Commun. Chem. 2023 | 基于逆折叠与结构预测的肽结合物设计
今天介绍的是发表在 Commun. Chem. 的一项研究,主题为基于逆折叠与结构预测的肽结合物设计。肽类药物在近几十年发展迅速,全球市场规模已超过500亿美元,但如何针对特定蛋白界面设计高效结合肽仍是重大挑战。传统依赖对接打分的方法成功率极低,而实验手段如定向进化与噬菌体展示虽能改进,却常导致非特异性结合。
随着深度学习和AlphaFold2在结构预测上的突破,研究者提出了一个自动化框架:利用Foldseek生成种子、ESM-IF1逆折叠设计序列,并结合AlphaFold2预测复合物结构。这一流程不仅能够区分真实结合物与突变体,还能通过定制损失函数筛选出高置信度的结合物。在零样本条件下,ROC AUC达到0.96;在多样本设计中,成功率进一步提升至6.5%,显著优于ProteinMPNN等方法。
研究还发现,接触密度是影响设计成功的关键因素,高接触密度可将成功率从不足1%提升至67%。该成果凸显了AI在肽结合物设计中的潜力,为未来大规模、精准的药物研发提供了新的路径。

获取详情及资源:
0 摘要
针对特定蛋白界面的计算机辅助肽结合物设计,对于诊断与治疗具有重要意义。该研究提出了一种整合Foldseek、ESM-IF1以及AlphaFold2(AF)的联合框架。Foldseek用于生成主链种子,经过适配蛋白复合物的改进版ESM-IF1处理后得到候选序列,再由AF进行评估,其中受体采用MSA表示,结合肽使用单序列表示。结果表明,AF能够准确评估结合肽的结合能力,并通过设定的bind score进行筛选,在异源二聚体任务中取得了ROC AUC=0.96的表现。研究发现,来源于具有更多残基接触的种子所生成的设计更成功,且往往较短。同时,界面位点的序列恢复率与设计的plDDT高度相关,当恢复率≥80%时,平均plDDT可达84,而在0%时仅为55。设计的序列在针对目标受体时的plDDT中位数比非目标受体高出约60%。成功的结合物(预测界面RMSD≤2 Å)包括185个(6.5%)异源界面和42个(3.6%)同源界面,显著优于ProteinMPNN在相同采样数下的结果(仅18个,1.5%)。
引言
在药物开发领域,针对特定蛋白界面的肽结合物设计一直是备受追捧的目标。过去六十年间,获批的肽类药物年均增长约8%,全球销售额已超过500亿美元。早期的计算机设计主要依赖对接打分,但其成功率极低,约十万分之一。相比之下,实验方法如定向进化与噬菌体展示虽可结合机器学习提升效果,却难以限定结合区域,易导致非预期结合。近年的深度学习方法,尤其是基于AF的预测,大幅提升了蛋白–蛋白、蛋白–肽相互作用的建模精度,并推动了蛋白设计的突破。例如ProteinMPNN不仅提升了设计准确性,还改善了溶解性。但若以整体序列恢复作为评价指标,并不适合结合物设计,因为仅残基错位即可导致恢复率为0%,而关键界面残基却可能仍被正确预测。因此,通过结构预测评估界面残基恢复成为更具价值的指标。已有研究表明,以AF作为打分函数重新评估旧设计,实验成功率可接近90%,远超基于物理计算的0–5%。此外,方法如MaSIF利用学习到的相互作用势与骨架搜索辅助设计。ESM-IF1通过逆折叠生成与骨架匹配的序列,尽管未针对复合物训练,却显示出良好泛化性。
该研究探索了在2843个异源二聚体界面与1172个同源界面上,利用ESM-IF1生成序列并结合AF评估的设计策略。通过Foldseek生成种子结构,整体方法形成了一套自动化且可扩展的肽结合物设计流程,为大规模靶向不同蛋白的设计提供了可行路径。
2 结果
研究首先探讨了预测蛋白–肽复合物结构的最佳方式,以及如何筛选能够被高精度预测的结合物。结果表明,AlphaFold2能够区分真实结合残基,并由此构建出一个损失函数,可用于从实验数据中识别真实结合物。当预测肽与天然肽的界面RMSD≤2 Å时,结合物被视为成功。该损失函数(Eq.1)贯穿整个研究,用于结合物的评估。研究者提出了结合物设计的流程,并以零样本方式在PDB中所有异源界面上进行验证,进一步分析了成功设计的标准,包括高置信度预测、收敛性与潜在的非靶向效应,并将性能与其他蛋白设计方法进行了对比。值得注意的是,研究中应用了原本仅用于单链预测的方法,从而实现了对大量蛋白复合物的泛化能力评估。
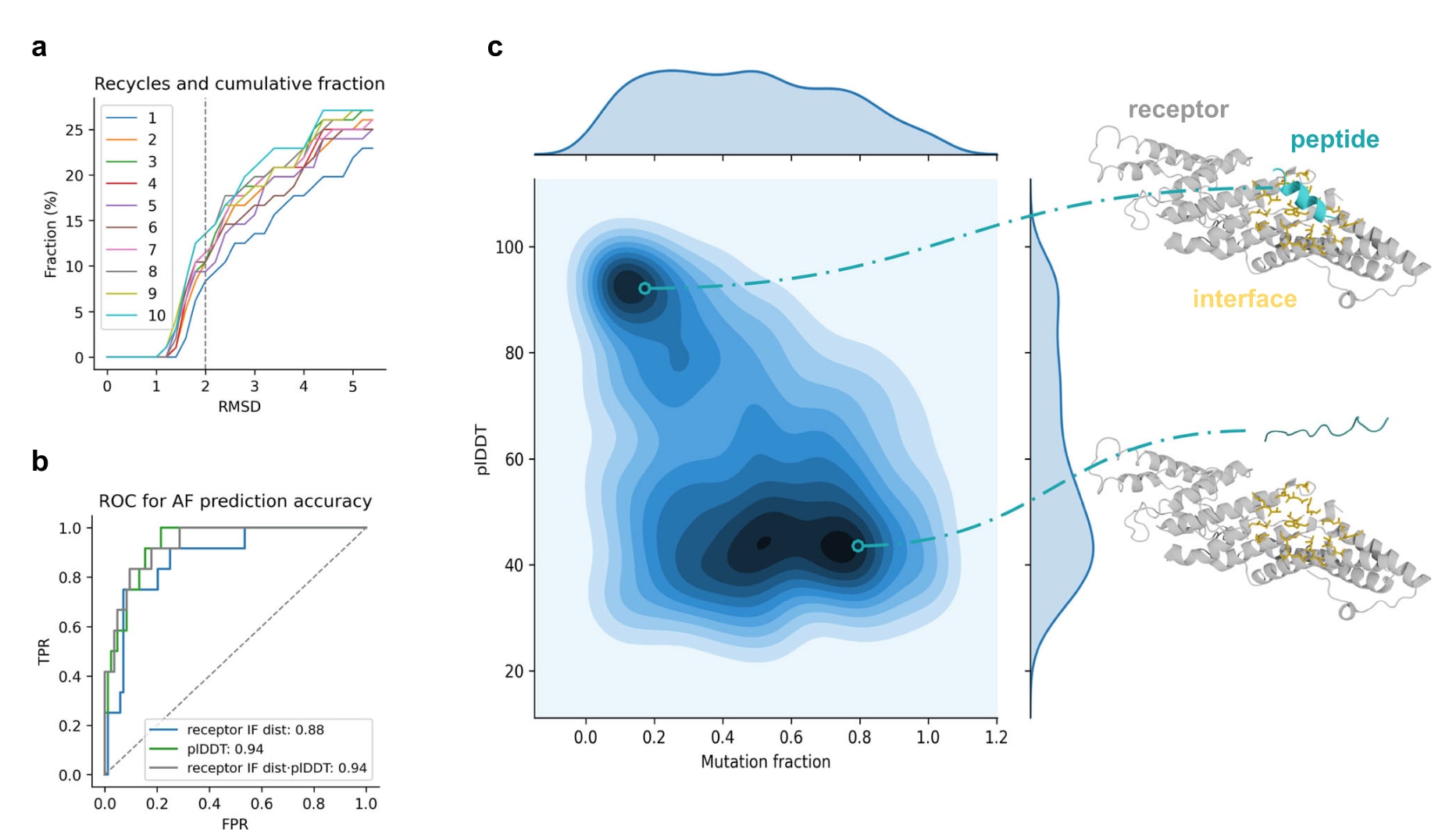
图1 | AlphaFold2预测准确性的概览 a. 循环次数对预测结果的影响。通过平均肽–受体界面(IF) RMSD进行评估。界面(IF)定义为肽与受体之间8 Å范围内的所有Cβ原子。界面RMSD是在将预测与天然受体结构的Cα原子对齐后计算的。图中展示了累计比例与RMSD阈值(截断在5 Å)的关系。结果表明,当循环次数≥8时,在2 Å阈值(灰色虚线)下不再有改进,不过1次与8次循环的差异仍然存在(分别为6.25%和12.5%)。b. ROC曲线。在8次循环的条件下,RMSD≤2 Å的预测作为正样本(n=12),其余为负样本(n=84)。结果显示,肽的plDDT达到最高AUC=0.94。当将plDDT与受体目标残基到肽的距离结合时,在低假阳性率下真阳性率略有提升,AUC并未降低。c. plDDT与突变比例的关系。以能够在2 Å RMSD内预测的12条肽为对象,考察界面残基突变比例(相对肽总长度)。总计得到1160个样本,每个界面残基生成10条突变序列。结果显示,突变比例较低的肽往往具有更高的plDDT。图中给出PDB ID 3c3o的示例(灰色):低突变比例的肽(青色,上方)结构有序且靠近界面(橙色),而高突变比例的肽(下方)则无序且远离界面。
2.1 蛋白–肽设计中的损失函数确定与验证
2.1.1 最优的蛋白–肽结构预测
AlphaFold2(Af)能够通过多序列比对(MSA)表示靶蛋白结构,单序列表示肽,从而预测蛋白–肽复合物的三维结构。已有研究表明,当循环次数增加至9次并取10个模型中的最佳结果时,该方法表现最佳。然而,若将AF作为序列搜索的目标函数,增加循环次数并不高效,而“取前10”策略也不现实。
研究者以96条非冗余肽为测试集,对1–10次循环下的top-1预测结果进行了评估。界面被定义为肽与受体蛋白之间所有在8 Å以内的Cβ原子集合,界面RMSD通过对齐预测结构与天然结构的Cα原子计算。结果显示,当循环次数超过8次后,预测精度不再提升,在96条肽中仅有12条(12.5%)达到2 Å RMSD以内,而在1次循环下只有6条达到该标准。
由于并非所有肽序列都能在真实位置被准确预测,研究进一步考察了能否通过指标区分预测是否可靠。分析中采用了预测的lDDT分数(plDDT),该指标衡量AF对每个残基预测的准确度,同时计算靶界面残基到肽原子间的最短平均距离。ROC曲线显示,若以RMSD≤2 Å作为正样本,plDDT在区分真实结合物时的AUC可达0.94。当plDDT与受体界面距离结合使用时,真阳性率在低假阳性率下有所提升,但仍遗漏了大量真实结合物(96条中仅12条被正确识别)。
总体来看,这表明虽然AlphaFold2在蛋白–肽相互作用预测上整体准确性有限,但一旦预测准确,肽通常位于目标界面附近并伴随较高的plDDT分数。
2.1.2 AlphaFold2区分真实结合物与突变结合物
AlphaFold2可能已经学习到某些结合口袋或潜在界面区域,因此任意序列都有可能被预测与这些区域发生作用。为了验证其是否能够区分不同的潜在肽序列,研究者对先前能在2 Å RMSD内被准确预测的12条肽进行了残基突变。具体做法是随机改变这些肽在天然状态下与受体接触的氨基酸残基,并使用8次循环的AF重新预测其结构。对每个接触残基数目,引入10条新的序列,最终得到1160条突变肽(平均长度12个残基)。
结果显示,低突变比例的肽普遍保持较高plDDT,其中大量样本的plDDT高于80且突变率低于20%。与此同时,受体界面距离随突变比例增加而上升。那些突变比例较低、且与受体界面平均距离较小的肽往往拥有高plDDT。部分突变肽可能仍能在一定程度上结合靶界面,这解释了为何部分突变体仍接近目标界面,但AF对其结合位置的置信度明显下降,表现为plDDT降低。
基于这些观察,研究者提出了一个新的损失函数(Eq.1),用于后续结合物的评价:
其中,
这一损失函数通过综合考虑预测置信度、界面距离以及质心对齐偏差,能够更有效地筛选出真实结合物。
2.1.3 从实验数据中筛选真实结合物
小型蛋白(miniproteins)是由不超过70个氨基酸残基组成的蛋白骨架,这类蛋白曾被设计用于结合特定靶点界面,但成功率有限。为了验证经过修改的AlphaFold2预测流程是否能够区分这些结合物,研究者分析了针对四种不同受体蛋白的序列,这些复合物的结构已被解析(FGFR2、TrkA、IL7Ra和VirB8)。这些结合物在实验中通过流式细胞术(FACS)检测其在酵母表面与受体结合的次数,并以新一代测序(NGS计数)的方式量化。研究对NGS计数低于1000的1000条序列(为降低计算成本)以及所有高于1000的序列进行了采样,并采用相同的AlphaFold2预测流程生成受体–小型蛋白复合物结构。
结果显示,在NGS计数为零时,结合物更倾向于产生高损失值,而在计数约为0.04时损失值接近0。因此,当以归一化NGS计数的0.01为成功阈值时,可以在10%的假阳性率下选出约20%的结合物。尽管这一比例并不理想,但仍提高了成功筛选的可能性。与突变肽结合物相比,小型蛋白的平均plDDT更高(84对比58),这表明AlphaFold2对于游离状态下肽的残基位置不够确定,而对于小型蛋白则更加稳定。这与肽的高柔性相吻合,肽往往只有在结合后才稳定成天然构象。因此,单独依赖plDDT不足以评估结合物,损失函数(Eq.1)的引入显得尤为必要。
2.1.4 损失函数的实验验证
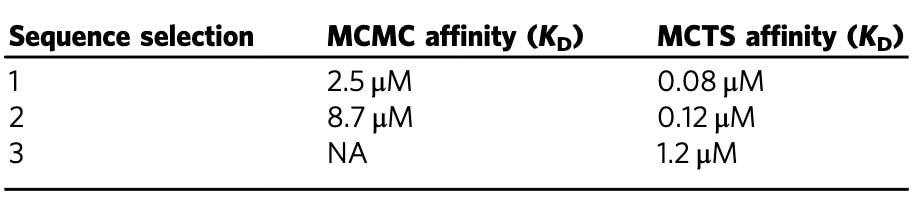
该损失函数(Eq.1)还在独立研究中得到验证,例如针对PDB ID 1ssc的目标。结果显示该损失函数在筛选真实结合物方面具有极高准确性,并被用于比较两种序列设计搜索方法:马尔科夫链蒙特卡洛(MCMC)与蒙特卡洛树搜索(MCTS)。结果表明,关键在于找到能够产生低损失的序列。使用MCMC时,三条设计序列中有两条表现出微摩尔(μM)级亲和力,最佳设计的KD为2.5 μM;而使用MCTS时,最佳设计的KD达到0.08 μM,且所有设计均表现出μM级亲和力。这进一步证明了损失函数的有效性,并凸显其在结合物设计中的核心作用。
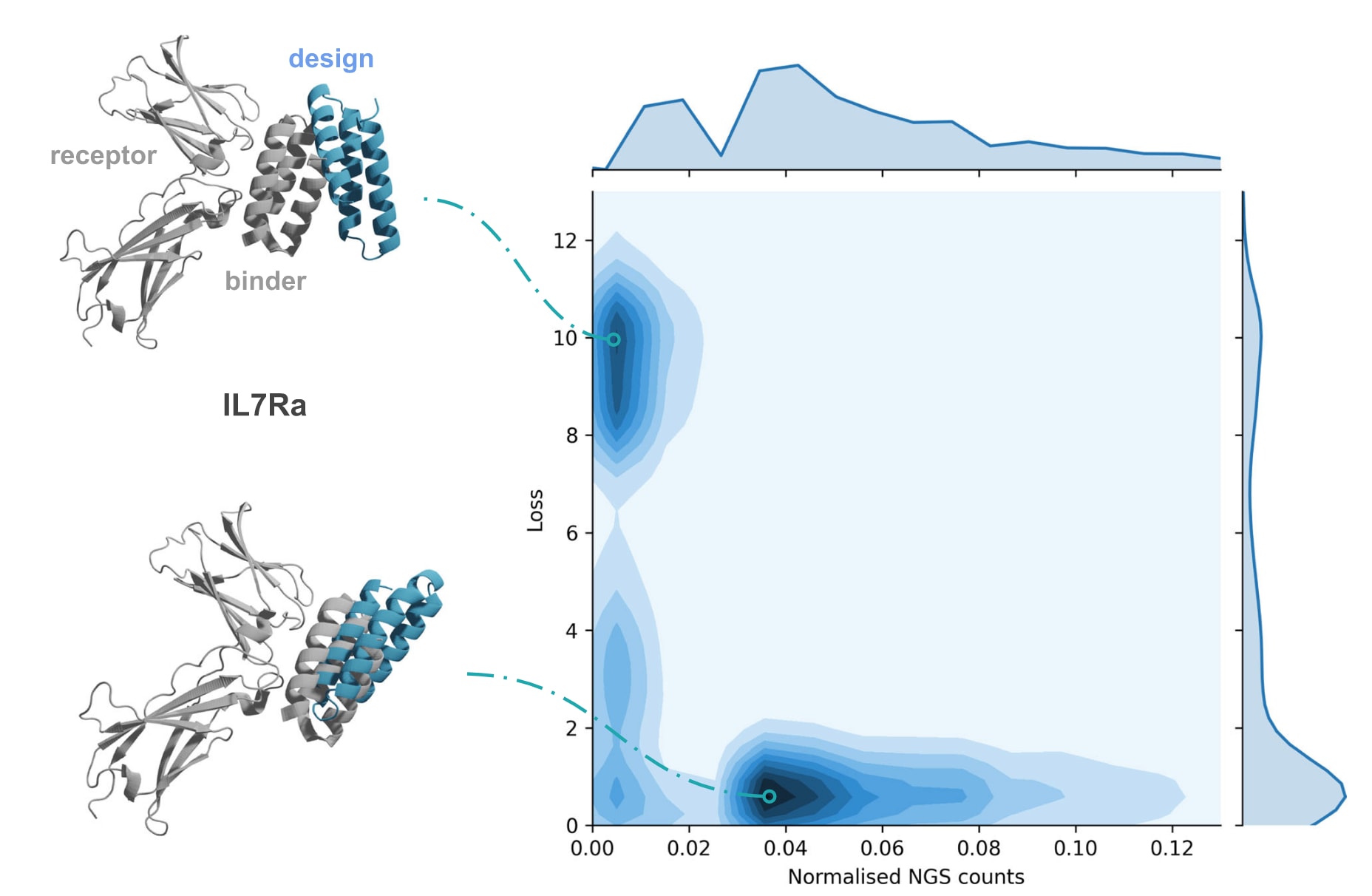
图2 | 损失函数与标准化NGS计数的关系 图中展示了四个已解析结构体系下的小型蛋白结合物设计结果(总计5578个,其中2782个的NGS计数为0)。横轴为标准化NGS计数(以最大值归一化),纵轴为损失值。坐标范围分别截断在13(损失值)和0.13(标准化计数),完整范围见补充图4。以IL7Ra为例,图中提供了高损失与低损失设计的结构示例:天然结构以灰色表示,设计结构以蓝色表示。结果显示,低损失值与较高的NGS计数相关,而高损失值往往对应NGS计数为零。基于损失函数筛选结合物的ROC曲线见补充图3,相关数据见补充数据2。
2.2 结合物设计
目前PDB中约有20万个结构。研究者构建了一个联合框架(图3),利用Foldseek在已知结构中搜索并生成种子,再结合改进后的蛋白设计方法ESM-IF1,生成受体–结合肽序列,并通过AlphaFold2进行评估(详细数据见补充数据3)。在实验中,研究者选取了1463个非冗余蛋白复合物,覆盖PDB中的2926个独特异源界面。每个靶点仅使用一个种子进行评估,尽管在实际应用中可以使用更多。值得强调的是,ESM-IF1与AF均未见过这些复合物,因为它们只在单链蛋白上训练。
在具体操作中,研究者从每个相互作用伙伴中截取连续的10–50个残基片段,类似于Foldseek在PDB中的搜索结果。截取片段与目标蛋白拼接,中间插入10个残基的mask,再将所得主链输入ESM-IF1以生成结合序列。每个界面仅生成一个序列,用于验证零样本设计(zero-shot)的能力。结果如图4a所示,通过定制的损失函数(Eq.1)作为结合评分,能够筛选出界面RMSD较低、接触恢复率大于80%的结合物,表明设计效果显著。如果AF不是一个有效的评估器,所有序列都可能集中或完全不落在界面,接触恢复率也会很低。而AF能够成功区分真实结合序列(已有实验验证),体现了其预测蛋白复合物的强大泛化能力。
在评估表现上,使用损失函数挑选2 Å RMSD以内设计的AUC ROC达到0.96(图4b)。在10%的假阳性率下,可以识别出87%的成功设计(RMSD≤2 Å)。成功率在10残基设计时最高(1.9%),随着片段长度增加而略有下降(图4c),这可能源于短序列中结合信号更强(图4d)。许多短肽只有在与受体结合时才获得稳定结构,因此更依赖链间作用。另一方面,短序列的界面序列恢复率低于长序列(图4e),这凸显了评估函数的重要性,以及AF在预测设计时的准确性。
总体而言,研究最终针对137个独特靶点界面生成了205个成功设计。
表1 针对PDB ID 1ssc的三种序列设计的结合亲和力(KD)
2.3 结合物设计的收敛性
零样本分析(图4)表明,10个残基长度最容易产生成功设计。在蛋白设计中通常会评估多个序列以提升成功率,而采样温度(或噪声)调节了生成的多样性:温度越低,结果越确定性。在零样本分析中,采样温度设为
为了分析生成多少样本才能获得成功设计(RMSD≤2 Å),研究者针对每个种子在10个残基长度和采样温度为1的条件下生成100个多样化样本。结果显示,平均成功率在80个样本时达到约6.4%并趋于稳定(图5a),整体成功率为6.5%,覆盖2843个界面中的185个,相较零样本方法(1.9%)提升约3.4倍。若使用损失函数阈值1并结合plDDT>80的标准,可筛选出131个成功设计,真阳性率(TPR)高达95%。损失值、RMSD与接触恢复率之间的关系与图4a一致(Supplementary Fig. 5),结合物界面RMSD与损失函数的Spearman相关系数为0.87。
随着采样序列数增加,成功率提高,且可通过损失函数区分准确设计,就像零样本评估时一样。进一步比较成功与失败设计(n=185)在界面残基的序列恢复率,发现平均plDDT随界面残基恢复率增加而升高(图5b)。当序列恢复率超过80%时,平均plDDT为84,平均界面RMSD仅0.43。这与图1c中已知结构肽的结果一致,说明高plDDT值对应低界面RMSD和高序列恢复率。
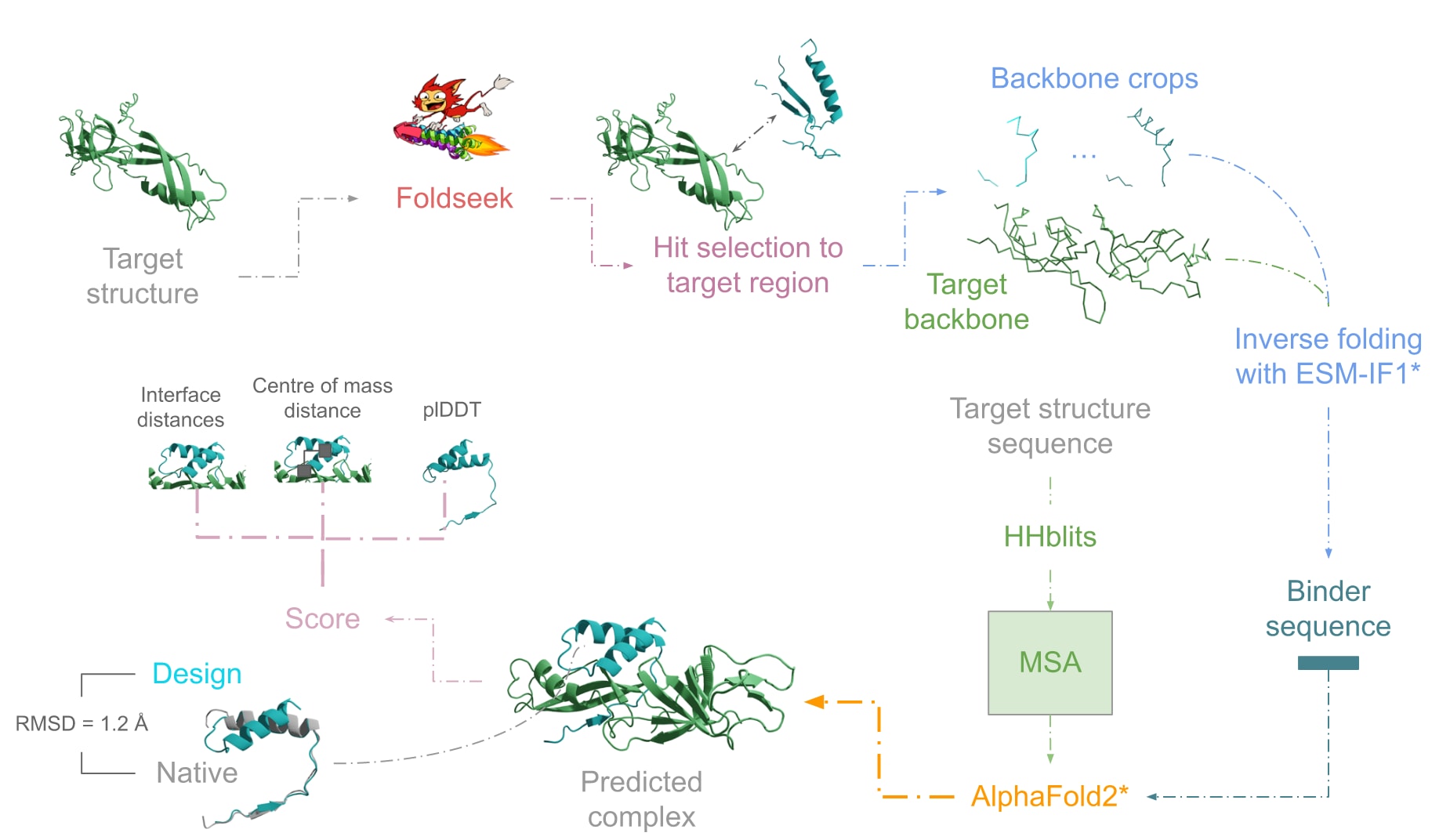
图3 | 设计流程示意图 设计流程以目标结构(预测或实验解析)作为输入,首先利用Foldseek进行搜索以获得候选结构。随后选择与目标界面接触密度最高的匹配结果,并通过ESM-IF1逆折叠生成结合物序列。接着,将目标结构序列用HHblits在Uniclust30数据库中进行搜索,得到的MSA与设计的序列一起输入,用于预测蛋白复合物结构。生成的设计通过定制损失函数进行评估,该损失结合了界面距离、质心偏移与plDDT三个指标。图中展示了2RF4_A–2RF4_B的实例,最终设计出的结合物与天然种子在结构上进行叠合,在界面位置的RMSD为1.2 Å,表明设计结果与天然结合物高度一致。
2.4 设计结合物的特异性
在结合物设计中,理想情况是仅结合目标界面而不与其他蛋白发生作用。为评估特异性,研究者选择了在2 Å RMSD内成功预测的185条最佳设计序列,并将其与100个随机挑选的受体蛋白进行交叉预测,这些受体来自其他非冗余异源二聚体复合物。尽管这些随机受体与目标受体差异显著,但该分析有助于验证结合物对特定界面类别的选择性。
结果显示(图5c),在与目标受体结合时,结合物的plDDT分布集中在60–90之间,而在随机受体上的plDDT则持续较低,分布在30–60之间。中位值方面,非目标受体的plDDT为45,而目标受体为72,高出约60%。这表明AF不会为非预期的相互作用给出高置信度预测。以plDDT作为判别指标时,ROC曲线的AUC达到0.96,在10%假阳性率下可实现100%真阳性率(补充图6)。
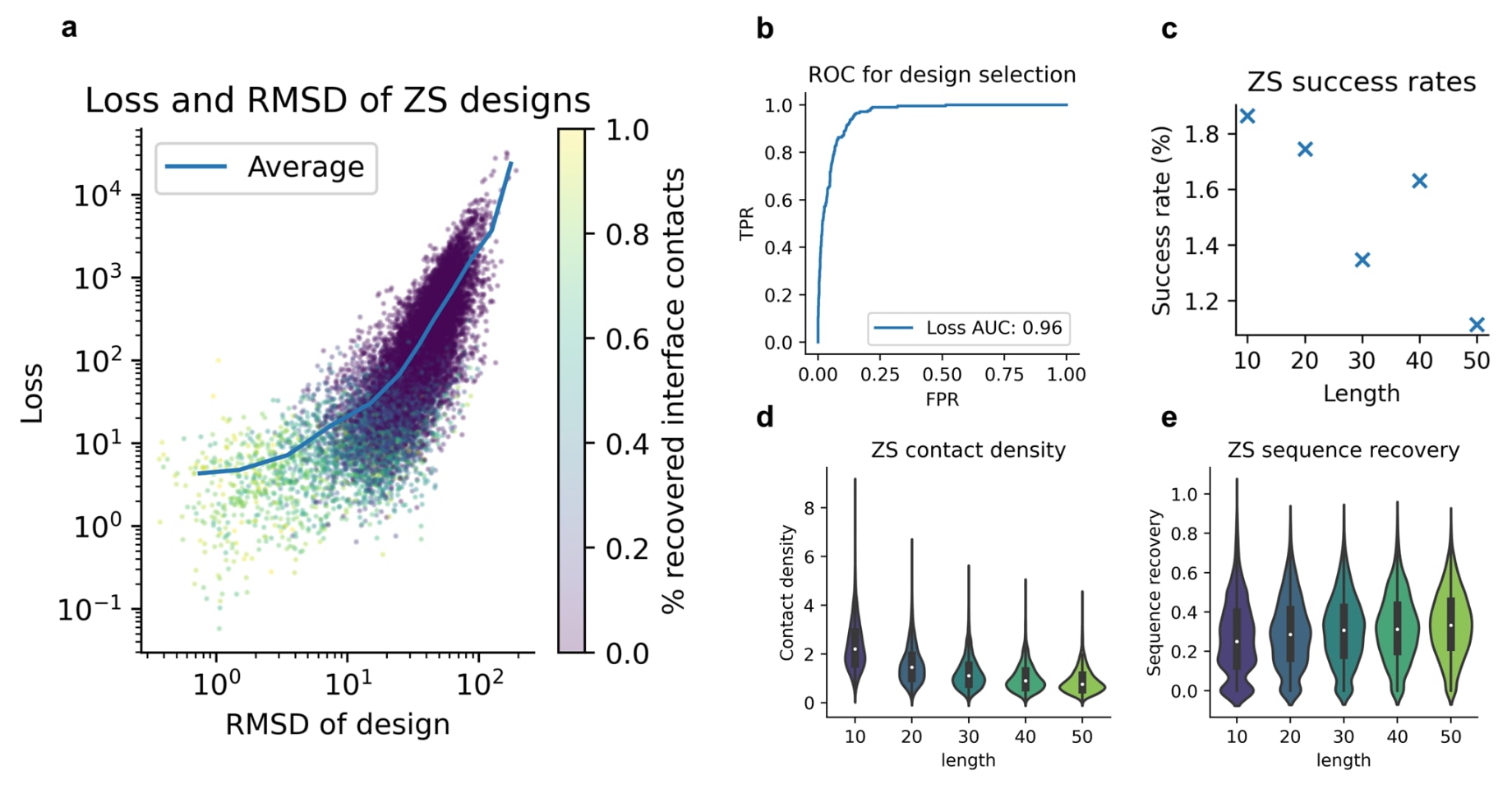
图4 | 零样本(ZS)设计分析 a. 损失值与界面RMSD的关系。展示了AlphaFold2预测的设计结合物与天然结合物在界面上的差异(n=13,216),Spearman相关系数为0.81。点的颜色表示界面接触恢复率。当损失值较低时,设计的RMSD也低,且接触恢复率较高(>80%)。b. ROC曲线。用于筛选界面RMSD≤2 Å的设计结合物。结果表明,利用损失函数时ROC AUC为0.96。在10%假阳性率下,可以选出87%的成功设计(损失阈值=0.11)。c. 成功率与设计长度的关系。考察不同长度设计的成功率(n=13,156,其中n10=2895, n20=2805, n30=2672, n40=2451, n50=2333)。结果显示,最短设计(10个残基)的成功率最高(1.9%),最长设计(50个残基)最低(1.1%)。总体上,共获得205个成功设计,覆盖136个独特靶点界面(更多数据见补充表1)。d. 接触密度与长度。结果表明,短序列的单位残基接触数更高,使其相互作用更容易被预测,从而解释了较高的成功率。黑框为四分位数范围,白点表示中位数。e. 界面序列恢复率与长度。短序列的中位恢复率最低,但差异仅为几个百分点。黑框为四分位数范围,白点表示中位数。
2.5 失败设计的原因分析
前文已表明AF能够准确评估可预测的结合物,因此设计任务的关键在于生成AF能够稳定预测的序列。结果显示,成功设计率为6.5%,意味着仍有93.5%的界面未成功。为探讨失败原因,研究者考察了界面是否与特定二级结构相关(图6a),结果发现界面二级结构比例与RMSD无关,表明并不存在针对特定结构界面的偏好。
进一步分析发现,影响成败的关键因素在于接触密度以及目标蛋白MSA中的进化信息(以有效序列数Neff衡量)。结果表明,接触密度与设计成功率高度相关(Spearman R=0.93)。在接触密度为8.8时,成功率可达67%,而在密度仅为1时成功率不足1%。这说明获得合适的种子对设计至关重要,且短序列更容易满足该条件(图4d)。相比之下,Neff与RMSD之间没有明显关系,表明无论进化信息丰富与否,都有可能生成成功设计。
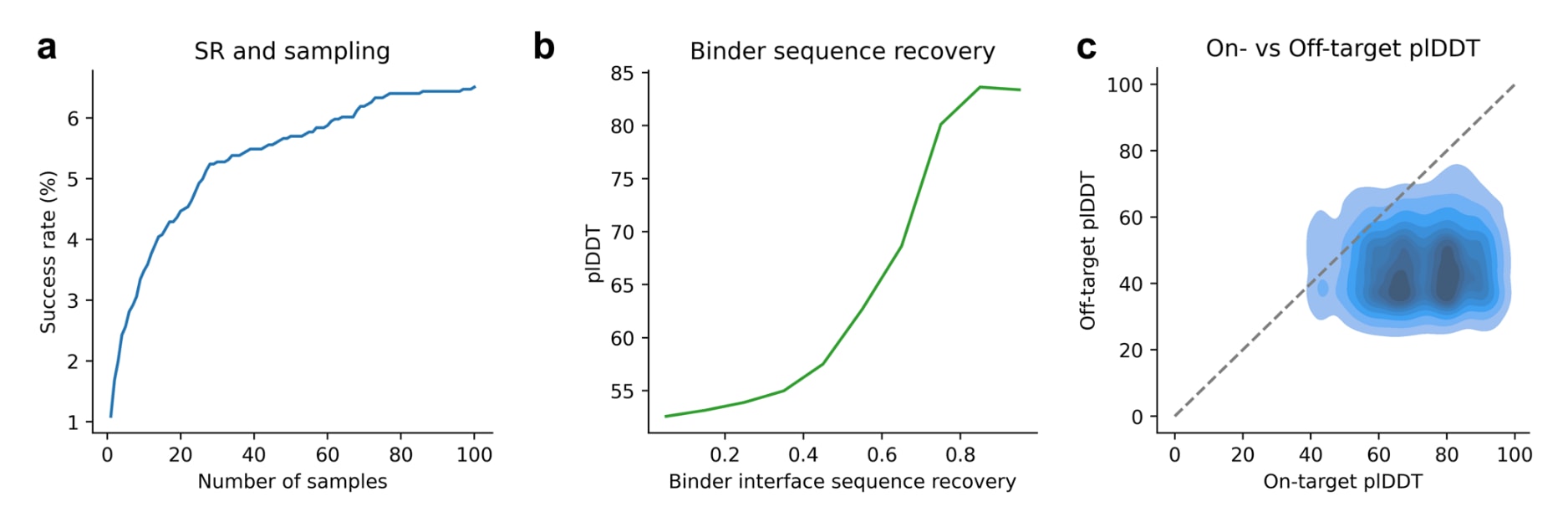
图5 | 成功率与序列恢复的概览 a. 成功率与设计数量的关系。在2843个界面上生成1–100个设计后,若至少有一个设计的界面RMSD≤2 Å,则视为成功。总体成功率为6.5%,共获得185个独特界面的成功设计(详细数据见补充表1)。b. 界面序列恢复率与平均plDDT的关系。在成功设计(n=18,500)中,低序列恢复率对应低plDDT(约50)。随着序列恢复率增加,平均plDDT逐渐升高。当恢复率超过80%时,平均plDDT达到84,说明高恢复率对应高预测置信度(见补充表1)。c. 结合特异性分析。在185个成功设计中,将其与100个随机选择的受体蛋白进行交叉预测(n=18,500)。结果表明,与非目标受体相比,设计在目标受体上的plDDT始终更高:目标受体中位plDDT为72,而非目标为45。相关数据见补充数据4。
2.6 与其他蛋白设计方法的比较
研究将该方法与最新的ProteinMPNN进行对比。ProteinMPNN已被证明显著优于传统的蛋白设计基础方法Rosetta。需要注意的是,ESM-IF1主要在更具挑战性的异源界面上进行评估,而ProteinMPNN的测试集中绝大多数(99.3%,n=677)为同源界面,这些界面往往可以从受体序列中推断出来。
在对比实验中,两种方法均以10残基的种子长度为条件,每个界面采样100条序列,共对1172个界面进行了100个设计的评估(图6f)。结果显示,当以界面RMSD≤2 Å作为成功标准时,ESM-IF1的成功率为3.6%,ProteinMPNN为1.5%,分别获得42个与18个准确设计。由此可见,ESM-IF1在成功率上比ProteinMPNN高出约2.3倍。
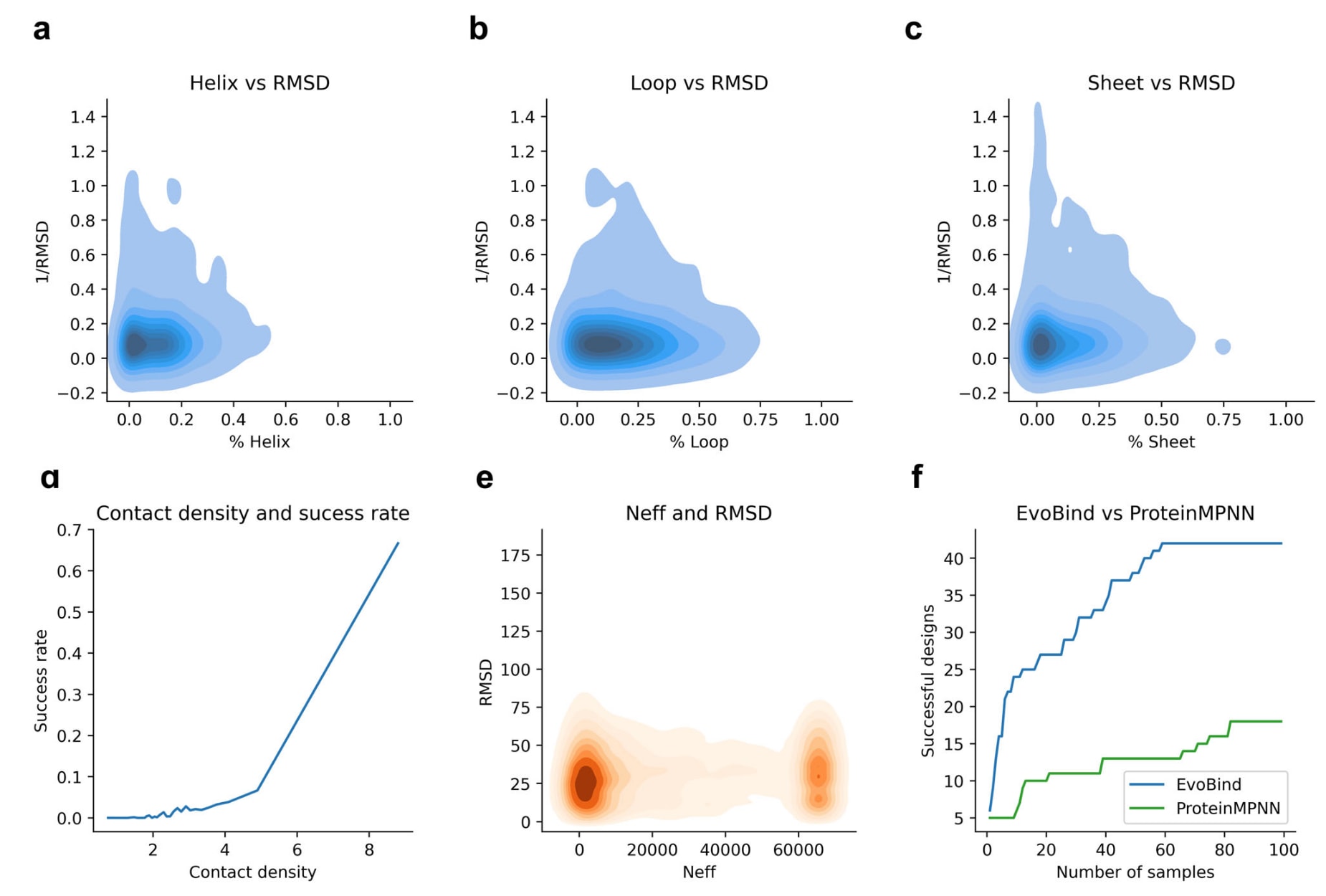
图6 | 不同结构组别的成功率概览 a–c. 受体界面类型与界面RMSD⁻¹。横轴为不同界面类型比例,纵轴为界面RMSD⁻¹(取倒数,因此数值越高表示RMSD越低)。结果显示界面类型与RMSD无相关性,说明设计过程对特定界面没有偏好。d. 接触密度与成功率。以所有设计(n=280,553)为基础,按照接触密度划分为30个等份(每份n=9345),计算平均接触密度与成功率的关系。结果显示二者高度相关(Spearman R=0.93)。在最高接触密度(8.8)时,成功率达到67%(详细结果见补充表1)。e. 有效序列数(Neff)与RMSD。分析MSA中的Neff与设计RMSD之间的关系,结果显示二者无显著相关性,表明无论进化信息多寡,均可能产生成功设计。f. ESM-IF1与ProteinMPNN的对比。在1172个界面上,每个界面生成1–100条序列进行比较。当以界面RMSD≤2 Å为成功标准时,ESM-IF1的成功率为3.6%,ProteinMPNN为1.5%,分别得到42与18个准确设计。ESM-IF1的表现约为ProteinMPNN的2.3倍。相关数据见补充数据5。
3 结论
研究表明,AlphaFold2能够区分真实结合物与突变结合物,并由此构建出一个损失函数,可从实验数据中筛选出真实的小型蛋白结合物,这与近期研究结果高度一致。基于PDB中所有非冗余异源界面评估显示,在零样本条件下(每个靶点仅设计一个序列),成功结合物(界面RMSD≤2 Å)的筛选ROC AUC达到0.96。当针对每个靶点生成100条序列时,ESM-IF1能够在185个异源界面上设计出成功结合物,占已知界面的6.5%。
设计结果显示,结合物具有较强的界面特异性:在目标受体上的中位plDDT为72,而在非目标受体上仅为45。进一步分析发现,设计成功与否并不依赖于界面类型或MSA深度(Neff),而是由接触密度决定。当接触密度较低时,成功率不足1%;而在接触密度达到8.8时,成功率可达67%。在对比实验中,ESM-IF1在1172个同源界面上的成功率为3.6%,ProteinMPNN为1.5%(分别得到42和18个成功设计),表现出2.3倍优势,预计在异源界面上的性能几乎是同源界面的两倍。
人工智能的快速发展正在推动结构生物学和蛋白设计的革命。然而,大部分界面的准确结合物仍难以生成(异源界面失败率93.5%,同源界面失败率96.4%)。这一挑战有望通过获得更优质的支架来解决:当支架具有高接触密度和低损失值时,ESM-IF1能够生成更精确的设计,并以较高置信度被筛选出来。但需要注意,预测结果仍可能受到AlphaFold2错误构象预测的影响。随着更多蛋白结构被快速解析,未来有望获得更高质量、包含多重构象的支架,从而进一步提升结合物设计的成功率。