arXiv 2025 | SimpleFold: 蛋白折叠,大道至简

获取详情及资源:
0 摘要
蛋白质折叠模型的突破性成果,通常依赖于将领域知识融入模型架构与训练流程的组合方式。然而,随着生成模型在多个相关任务上的成功,便引出了一个自然的问题:这些复杂的架构设计是否真的是高性能模型的必要条件?
在这项研究中,提出了SimpleFold,这是首个基于流匹配(flow-matching)的蛋白质折叠模型,并且完全由通用的Transformer模块构成。传统的蛋白质折叠模型往往需要计算量庞大的组件,例如三角更新、显式的残基对表示,或是为该领域特别设计的多重训练目标。而SimpleFold仅依赖标准的Transformer模块,配合自适应层,并通过生成式的流匹配目标函数进行训练,同时引入一个额外的结构项。
研究将SimpleFold扩展到30亿参数规模,并在约900万条蒸馏蛋白质结构数据与实验PDB数据上进行训练。在标准折叠基准测试中,SimpleFold-3B取得了与当前最先进方法相当的性能,同时在集成预测(ensemble prediction)任务上展现出强大优势——这一点对基于确定性重建目标训练的模型而言尤为困难。得益于其通用化的架构,SimpleFold在消费级硬件上的部署与推理也表现出高效性。
总体而言,SimpleFold挑战了蛋白质折叠领域对复杂、特定架构的依赖,为未来的模型设计开辟了新的思路与空间。
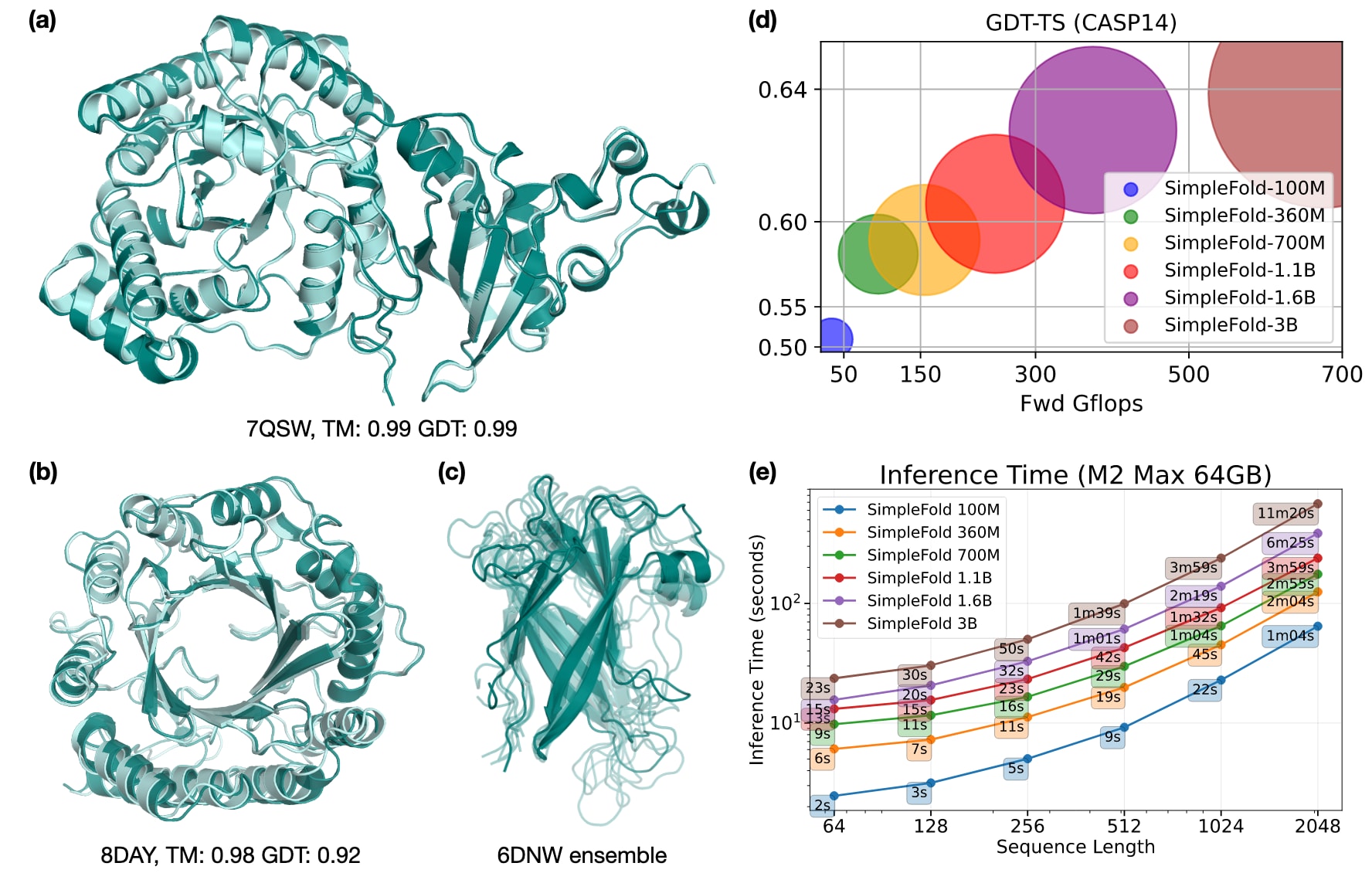
图1 | SimpleFold在不同目标上的预测示例 (a) 7QSW的A链(大亚基RubisCO),(b) 8DAY的A链(二甲基烯丙基色氨酸合酶1),其中真实结构以浅青色表示,预测结构以深青色表示。(c) 基于MD集成数据微调的SimpleFold在6NDW的B链(鞭毛钩蛋白FlgE)上生成的构象集合。(d) SimpleFold在CASP14上的表现,随着模型规模从1亿到30亿参数逐步提升。(e) 不同规模的SimpleFold在消费级硬件上的推理耗时示例,即M2 Max 64GB Macbook Pro。
1 引言
蛋白质折叠,即根据氨基酸序列预测其三维原子结构,是计算生物学中一项长期且极具挑战性的任务,对药物发现等领域有着深远影响。该研究从生成式建模视角切入,不对蛋白结构的自然生成过程作过强假设,而是借鉴视觉生成模型(如文本生成图像、文本生成3D)的思路,将氨基酸序列视为“文本提示”,由生成模型直接输出全原子三维坐标。
传统折叠模型如AlphaFold2与RoseTTAFold依赖复杂且高成本的领域专用模块,例如多序列比对(MSA)、残基对表示及三角更新,这些设计旨在硬编码现有结构生成知识。然而研究表明,对于缺乏同源蛋白的孤儿蛋白,基于蛋白语言模型的策略甚至优于依赖MSA的方法。因此,该研究提出一种彻底摆脱领域特定组件的思路,转向通用的Transformer骨干架构,并通过流匹配(flow matching)生成目标进行端到端训练。
生成模型天然能够捕捉结构的不确定性,不仅可输出单一解构,还能生成可行构象的集合,更符合自然状态下的蛋白折叠过程。尽管近年来已有基于扩散或流的折叠尝试,但大多仍沿用了昂贵的AlphaFold2式模块。SimpleFold则在方法上实现了突破:不依赖MSA、残基对表示、三角更新或几何等变模块,而是基于标准Transformer与自适应层,直接将序列映射到三维结构。
主要贡献包括:
- 将蛋白质折叠重新表述为条件生成任务,提出SimpleFold——首个基于流匹配的Transformer折叠模型。
- 将模型规模扩展至30亿参数,并利用约900万条蒸馏结构与PDB实验数据进行训练。
- SimpleFold-3B在折叠基准测试中取得与依赖启发式设计的先进方法相当的性能,并在集成预测中表现突出。
- 发布从1亿参数到30亿参数的完整模型族,其中SimpleFold-100M在主流基准上恢复了约90%的性能,同时在消费级硬件上推理效率极高。
综上,SimpleFold打破了蛋白质折叠模型必须依赖复杂架构设计的传统观念,展现出一种更简洁而高效的替代路径。
2 SimpleFold
2.1 流匹配(Flow-Matching)预备知识
流匹配生成模型将生成过程视为一个随时间演化的过程,通过对常微分方程(ODE)的积分,将噪声逐步映射为数据。在时间区间
在实际中,这一变换由一个可学习的时间依赖速度场
其作用是将噪声逐渐映射为数据。
在训练时,采用线性插值路径(linear interpolant path),也称为校正流(rectified flow)。具体做法是,在经验数据分布
此时目标速度定义为
这一方法能够提供与真实(不可解)边际得分一致的梯度。已有研究表明,在高斯边际分布下,扩散模型与流匹配在超参数变换的意义下等价。
2.2 基于流匹配的折叠
SimpleFold将蛋白质折叠建模为一个条件流匹配生成模型,从噪声中生成结构,条件输入为氨基酸序列。这种“氨基酸序列→蛋白质结构”的生成方式,在概念上与计算机视觉中的“文本生成图像”或“文本生成3D”模型非常相似。
具体而言,假设蛋白含有
训练目标 网络
其中
此外,引入了一个局部距离差异测试(Local Distance Difference Test, LDDT)损失,用于度量生成结构
LDDT损失定义为:
其中
最终损失函数为流匹配项与LDDT项的加权组合:
其中
时间步重采样 为了提升训练效率,并促使模型生成更精细的结构,研究在时间步
其中,
这种偏移能够显著改善生成样本的质量,尤其是在对侧链原子等精细结构进行建模时效果突出。原因在于,蛋白质结构本身存在明显的由粗到细的层级关系:二级结构→Cα骨架→侧链。通过在接近数据流形的区域进行过采样,模型能够更好地学习并还原这些细致的原子位置。
关于LDDT损失与时间步重采样的更多细节可见附录C.1。
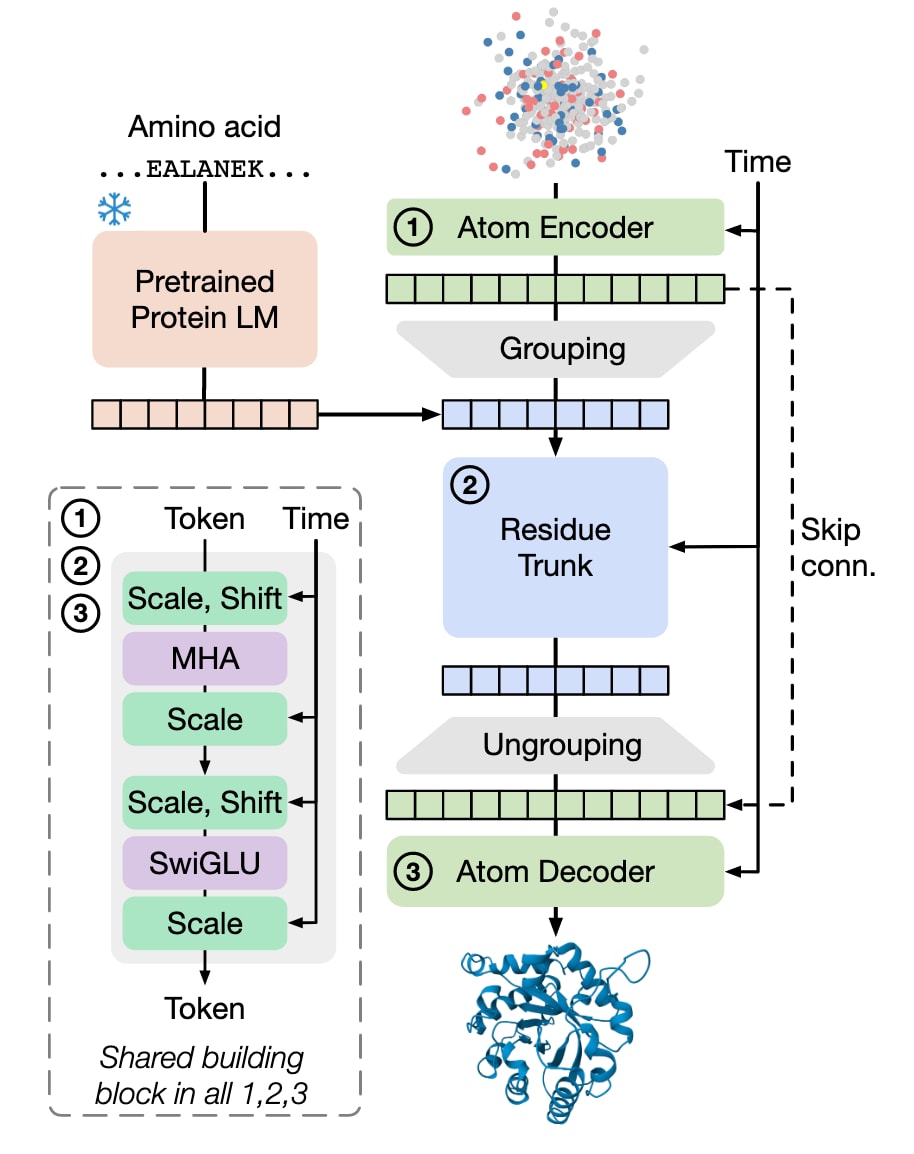
图2 | SimpleFold架构概览。 整体基于通用的标准Transformer模块,并结合自适应层构建。原子编码器、残基主干网络与原子解码器均共享相同的基础模块。该设计避免了对成对表示与三角更新的依赖,实现了更简洁高效的结构建模。
2.3 架构
自AlphaFold2提出以来,诸如三角更新(triangle updates)以及单一表示与成对表示的显式交互建模等组件,几乎成为蛋白折叠模型的标准设计。但这些架构选择是否是构建高性能模型的必要条件,仍然是一个开放性问题。与以往方法形成鲜明对比,SimpleFold完全基于通用的Transformer模块来搭建架构(见图5对比)。
如图2所示,SimpleFold包含三大模块:轻量化的原子编码器与解码器(对称设置,块数与隐藏维度一致)以及一个计算量更重的残基主干网络。所有模块均采用带有时间步
原子编码器输入“带噪”的原子坐标
与文本生成图像/3D的做法类似,模型引入预训练蛋白语言模型(PLM)对氨基酸序列进行嵌入。所有实验均采用ESM2-3B将序列
最后,原子解码器更新原子token并输出预测速度场
在位置编码上,残基主干采用旋转位置嵌入(RoPE):在注意力块中,序列第
与以往工作显著不同,SimpleFold摒弃了显式成对表示。不像AlphaFold2或ESMFold那样依赖昂贵的MSA嵌入或PLM注意力得分来初始化pair representation,SimpleFold只保留单序列表示,既不需要三角更新,效率也更高。并且,它没有使用等变架构来保证物理意义的几何约束,而是直接建立在非等变Transformer上。为了处理蛋白结构的旋转对称性,训练时采用SO(3)数据增强,通过随机旋转结构目标,并依赖模型自身去学习对称性。
2.4 采样
在推理阶段,给定氨基酸序列
具体而言,采用Euler–Maruyama积分器:
其中
并引入小常数
2.5 置信度模块
为更好地评估生成结构的质量,设计了一个预测LDDT(pLDDT)模块,输出逐残基的LDDT值(范围0–100)作为置信度分数。在折叠模型完全训练后,冻结其参数,再单独训练该模块(见图8)。训练过程中,实时采样结构
pLDDT模块由4层标准Transformer块组成,不含自适应层,以
2.6 基于蒸馏数据的训练
近年来,计算预测的蛋白质结构数量呈爆炸式增长,典型资源包括AlphaFold蛋白质结构数据库(AFDB)与ESM宏基因组图谱,其规模均已达到上亿条。近期提出的AFESM进一步整合了这两大资源,并将其中的蒸馏结构划分为约512万个非单例结构簇。尽管已有部分折叠模型利用蒸馏数据进行训练(通常通过自蒸馏实现),但相比目前公开可得的庞大数据规模,实际应用仍然有限。因此,如何有效利用这些大规模蒸馏数据以训练更强大的折叠模型,仍是一个未被充分研究的问题。
SimpleFold的训练采用三类数据源的混合:
- PDB实验数据:约16万条结构,时间截断至2020年5月,与ESMFold保持一致。
- AFDB的SwissProt子集:筛选平均pLDDT大于85且标准差小于15的样本,得到约27万条蒸馏结构。
- AFESM代表性结构:为每个簇选择代表性结构,并过滤pLDDT大于0.8的样本,共约190万条。
除最大规模模型外,所有SimpleFold均基于以上三类数据进行训练,总量约200万条。
对于SimpleFold-3B,训练集进一步扩展为AFESM-E:在每个簇的代表性结构之外,额外随机采样最多10条平均pLDDT大于80的结构,总计约860万条蒸馏数据。结合PDB与SwissProt,这一更大规模的训练集充分发挥了大模型的容量优势,因此成为SimpleFold-3B的训练基座。
3 相关工作
蛋白质折叠 自AlphaFold2与RoseTTAFold提出以来,基于学习的方法在蛋白质折叠任务上取得了突破性成果,引发了持续研究。AlphaFold2引入了特定领域的网络模块,如三角注意力,并在架构设计上显式建模了单一表示与成对表示的交互,同时依赖MSA提取蛋白序列的进化信息,以贴近生物学专家对数据生成过程的理解。后续工作如OmegaFold与ESMFold用预训练蛋白语言模型的嵌入替代MSA,在推理效率与孤儿蛋白预测上表现尤为突出。部分研究也致力于通过高效实现加速AlphaFold2的模块,如FastFold与MiniFold。这类模型大多基于局部坐标框架的回归目标,而非直接建模全原子坐标,因此在**构象集成预测(ensemble generation)**方面缺乏多样性。
蛋白质的流匹配方法 生成模型,尤其是扩散与流匹配方法,因能生成高质量合理样本而被引入蛋白质折叠。AlphaFlow/ESMFlow通过在AlphaFold2或ESMFold上调优流匹配目标,展现了集成预测的优势。然而,这些方法并非从零开始设计的生成模型,而是依赖于预训练的、基于确定性回归目标训练的折叠模型。AlphaFold3及其一系列复现模型(如Boltz-1、Protenix、Chai-1)则采用扩散方法,用于蛋白复合物及生物分子相互作用的生成。此外,已有工作将扩散或流匹配方法用于从头蛋白质结构生成,如RFDiffusion、Genie-2、P(all-atom)等。但这些方法大多仍依赖AlphaFold系列的启发式架构设计,如昂贵的三角注意力与显式pair表示,有些甚至基于手工构造的等变扩散过程。Proteina尝试简化架构,但仍需显式pair表示,且仅限于Cα结构的生成。
在小分子体系中,MCF曾探索了利用通用Transformer骨干进行构象生成。相比之下,SimpleFold与以往折叠模型形成鲜明对照:它旨在用通用的Transformer骨干网络解决折叠问题,并通过数据直接学习结构生成过程中的对称性,而非依赖繁复的特定领域架构。
4 实验
4.1 实验设置
为验证所提框架在蛋白质折叠任务中的可扩展性,训练了不同规模的SimpleFold模型(100M、360M、700M、1.1B、1.6B与3B)。随着模型规模的增大,同时提升原子编码器、解码器及残基主干网络的深度与隐藏维度(具体参数见表5)。训练时,单GPU上会对同一蛋白复制
预训练 分为预训练与微调两个阶段,主要区别在于数据选择。预训练阶段使用尽可能大的数据集(约200万结构,3B模型约870万),涵盖PDB、AFDB的SwissProt子集及AFESM。最大序列长度设为256,短序列不做填充,长序列裁剪为256残基。损失函数中
微调 阶段在高质量数据(PDB与SwissProt子集)上进行,以提升生成结构的保真度。最大序列长度扩展至512,便于利用更大蛋白结构。为适配显存,批次中的
即在接近真实数据(
pLDDT模块训练 在SimpleFold完成预训练与微调后,冻结折叠模型参数,仅训练pLDDT模块。训练数据为PDB与SwissProt的组合,包含实验与高质量蒸馏结构。训练时
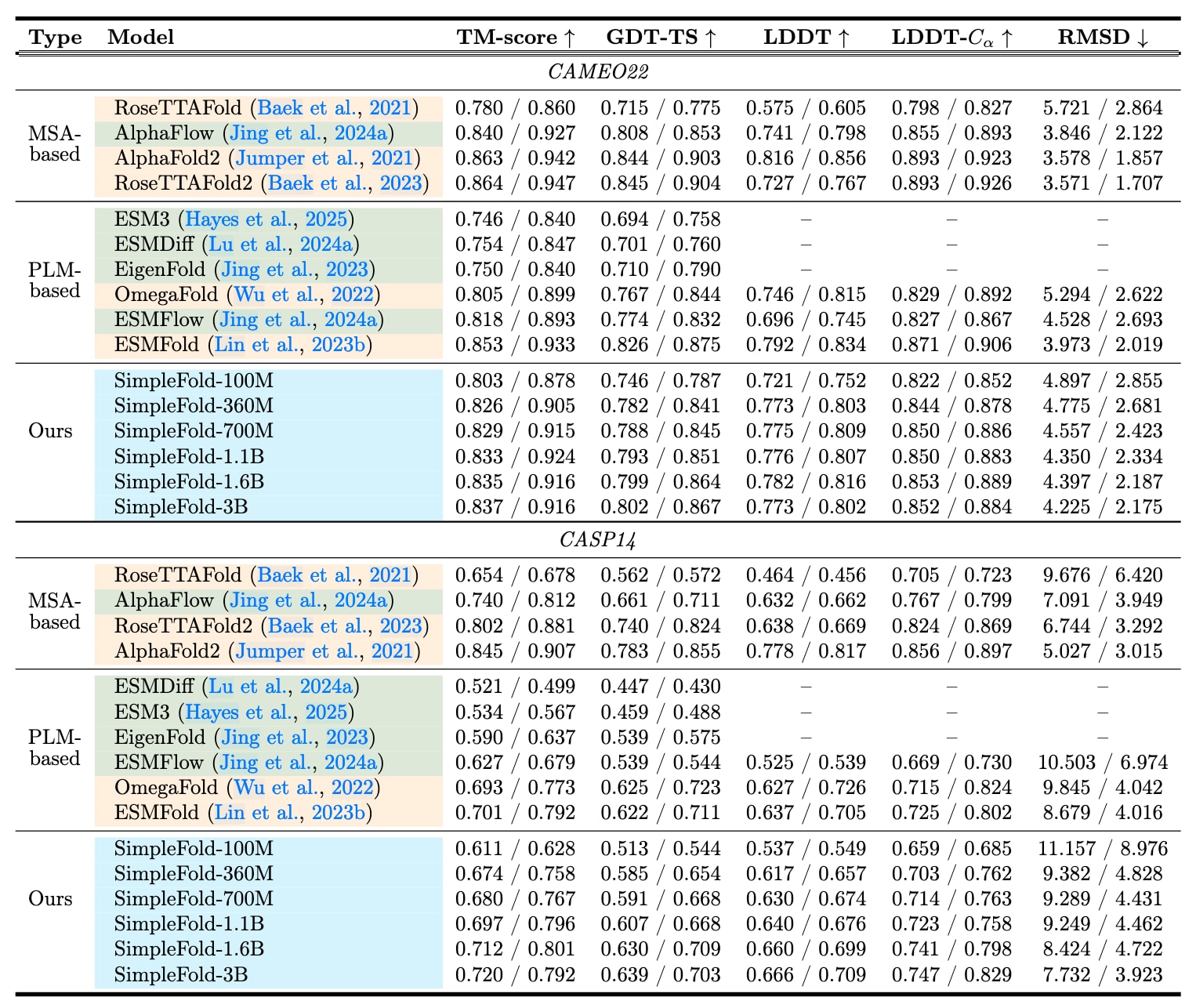
表1 | 蛋白质折叠在CAMEO22(上)与CASP14(下)基准上的性能表现。 各指标均报告所有样本的均值/中位数。其中,橙色表示基于回归目标训练的基线模型,绿色表示基于生成式目标(如扩散/流匹配或自回归)训练的基线模型,蓝色表示SimpleFold,其同样采用生成式目标进行训练,但不依赖MSA。
4.2 蛋白质折叠
研究者在两个被广泛采用的蛋白质结构预测基准上评估了SimpleFold:CAMEO22与CASP14。这两个基准都对折叠模型的泛化性、鲁棒性以及原子级精度提出了严格要求。CAMEO22包含183个目标结构,长度在100至750个残基之间;CASP14则是更具挑战性的盲测平台,此处选取了70个单链蛋白,长度在50至1000个氨基酸之间。在推理中,SimpleFold采用
评估指标包括:TM-score与GDT-TS(全局拓扑相似性)、LDDT与LDDT-Cα(局部原子精度)、以及RMSD(原子坐标均方根偏差)。对于每个指标,报告均值与中位数。对全原子模型报告全部指标,而对仅预测骨架的模型则仅报告TM-score与GDT-TS。表1给出了在两个基准上的结果。
基线方法按序列信息获取方式分组:一类依赖MSA搜索(如RoseTTAFold、RoseTTAFold2、AlphaFold2),另一类使用预训练蛋白语言模型(PLM)嵌入(如ESMFold与OmegaFold)。此外,还区分是否采用生成式目标(扩散、流匹配或自回归),而非直接回归到真实结构。例如,AlphaFlow与ESMFlow即为基于AlphaFold2和ESMFold微调的流匹配模型。值得注意的是,这些微调模型在全部指标上反而落后于原始的回归模型,原因在于CAMEO22与CASP14通常仅提供单一“真实”结构,从而更有利于点对点预测的回归模型。
尽管架构更为简洁,SimpleFold依然取得了与现有基线相当甚至更优的表现。在CAMEO22上,SimpleFold表现接近最优模型(如ESMFold、RoseTTAFold2与AlphaFold2),在多数指标上达到后两者的95%以上,但无需昂贵的三角注意力与MSA。在更具挑战性的CASP14中,SimpleFold表现甚至超过了ESMFold。例如,SimpleFold-3B的TM-score为0.720/0.792、GDT-TS为0.639/0.703,而ESMFold对应结果为0.701/0.792与0.622/0.711。同时,SimpleFold在性能上接近甚至超过一些依赖MSA的模型如RoseTTAFold与AlphaFlow。值得注意的是,除了AlphaFold2外,所有模型在CASP14上的表现都相较CAMEO22有明显下降,但SimpleFold的性能下降幅度更小,显示出其在复杂任务上的鲁棒性。
进一步分析不同规模模型的表现发现:最小的SimpleFold-100M在保持高效率的同时,性能已能达到ESMFold在CAMEO22上的90%以上,证明了通用Transformer架构的有效性。而随着模型规模的扩大,性能整体上持续提升,尤其在CASP14这一更复杂的基准上提升更为显著。这为通用架构在大规模下的潜力提供了有力实证。
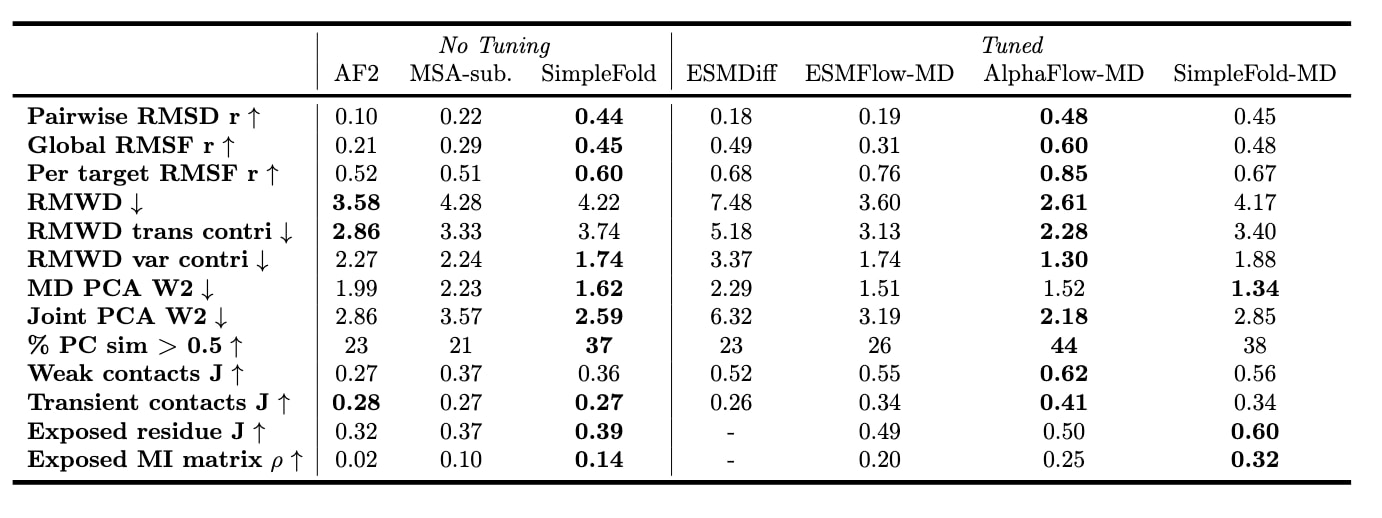
表2 | 在MD构象集上的评估结果。 基线模型的结果取自(Jing et al., 2024a; Lu et al., 2024a),SimpleFold(SF)与SimpleFold-MD(SF-MD)遵循相同的评估流程。
4.3 基于pLDDT的置信度评估
图3(a)展示了一个带有pLDDT评分的预测结构,其中红色与橙色表示低pLDDT,蓝色表示高pLDDT。如图所示,SimpleFold在二级结构区域的预测具有较高置信度,而在柔性环(loop)区域则表现出不确定性。图3(b)与(c)对比了pLDDT与真实LDDT-Cα的关系,评估对象包括CAMEO22的目标以及自2023年1月以来从PDB随机选取的1000条蛋白链。结果表明,pLDDT与LDDT-Cα之间的Pearson相关系数达到0.77,说明SimpleFold的pLDDT模块能够较好地刻画预测结构的整体质量。
需要强调的是,pLDDT模块的输出并不依赖于生成流过程,因此它也可以无缝应用于其他模型的预测质量评估。这一方向将在未来研究中进一步探索。
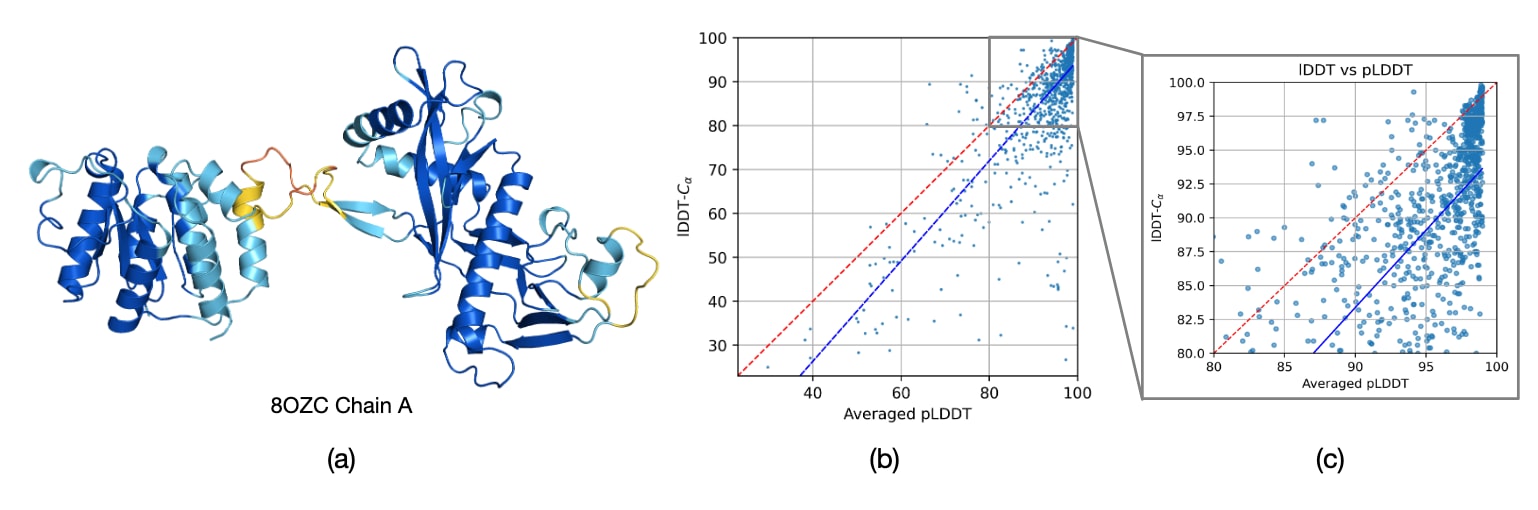
图3 (a) SimpleFold预测结构及其pLDDT评分示例(颜色从红到深蓝分别表示pLDDT由低到高,可视化方式参考Chakravarty and Porter (2022))。(b)与(c) 对比了pLDDT与LDDT-Cα的关系。
4.4 集成生成
4.4.1 分子动力学集成
由于采用了生成式训练目标,SimpleFold天然能够建模蛋白质结构的分布,不仅能针对给定氨基酸序列生成一个确定性结构,还能生成不同构象的集成。为验证这一能力,研究者在ATLAS数据集上进行了基准测试。ATLAS包含1390个蛋白的全原子分子动力学(MD)模拟。按照AlphaFlow的划分方法,将数据集分为训练、验证与测试集,并在测试集上对每个蛋白生成250个构象。表2比较了SimpleFold与基线模型在ATLAS上的表现(不同规模SimpleFold结果见表9)。评估指标涵盖了灵活性预测(RMSDᵣ与RMSFᵣ)、分布精度(RMWD)以及集成可观测量(如暴露残基与暴露MI矩阵)。
首先,直接评估未经MD数据微调的SimpleFold-3B。在推理中设
此外,还训练了SimpleFold-MD,即在ATLAS训练集上对完整训练过的SimpleFold额外微调2万步(损失中
表3 | 双态构象结果。 对于最后两个指标,同时报告所有目标的均值与中位数。基线结果取自ESMDiff论文(Lu et al., 2024a),其余模型的评估流程保持一致。
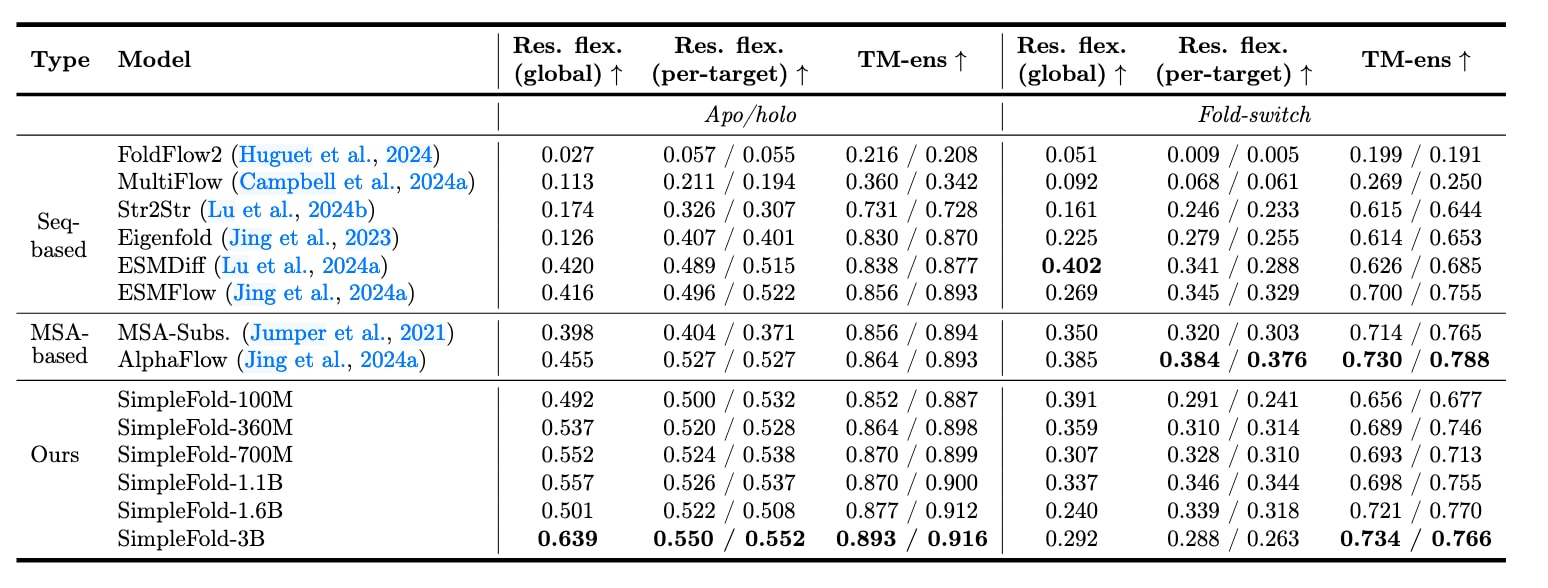
4.4.2 多态结构预测
研究者进一步评估了SimpleFold在生成具有多种天然构象的蛋白结构方面的能力。采用的基准包括apo-holo构象变化数据集(Saldaño et al., 2022)以及fold-switchers数据集(Chakravarty and Porter, 2022),评估流程与EigenFold (Jing et al., 2023)保持一致。每个数据集中的目标由(1)氨基酸序列和(2)两种不同的真实结构构成,要求模型能够生成一组多样且准确的样本,既能覆盖两种构象状态,又能反映正确的局部柔性。
对比方法包括:(1) 基于序列的方法:FoldFlow2、MultiFlow、Str2Str、EigenFold、ESMDiff、ESMFlow;(2) 基于MSA的方法:MSA subsampling与AlphaFlow。评估指标包括全局与逐残基柔性(res. flex.),以及集成TM分数(TM-ens)。具体评估流程为:对每个目标生成5个样本,并分别与两种真实构象进行比较以计算指标。在推理中,将SimpleFold的
结果如表3所示,SimpleFold在Apo/holo数据集上取得了当前最优表现,显著超过了强基线方法AlphaFlow等MSA类模型;在Fold-switch数据集上,SimpleFold表现与ESMFlow相当甚至更优,而ESMFlow同样采用流匹配目标并基于ESM嵌入。结果验证了SimpleFold在**高质量结构生成(集成TM分数)以及结构柔性建模(残基柔性)**上的能力。同时,随着模型规模的增加,SimpleFold整体性能进一步提升,展示了该框架在蛋白质集成生成上的潜力。
综上,在MD构象集与多态结构基准上的实验结果均表明,SimpleFold能够有效建模蛋白质结构的集成,这对于需要蛋白柔性建模的应用(如分子对接)具有重要价值。
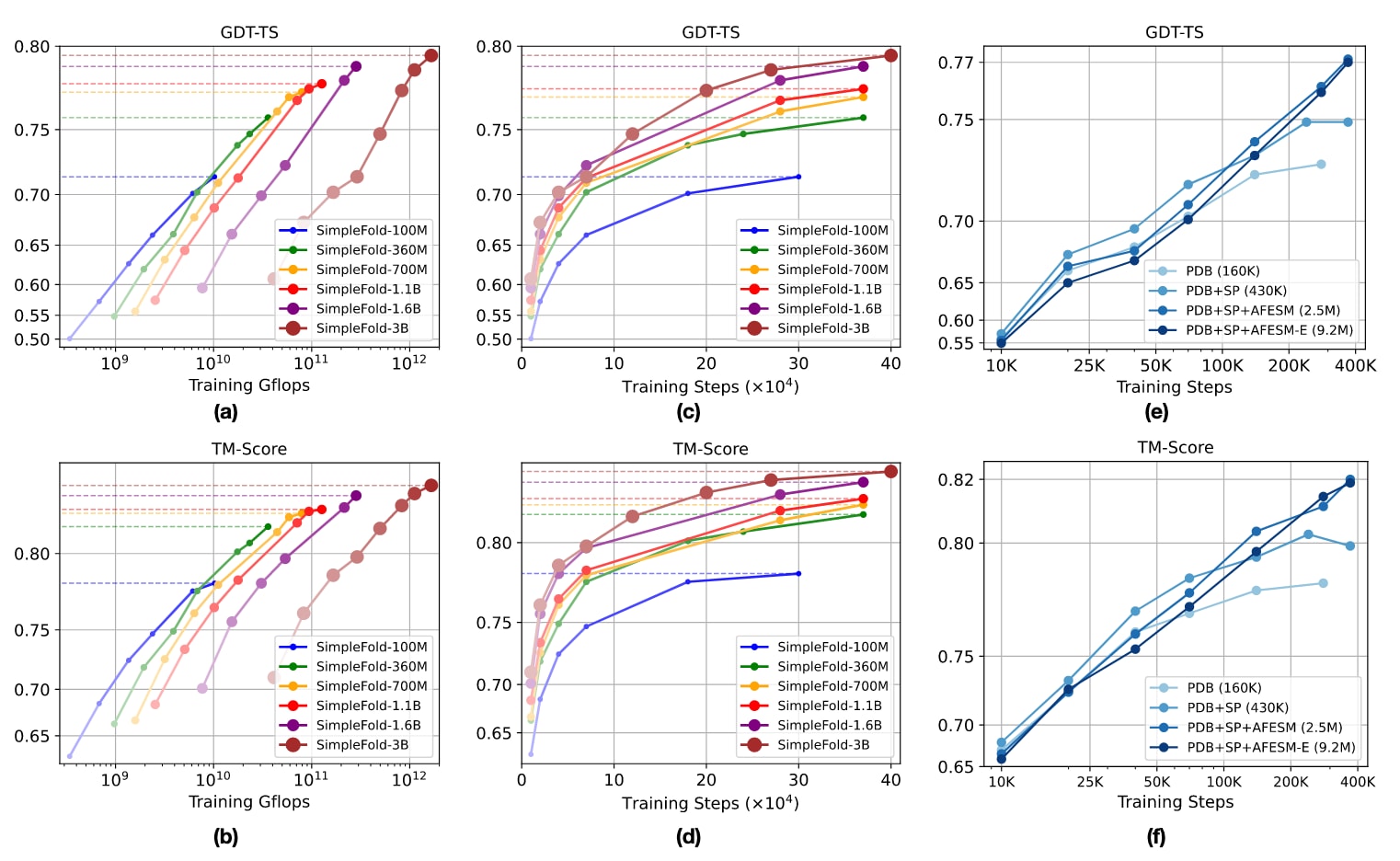
图4 | SimpleFold的可扩展性表现。 (a)(b) 展示训练计算量(Gflops)与折叠性能(GDT-TS、TM-score)的关系。(c)(d) 展示训练步数与折叠性能(GDT-TS、TM-score)的关系。(e)(f) 展示数据规模对性能(GDT-TS、TM-score)的影响。所有模型均在CAMEO22基准上进行评测。
4.5 蛋白质折叠中的规模效应
SimpleFold在扩展模型规模时获益显著,这与生成模型在视觉、语言等领域的成功经验相一致。然而,在蛋白质折叠任务中,训练数据与模型规模扩展的效果尚未得到系统研究。本节通过实证分析,从模型规模与数据规模两方面展示了SimpleFold的扩展特性,并强调了构建强大生物生成模型的重要考量。
模型规模扩展 为评估模型大小的作用,训练了从1亿参数(SimpleFold-100M)到30亿参数(SimpleFold-3B)的一系列模型。所有模型均使用完整的预训练数据(PDB、AFDB的SwissProt以及筛选后的AFESM)。结果如图4(a)-(d)所示,模型规模越大、训练预算(计算量与迭代步数)越充足,性能提升越明显。这表明SimpleFold展现出良好的正向扩展趋势,为进一步发展更强大的生物生成模型提供了方向。
数据规模扩展 进一步使用SimpleFold-700M考察训练数据规模的影响,依次采用:(1) PDB(16万条结构),(2) PDB+AFDB的SwissProt(27万条结构),(3) PDB+SwissProt+AFESM代表性结构(190万条),(4) 扩展AFESM-E(约860万条结构)。如图4(e)(f)所示,随着数据源多样化与结构数量增加,SimpleFold在40万步训练后的性能逐步提升。
综上,这些结果支持了SimpleFold的核心贡献:通过简化且可扩展的架构,充分利用不断增长的实验与蒸馏蛋白数据,实现性能持续提升。
5 结论与未来工作
该研究提出了SimpleFold,一种基于流匹配的蛋白质折叠生成模型,代表着与以往方法架构设计的显著背离。SimpleFold完全由通用的Transformer模块与自适应层构成,摒弃了AlphaFold2中昂贵的成对表示与三角更新等启发式设计;训练目标也仅由简洁的流匹配损失与附加的LDDT损失组成,而非多种复杂的蛋白质专用损失函数。这一简化框架使得SimpleFold能够在模型规模与数据规模上实现扩展。其中最大模型SimpleFold-3B在标准折叠任务中表现出与现有方法竞争的性能,同时凭借生成式训练目标,在多个集成生成任务中达到甚至超越了最新水平。SimpleFold是首个在蛋白质折叠中系统展现良好扩展特性的工作,凸显了通过大幅简化架构即可实现高效折叠预测的潜力,减少了对计算复杂网络模块的依赖。
由于代码与模型检查点已公开,SimpleFold有望被应用与扩展至更多蛋白质相关任务。其简化架构基于标准Transformer模块,便于结合常见微调方法(如Adapter与LoRA)在特定蛋白结构数据与下游任务中发挥作用。此外,SimpleFold也可通过蒸馏加速推理与提升3B大模型的部署效率。除大规模模型外,研究者还发布了轻量级版本SimpleFold-100M,其具备更高效的推理速度,适用于推理延迟受限的场景。
总而言之,SimpleFold展现了以通用架构扩展学习底层对称性的新范式,有望成为推动构建高效且强大的蛋白质生成模型的重要起点。