NMI | DrugGEN: 基于graph-transformer生成对抗网络的靶点特异性药物候选分子从头设计
今天介绍的是发表在Nature Machine Intelligence上的一项研究——DrugGEN: 基于graph-transformer生成对抗网络的靶点特异性药物候选分子从头设计。该工作聚焦于药物研发中的核心挑战:如何在庞大的化学空间中快速高效地发现具有靶点特异性的候选分子。研究团队提出了一个端到端生成系统DrugGEN,将graph-transformer与GAN架构相结合,并创新性地引入改进的注意力机制,更好地捕捉原子与化学键之间的长程依赖。模型在AKT1和CDK2两个关键蛋白上进行了训练与验证,结果显示DrugGEN不仅在基准指标上优于现有方法,还能够生成与真实抑制剂相似的类药分子。进一步的分子对接、分子动力学模拟及体外实验验证表明,其中两种新分子能在低微摩尔浓度下有效抑制AKT1。该研究为靶点中心化分子设计提供了新的工具,并已开源代码与数据,未来有望广泛应用于药物发现与生物技术领域。

获取详情及资源:
0 摘要
发现新的药物候选分子是药物研发中的基础步骤。生成式深度学习模型能够从学习到的概率分布中采样出新的分子结构,但其在药物发现中的实际应用关键在于能否生成针对特定靶点分子的化合物。该研究提出了一种端到端的生成系统DrugGEN,用于从头设计能够与特定蛋白相互作用的药物候选分子。该方法将分子表示为图结构,并通过包含graph-transformer层的生成对抗网络进行处理。DrugGEN在大规模类药化合物和靶点特异性活性分子数据集上训练,成功设计出针对AKT1的候选抑制剂,AKT1是一种在多种癌症中至关重要的激酶。分子对接和分子动力学模拟表明,这些生成的化合物能够有效结合AKT1,而注意力图则提供了模型推理过程的洞察。此外,部分从头生成的分子被合成并在体外酶学实验中验证,结果显示其在低微摩尔浓度下即可抑制AKT1。这些结果证明了DrugGEN在靶点特异性分子设计中的潜力。借助开源的DrugGEN代码库,研究人员只要具备相应靶点的已知活性分子数据集,就可以将模型重新训练应用于其他可成药蛋白。
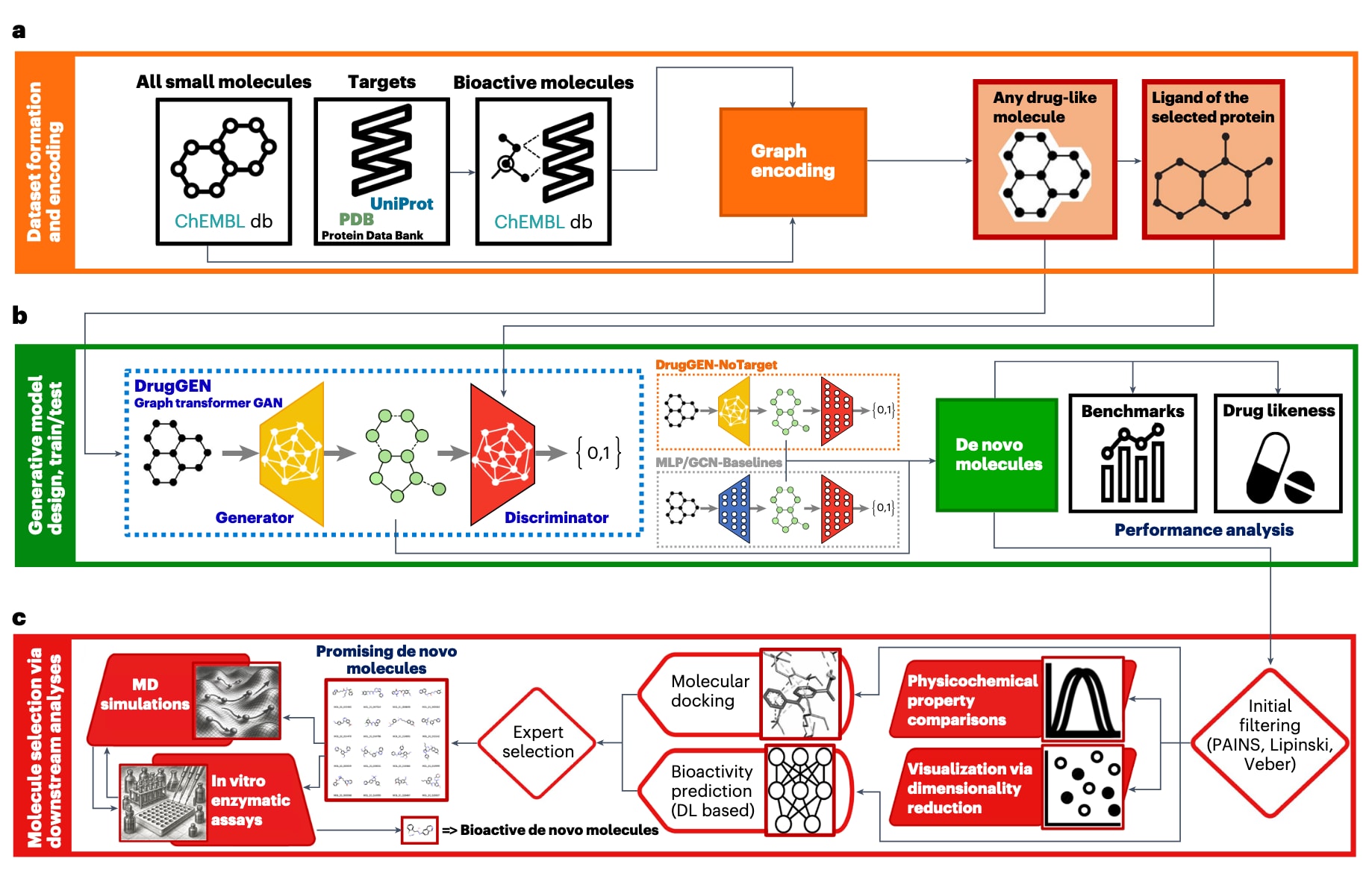
图1 | 研究流程。 a,小分子及其生物活性的数据集准备,并将样本进行基于图的编码。b,DrugGEN的图变换器-GAN架构,模型训练与评估包括对从头生成分子进行基础基准测试和类药性相关指标的检测。c,通过一系列计算机模拟实验对生成分子进行筛选,找出能够有效靶向所选蛋白的潜在候选物。
1 引言
新药研发是一个长期且高成本的过程,其中一个最初且最关键的步骤就是针对预定义的生物分子靶点识别具有生物活性的化合物。随着高通量筛选技术的进步,现在能够同时筛选数以万计的化合物。然而,由于化学空间和靶点空间的规模过于庞大,依然无法做到全面覆盖,这往往阻碍了最佳候选分子的发现。大多数“非理想”的药物候选分子会在研发后期(如临床试验阶段)因高毒性或低效性而被淘汰,这是近年来药物研发成功率低的主要原因之一。迄今为止开发的小分子药物在结构多样性方面较为有限,并且只能针对某些蛋白家族。同样的问题也存在于大型虚拟化学库中。因此,迫切需要具有真正结构新颖性和多样性的小分子候选药物,用于靶向人类蛋白质组中可成药的蛋白,包括其具有临床相关性的变体。
在理论上估计高达
深度生成模型已经被用于分子设计,通过在训练或预测过程中进行条件控制来获得具备期望性质的分子。多数模型采用条件向量作为在生成过程中注入分子性质的手段。常见方法包括变分自编码器(VAE)、生成对抗网络(GAN)、基于序列的语言模型、几何模型(如三维分子生成)以及扩散模型等。此外,强化学习也被引入,通过奖励-惩罚函数引导模型在潜在空间中向具备理想分子特性的方向优化。这些方法能够生成优化后的分子,但药物设计的根本目标之一是开发能够与特定靶点发生物理相互作用的小分子,仅仅获得性质优化的分子是不够的。虽然已有少量研究提出了原型模型,但AI驱动的靶点中心化药物设计仍然是一个新兴且研究不足的方向,具有为合理药物设计做出重大贡献的潜力。
该研究提出了一种新的从头小分子设计系统——DrugGEN,这是一个端到端框架,结合GAN与Transformer架构,并融合图表示学习,能够生成靶点中心化的类药分子。研究的整体流程如图1所示:(1)首先准备小分子数据集(包括其生物活性与相互作用),并将其编码为图结构作为训练和测试数据(图1a);(2)随后进行DrugGEN模型的设计与实现,并在性能上与最新方法进行比较,同时开展消融实验(图1b);(3)最后对生成的分子进行下游分析,筛选出最有前景的候选分子以有效靶向所选蛋白(图1c)。在步骤(3)中,该研究生成并评估了针对**RAC-α丝氨酸/苏氨酸蛋白激酶(AKT1)**的新型抑制剂。
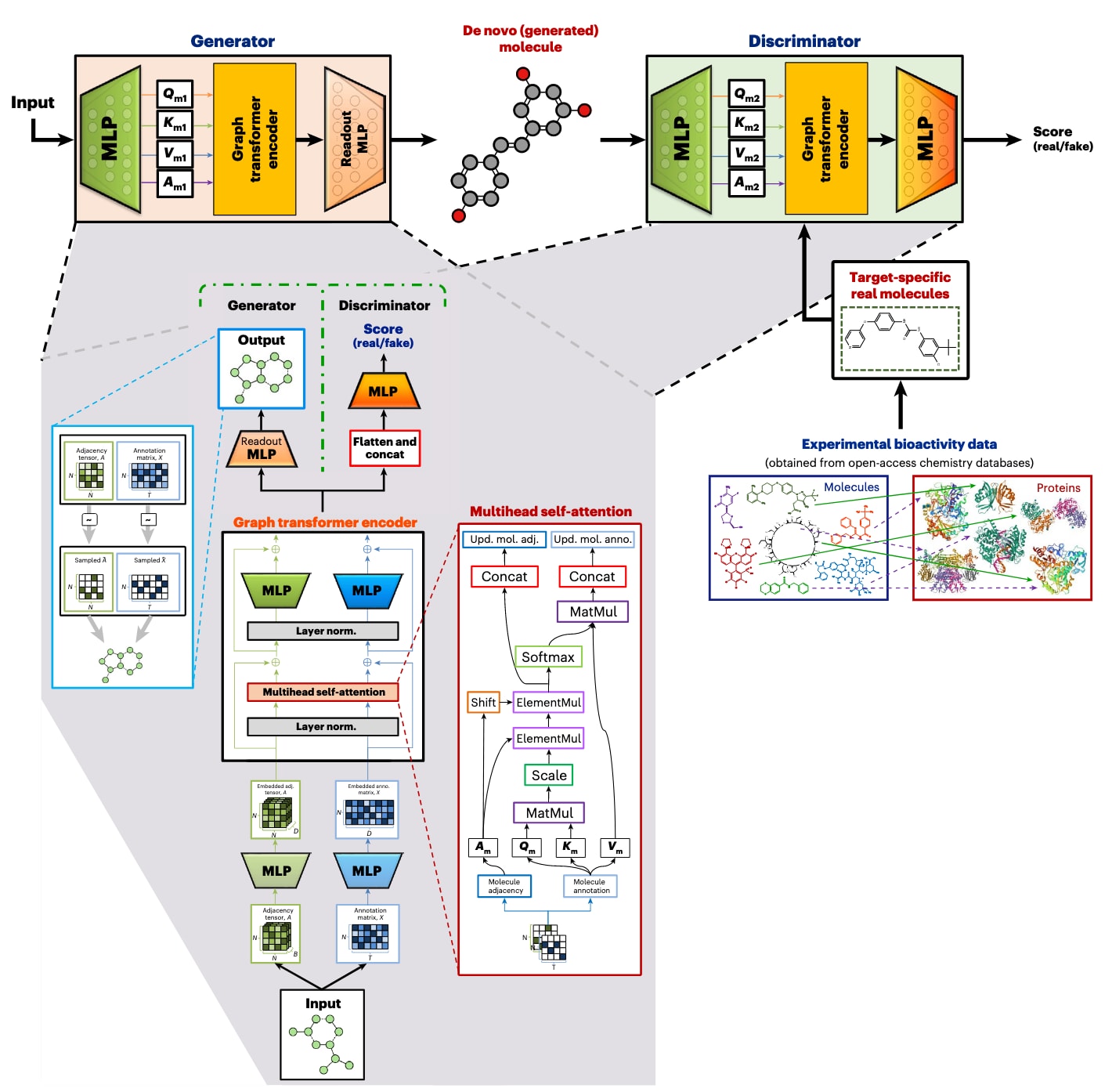
图2 | DrugGEN模型架构。 生成器与判别器网络均采用强大的graph-transformer编码模块。生成器将输入转化为新的分子表示;判别器则将生成的从头分子与已知的靶点蛋白抑制剂进行比较,并将其划分为“真实”或“伪造”分子并打分。
2 结果
2.1 DrugGEN概述
DrugGEN的目标是学习药物及候选分子的理化性质分布与拓扑特征,从而生成既合理又新颖的类药小分子。其生成器输出会与已知靶点抑制剂进行比较,判别结果再反馈至生成器,以引导生成能够与目标蛋白结合的配体。
研究者的主要创新在于将graph-transformer引入GAN的生成器和判别器。与传统MLP等架构相比,graph-transformer更能捕捉原子与化学键之间的长程依赖和相互作用,尤其适合大分子设计。据报道,graph-transformer此前多用于分子性质预测,而这是首次应用于靶点特异性分子生成。同时,研究者改进了注意力机制,加强了边信息的利用,提升了分子学习中对图连通性的表达能力。
与常规GAN不同,DrugGEN借鉴CycleGAN与DiscoGAN,将真实分子样本而非噪声作为输入,并在ChEMBL的类药分子与靶点抑制剂上训练,应用于AKT1与CDK2的从头配体设计。性能通过多维度评估,包括基准指标、理化性质分布和UMAP/t-SNE可视化,并辅以分子对接和基于深度学习的DTI预测验证靶点特异性,同时与多种生成模型进行了比较。
此外,研究者结合可解释AI,利用注意力图揭示关键原子作用位点,并从生成结果中筛选出30个候选分子,合成其中5个(纯度≥95%)。体外实验显示其中两者在低微摩尔浓度下对AKT1具有显著抑制活性,分子动力学模拟进一步阐释了其结合机制。
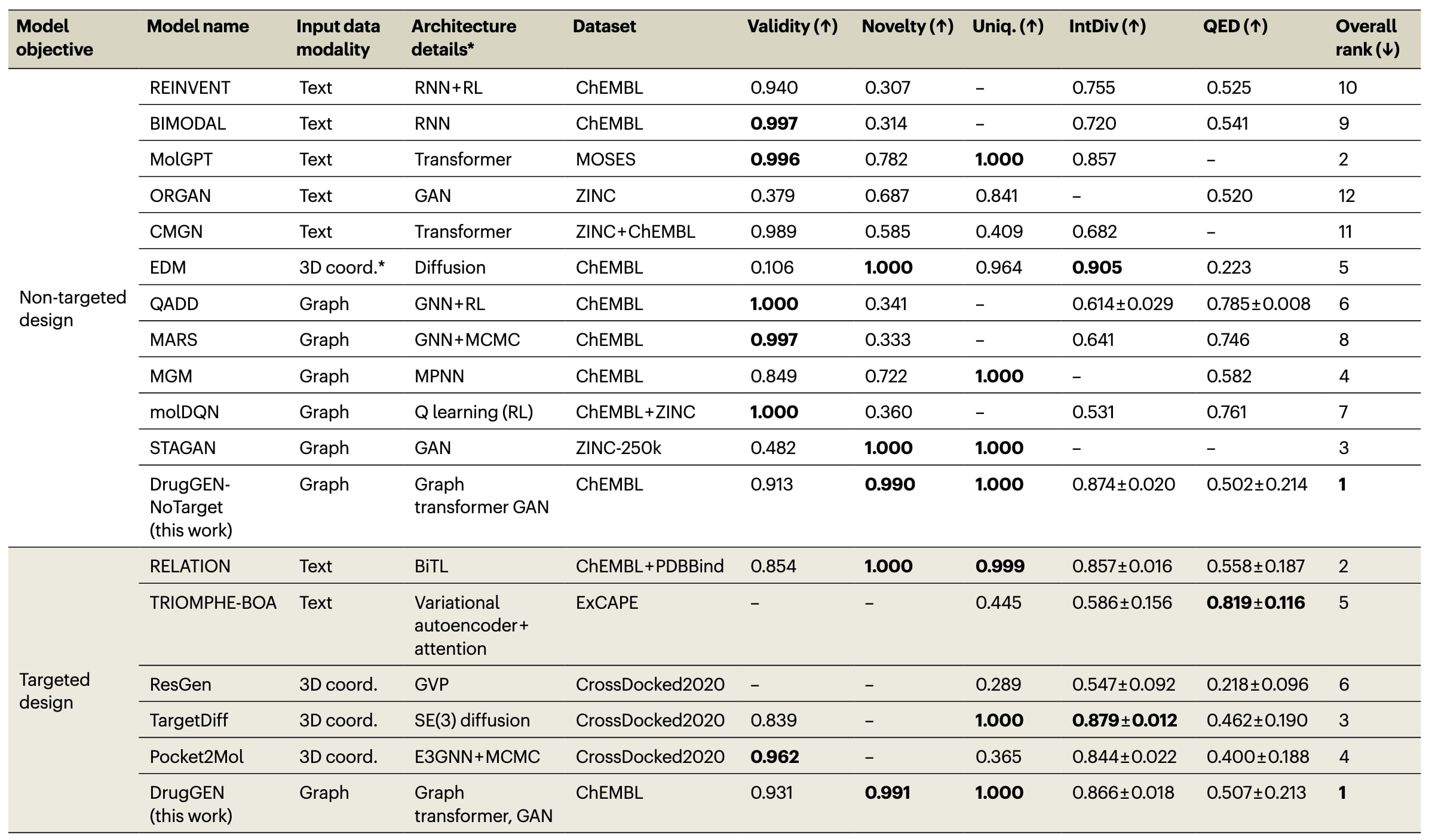
表1 | DrugGEN与其他方法的整体分子生成性能。
2.2 整体性能评估与比较
研究者将DrugGEN与分子生成式人工智能的多种方法进行系统对比。模型在推理模式下生成10,000个从头分子,并通过MOSES基准测试进行评估,涵盖有效性、独特性、新颖性、内部多样性、合成可及性(SA)与药物相似性(QED)等常用指标。这些指标仅能反映模型在基础层面的表现,无法直接衡量DrugGEN的核心目标——靶点特异性分子生成,因此研究者进一步结合机器学习的活性预测与物理学方法(分子对接、分子动力学)展开验证。
对比模型分为两类:非靶向设计(REINVENT、MolGPT、EDM等)与靶向设计(RELATION、TargetDiff、Pocket2Mol等)。除EDM外,其余均采用文献报告的结果。由于指标繁多,研究者采用综合排名方式,将各模型在不同指标下分为三档并取平均分,再依据连续数值解决并列问题。结果显示,DrugGEN在靶向模型中排名最高,而在非靶向模型中,DrugGEN-NoTarget同样位居首位,体现了其在不同任务场景下的领先性能。
2.3 DrugGEN生成靶点特异性的类药分子
研究者在AKT1和CDK2两个靶点上对DrugGEN进行了训练与测试,主要集中在AKT1,同时加入CDK2验证可重复性。CDK2在多种癌症中异常活跃,是重要的药物开发靶点,也便于与已有模型进行对比。
在对比分析中,研究者将DrugGEN与RELATION、TRIOMPHE-BOA、ResGen、TargetDiff、Pocket2Mol等模型进行评估,考察了对接得分及QED、SA、Lipinski、Veber、PAINS等类药性指标,并通过片段和骨架相似性分析验证与真实抑制剂的接近程度。
结果显示,DrugGEN在AKT1任务中表现最佳,平均结合能达**−8.386 kcal·mol⁻¹(99.98%)**,显著优于RELATION(P < 0.0001),同时在QED(0.507)、SA(3.510)等指标上保持平衡。TargetDiff和Pocket2Mol虽次之,但表现尚可。
在CDK2任务中,DrugGEN的中位结合能为**−8.747 kcal·mol⁻¹(86.99%)**,优于TargetDiff,接近Pocket2Mol。其生成分子的平均QED为0.547,且在Lipinski、Veber和PAINS规则下表现良好,体现了类药性优势。相比之下,Pocket2Mol因QED偏低、SA偏高而合成复杂度更高。
进一步对比临床候选药物,capivasertib、ipatasertib与uprosertib的结合能分别为**−9.517、−9.681、−8.569 kcal·mol⁻¹**,DrugGEN能生成40个结合能优于capivasertib的分子。此外,研究者还检验了不同输入(随机ChEMBL分子与真实抑制剂)对输出的影响,并在多个理化性质维度上对比了其他模型与真实分子。
综上,DrugGEN在不同靶点上均展现出稳定的生成能力与优异的类药性,证明了其在靶点特异性分子设计中的广泛适用性。
2.4 消融研究
在一项广泛的消融实验中,研究比较了多种模型变体,以评估图变换器架构与改进注意力机制在DrugGEN中的有效性,特别是其在靶点特异性分子生成中的作用。具体而言,对比了默认DrugGEN模型及其非靶向变体(DrugGEN-NoTarget),并与使用更简单架构的基线模型进行比较,这些基线模型在生成器和判别器模块中采用了多层感知机(MLP)或图卷积网络(GCN)模块。此外,还分析了不同的注意力机制策略,以研究如何引入边信息(即化学键)。每个模型变体均生成10,000个从头分子,并依据表1与扩展数据表1中的基础生成指标和类药性得分进行评估。结果表明,采用图变换器模块并结合改进注意力机制(即引入邻接矩阵平移乘积
2.5 分子可视化与降维分析
研究者进一步采用UMAP与t-SNE算法对真实分子及生成的从头分子进行可视化。如图3b所示,DrugGEN生成的分子与真实AKT1抑制剂聚集在同一区域,显示出较高的拓扑指纹相似性。相反,DrugGEN-NoTarget生成的分子因缺乏靶向性,几乎均匀分布于二维空间。结果表明,DrugGEN能够有效学习并重现AKT1抑制剂的结构分布特征。
2.6 基于深度学习的生物活性预测
为在分子对接之外进一步评估DrugGEN生成分子的靶点相互作用特性,研究者借助此前开发的DEEPScreen系统进行药物/化合物–靶点相互作用(DTI)预测。DEEPScreen是一个靶点中心的监督式深度学习框架,可将输入分子分类为活性(有相互作用)或非活性(无相互作用)(见图3c)。
在AKT1任务中,模型使用已验证的活性与非活性分子进行训练与测试。在包含484个已知结合物的scaffold-split保留数据集上,DEEPScreen准确预测出475个活性分子,在置信度阈值0.5下表现出精确率0.96、召回率0.94、F1=0.95、MCC=0.91的优异性能。其中421个分子被赋予高置信度(≥0.85),进一步验证了预测的可靠性。
研究者随后将10,000个DrugGEN分子输入DEEPScreen,得到1,382个活性预测,其中748个分子置信度≥0.85(补充图6)。虽然DrugGEN分子与训练集的平均分子相似度(Tanimoto系数0.309)低于保留数据集(0.637),导致总体阳性比例不算高,但依然展现出潜在优势。
对比来看,10,000个随机ChEMBL分子与10,000个DrugGEN-NoTarget分子仅得到122个与69个高置信度活性预测。相比之下,DrugGEN实现的748个高置信度预测凸显了其在靶点导向分子生成中的明显优势。
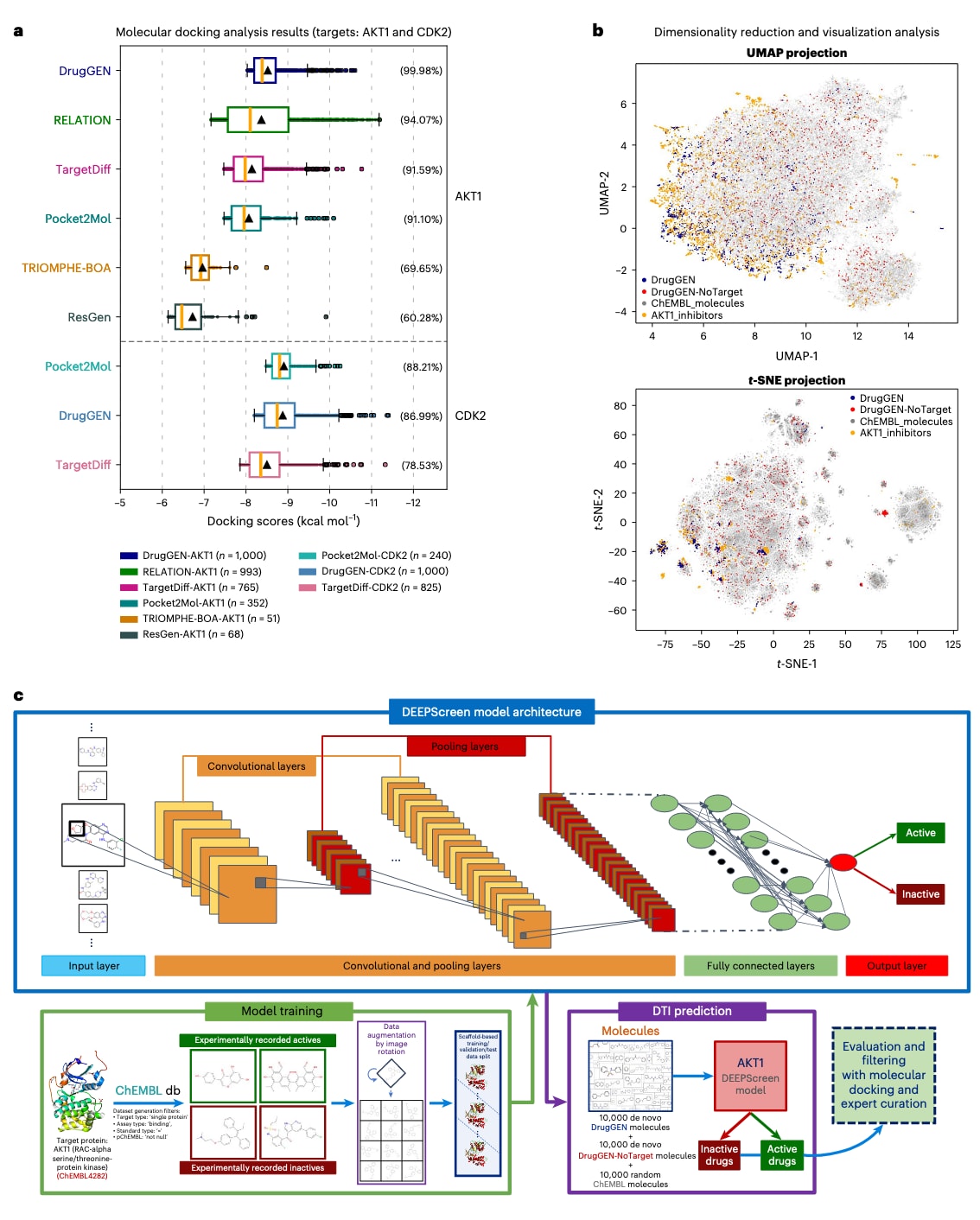
图3 | 从头生成分子的下游分析。 a,箱线图展示了多种模型在AKT1与CDK2上的对接结果:包括DrugGEN、RELATION、TargetDiff、Pocket2Mol、TRIOMPHE-BOA和ResGen在AKT1上的表现,以及DrugGEN、TargetDiff和Pocket2Mol在CDK2上的表现。图例标注了各模型使用的分子数量(n),若模型无法生成足够多的有效且唯一分子,则n值偏低。箱体表示四分位距(Q1–Q3),橙色线为中位数,黑色三角形为均值,须状线延伸至±1.5倍四分位距,超出范围的点以实心圆表示。箱线旁的百分比为对接得分相对真实抑制剂的归一化结果(真实抑制剂最高分设为100%)。模型根据中位结合自由能排序,数值越低表示预测亲和力越强。b,UMAP与t-SNE二维投影展示了不同来源分子的分布,包括DrugGEN与DrugGEN-NoTarget生成的分子、真实AKT1抑制剂以及随机选择的ChEMBL分子。t-SNE-1与t-SNE-2分别表示第一、第二维度。c,基于深度学习的生物活性预测分析。上方为DEEPScreen流程:输入分子的二维像素化结构图,经卷积神经网络处理。左下为AKT1模型的训练数据,来源于ChEMBL结合实验的二值化活性数据。右下展示了推理阶段,DrugGEN与DrugGEN-NoTarget生成的分子及随机ChEMBL分子被输入DEEPScreen AKT1模型进行预测。
2.7 筛选30个潜在分子
研究者通过专家筛选,从生成结果中挑选出30个最具前景的候选分子,主要依据为对接得分低于−8 kcal·mol⁻¹且DEEPScreen预测为“活性”(扩展数据图1)。其中大部分分子表现出良好的合成可及性与类药性,SA评分<6,QED评分>0.5;另一些分子则具有较高极性或分子量等非典型特征。
所有入选分子均符合Lipinski五规则,氢键供体(≤5)和受体(≤10)在合理范围内,进一步支持其药物潜力。补充图7展示了部分候选分子,并与训练集中最相似的真实AKT1抑制剂进行比较(每个从头分子匹配两个真实分子)。结果显示,即便与最接近的训练样本相比,这些分子仍保持明显的结构多样性,同时大多数在AKT1对接中取得了优于−8 kcal·mol⁻¹的结合能,验证了其作为潜在抑制剂的价值。
2.8 AKT1抑制的实验验证
在计算分析之后,研究者进一步开展了化学合成与体外酶学实验以验证结果。从扩展数据图1中的30个候选分子中,挑选出5个结构多样且合成复杂性较低的化合物进行合成(图4a),这些分子也具有较短的合成与纯化周期。对接分析显示,分子1(MOL_02_045762)、分子2(MOL_02_045795)、分子3(MOL_02_045758)、分子4(MOL_02_045609)和分子5(MOL_02_045627)的最佳结合自由能分别为**−8.948、−7.686、−8.655、−8.383和−5.341 kcal·mol⁻¹**,同时参考配体capivasertib的对接得分为**−9.517 kcal·mol⁻¹**(图4a,b)。相关细节见补充材料第9节。所有分子均由商业实验室合成,其纯度(≥95%)的分析证明见补充图12–16。
在后续的体外酶学实验中,这些分子被用于检测放射性³³P标记的磷酸基团从ATP向AKT1底物的转移。结果显示,5个化合物中有2个表现出明显活性:分子1的IC₅₀为1.89 μM,分子2的IC₅₀为48.6 μM(补充表3);其余分子3–5即使在100 μM的浓度下仍未表现出有效活性。对应的剂量–反应曲线见补充图17。为进一步阐释这些实验结果,研究者还进行了分子动力学(MD)模拟,相关分析将在后文展开。
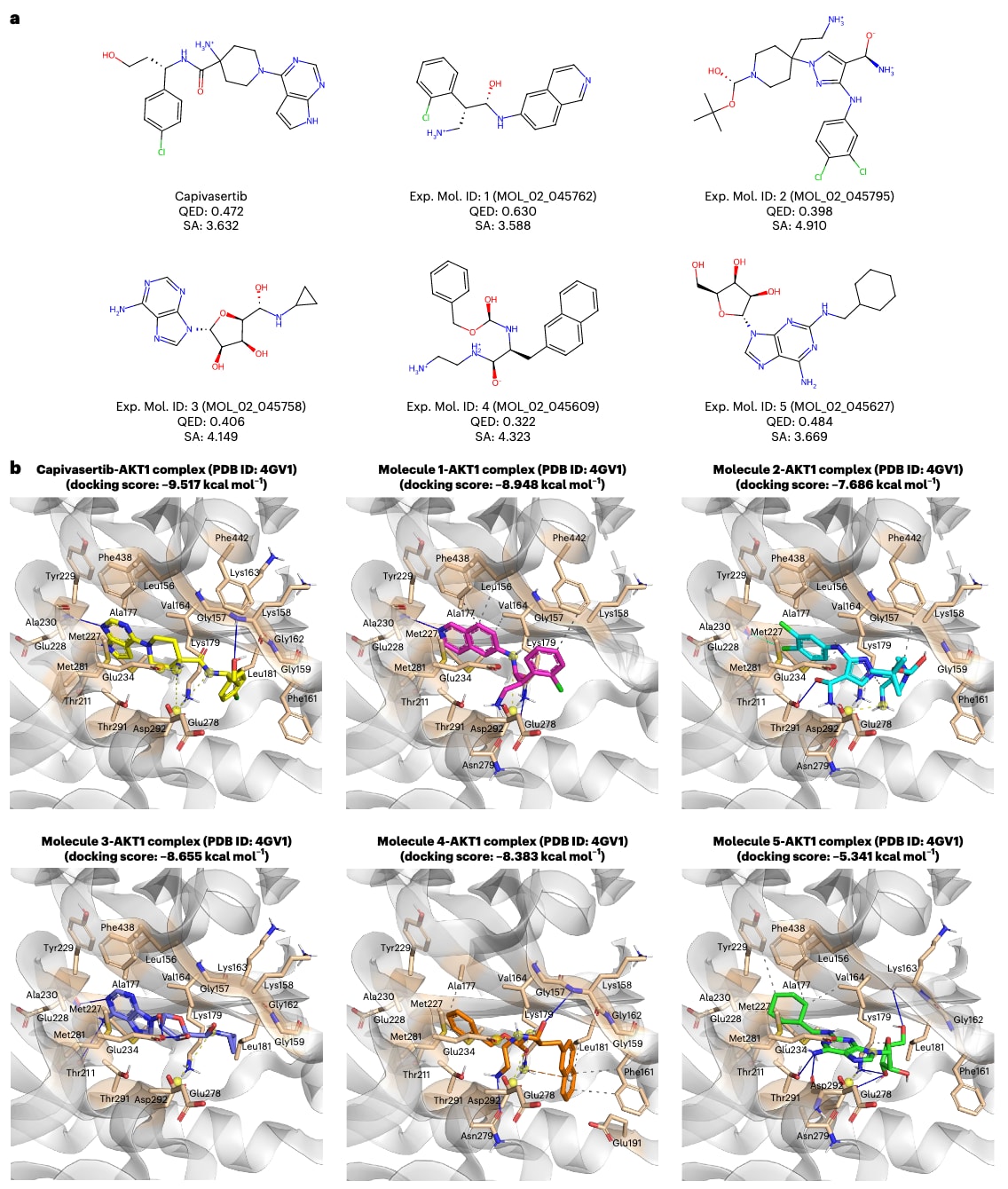
图4 | Capivasertib(结合配体,PDB: 4GV1)与五个从头生成分子的结构分析。 a,Capivasertib与分子1–5的二维可视化图,包括在分子生成过程中分配的唯一标识符、QED值与SA值。b,Capivasertib与分子1–5在与AKT1(PDB ID: 4GV1)对接后的三维蛋白-配体相互作用图。AKT1活性位点残基以米色高亮显示。不同的相互作用类型标注如下:蓝色线为氢键(H键),黄色虚线为盐桥,灰色虚线为疏水作用,橙色虚线为π–阳离子作用,绿色线为卤键。Exp. Mol. ID,实验分子编号。
2.9 分子结合特性的MD模拟研究
研究者利用分子动力学(MD)模拟评估了5个已合成的从头分子与AKT1的结合特性,对照对象为晶体结构中的共晶配体capivasertib以及分子1–5的对接构象。所有化合物的初始取向均定位于ATP辅因子结合位点(扩展数据图2a,补充图8)。模拟结果表明,所有分子在过程中均保持稳定(RMSD < 1.6 Å;扩展数据图2c,补充图8),且大多数情况下未出现显著构象波动(RMSF较低;扩展数据图2d,补充图8)。
在具体作用模式上,capivasertib在模拟中稳定维持与关键残基Tyr229和Ala230的氢键,并通过水分子介导与Lys158、Asn279和Asp292形成额外作用(扩展数据图2b)。分子1作为最强抑制剂(IC₅₀ = 1.89 μM),在铰链残基Ala230处形成稳定氢键(占比96%),并与周围酸性残基相互作用,显示出与capivasertib类似的结合网络,但由于缺乏额外铰链接触及稳固氢键网络,其效力可能略受限制(扩展数据图2b)。相比之下,分子2则在铰链残基Tyr229与Ala230处形成卤键,同时还与Lys179、Glu234、Glu278、Thr291和Asp292等关键残基形成氢键,从而解释了其作为次强抑制剂的效果(扩展数据图2b)。
2.10 注意力图可视化
为提升模型可解释性,研究者对DrugGEN生成器中的graph-transformer编码器进行了注意力得分可视化,以评估从头分子中的关键原子作用。分析结果显示,高注意力得分的配体原子与对接中鉴定的关键作用原子高度一致(即与AKT1残基直接相互作用的原子)。如图5所示,DrugGEN能将高注意力分配给参与氢键和盐桥的核心原子,准确识别约91%的相互作用位点。这表明,即使未基于成对相互作用数据进行训练,模型仍能有效捕捉对AKT1结合至关重要的分子亚结构,凸显其在靶点特异性分子设计中的潜力。
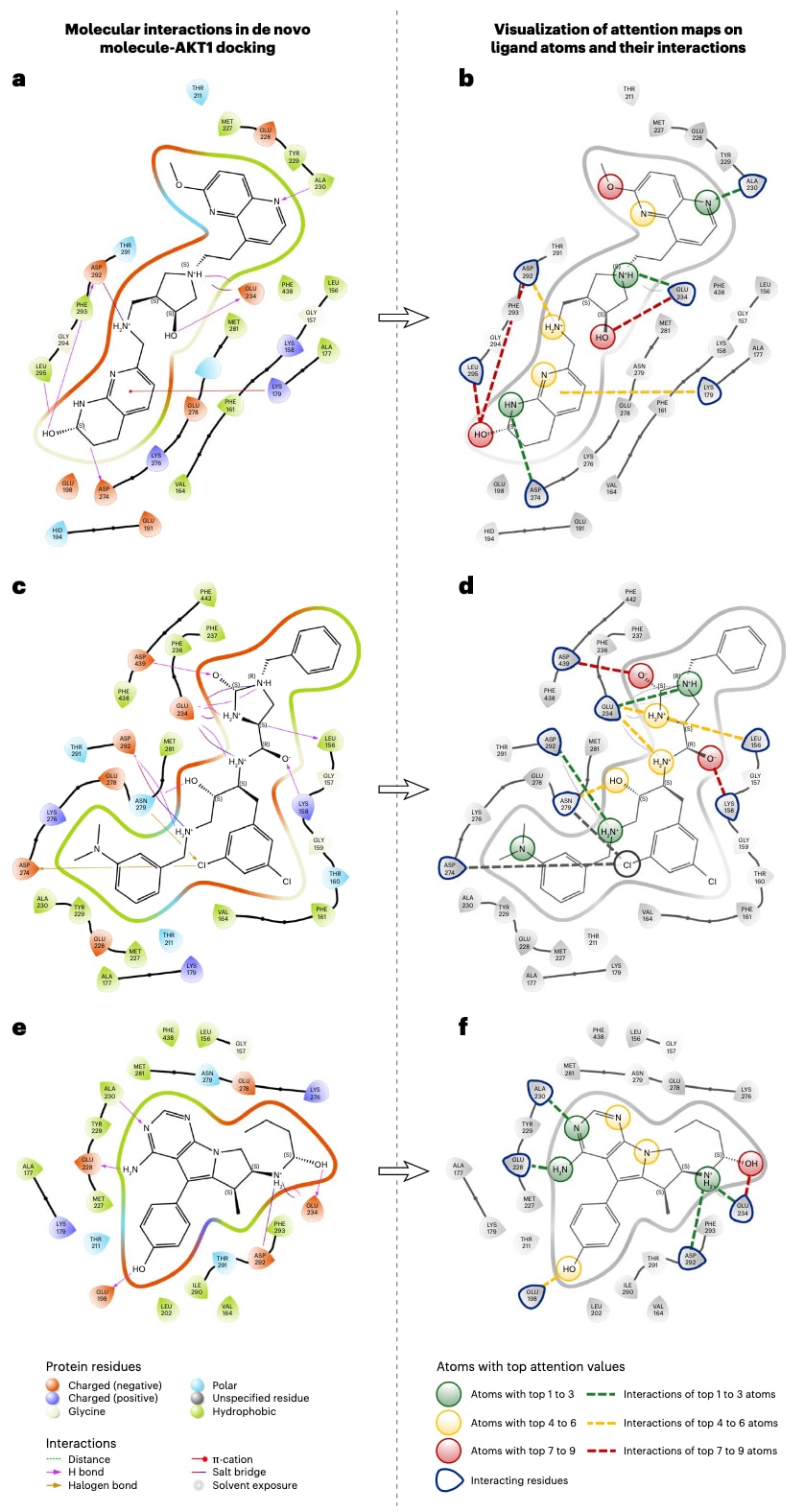
图5 | 三个从头生成分子的DrugGEN注意力图可视化。 a,c,e,三个DrugGEN生成分子与AKT1蛋白结构(PDB ID: 4GV1)的分子对接所得到的蛋白–配体相互作用图。对接配体位于结合口袋中,残基与配体的相互作用以线条表示,并按相互作用类型上色。蛋白残基根据理化性质着色。b,d,f,同三个分子的注意力图,来自DrugGEN生成器网络的图变换器模块。获得最高注意力分数的原子以颜色标出(绿色:第1至3个原子;黄色:第4至6个原子;红色:第7至9个原子,具有最高注意力分数),注意力较低的原子不着色。在b、d和f中,受体–配体相互作用(即对接分析得到的结果)以虚线表示,颜色与相应参与作用的分子原子注意力分数一致;若某作用原子未被捕获(注意力分数较低),则相互作用以灰色标注。a与b:分子编号MOL_02_045597,对接得分−9.803 kcal·mol⁻¹。c与d:分子编号MOL_02_000496,对接得分−9.693 kcal·mol⁻¹。e与f:分子编号MOL_02_008350,对接得分−9.619 kcal·mol⁻¹。
3 讨论与结论
研究者开发了DrugGEN系统,用于自动生成靶点中心的类药分子。该系统结合GAN与graph-transformer,并引入改进的注意力机制以更好地整合边信息,从而强化分子结构表示。在AKT1与CDK2两个靶点的训练和评估中,DrugGEN在基础指标上优于现有模型,能生成与真实AKT1抑制剂相似的从头分子。通过分子对接、分子动力学模拟和体外实验进一步验证了其有效性,其中两种合成分子表现出低微摩尔级活性(分子1:IC₅₀=1.89 μM;分子2:IC₅₀=48.6 μM)。
为促进应用,研究者已将代码、数据、结果与模型开源(GitHub与HuggingFace平台)。与多数早期生成模型不同,DrugGEN可处理接近真实药物规模的分子(平均33个、最多45个重原子),提升了实际价值。研究者还提出了一个结合计算模拟与实验验证的下游筛选流程,以加速候选分子的早期筛选与临床前开发。
在方法上,DrugGEN通过真实分子图而非噪声作为输入,利用WGAN-GP缓解模式崩溃与梯度消失,并通过惩罚不合理结构而非模仿训练集来保证新颖性和合理性。相比依赖外部预测模型的多目标优化方法,其架构降低了误差传播风险和训练复杂度。
未来,研究者计划:(1)扩展DrugGEN至更多蛋白靶点;(2)开发结合SELFIES与SELFormer的序列式Transformer GAN;(3)探索以分子片段为生成单元提高可合成性;(4)在GAN中引入约束优化以控制理化性质、靶点结合和合成可及性;(5)直接整合靶蛋白特征;(6)结合异构生物医学知识图谱,提升小分子的结构–功能学习与设计能力。