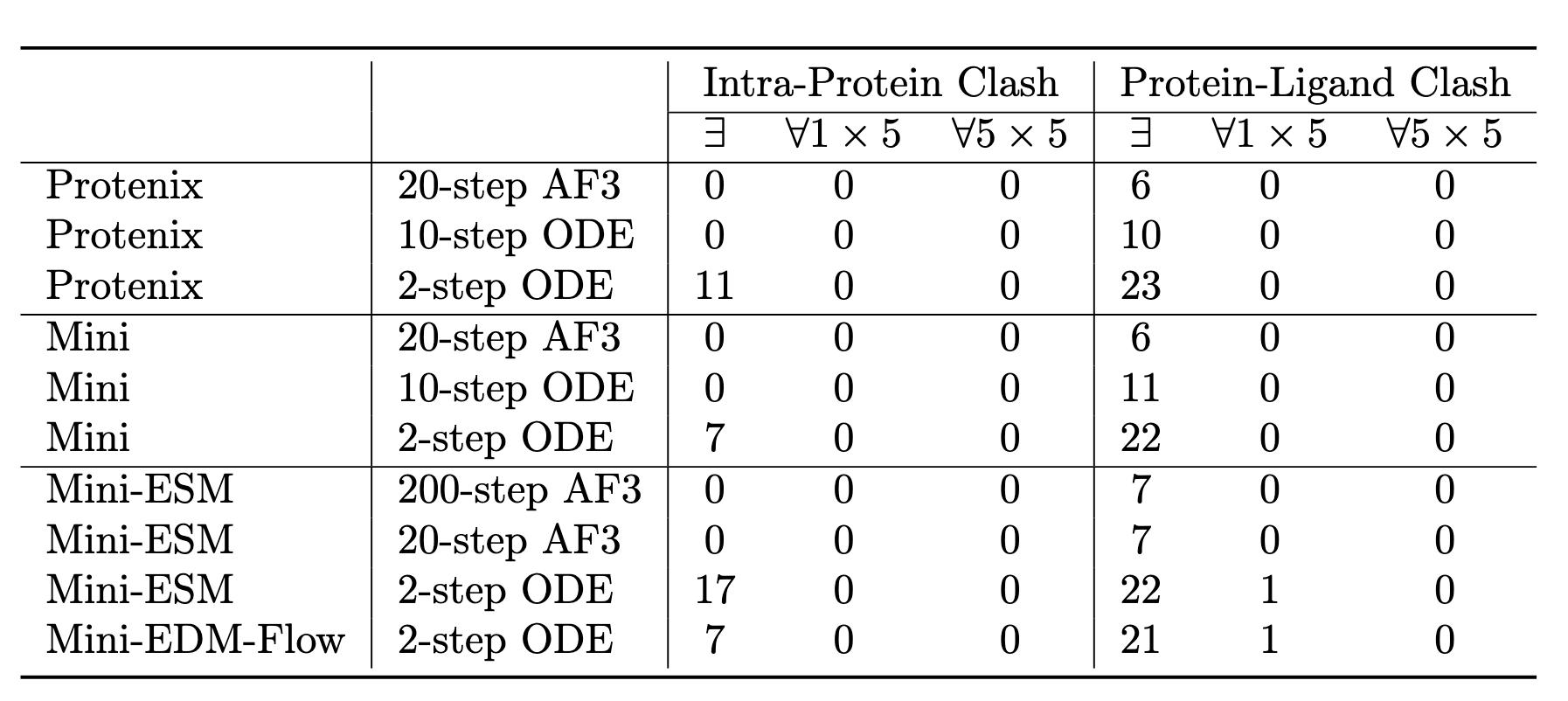
此外,在 2 步 ODE 与 多步 AF3 采样器对比中(5 个随机种子 × 每种 5 个扩散样本):
- Mini EDM 模型:两种采样器均会出现原子碰撞(clash),但增加采样数量可缓解;
- 完整 Protenix 模型:2 步 ODE 仍有碰撞问题,而多步 AF3 在这方面表现更好(见表6)。
4.2.3 案例分析
可视化对比发现,2 步 ODE 相比多步 AF3 更易生成塌陷的配体结构(见图6)。将 ODE 步数增至 10 可明显缓解此问题,推测原因是
在多样性评估中,两种采样器的最佳与最差样本差异几乎一致(见表7),说明少步 ODE 在保持采样多样性的同时,只需合理调节步长就能取得与多步采样相当的性能。

5 结论与未来方向
本文提出了 Protenix-Mini,一种将紧凑架构设计与 2 步 ODE 采样器相结合的轻量化框架,用于高效的生物分子结构预测。在关键基准上仅带来 1%–5% 的性能损失,却显著提升了推理效率。通过裁剪冗余 Transformer 组件并引入稳定的损失函数,验证了在扩散模型中平衡精度与速度的可行性,为其在真实应用中的部署提供了重要途径。同时,提出了基于 ESM 的变体以替代 MSA 搜索模块,在效果与效率间取得新的平衡。
未来的优化方向包括:
- 探索稀疏或自适应注意力机制,在处理大型生物分子复合物时优化 Transformer 主干,缓解其二次复杂度问题;
- 深入搜索架构细节配置中的最佳平衡点;
- 在蛋白质语言模型(pLM)预训练中引入结构信息。
这些工作旨在实现可扩展、低延迟的长序列与多组分系统结构预测,推动药物发现与合成生物学的进一步发展。