JACS 2025 | SO3LR: 利用预训练神经网络与通用成对力场的分子模拟
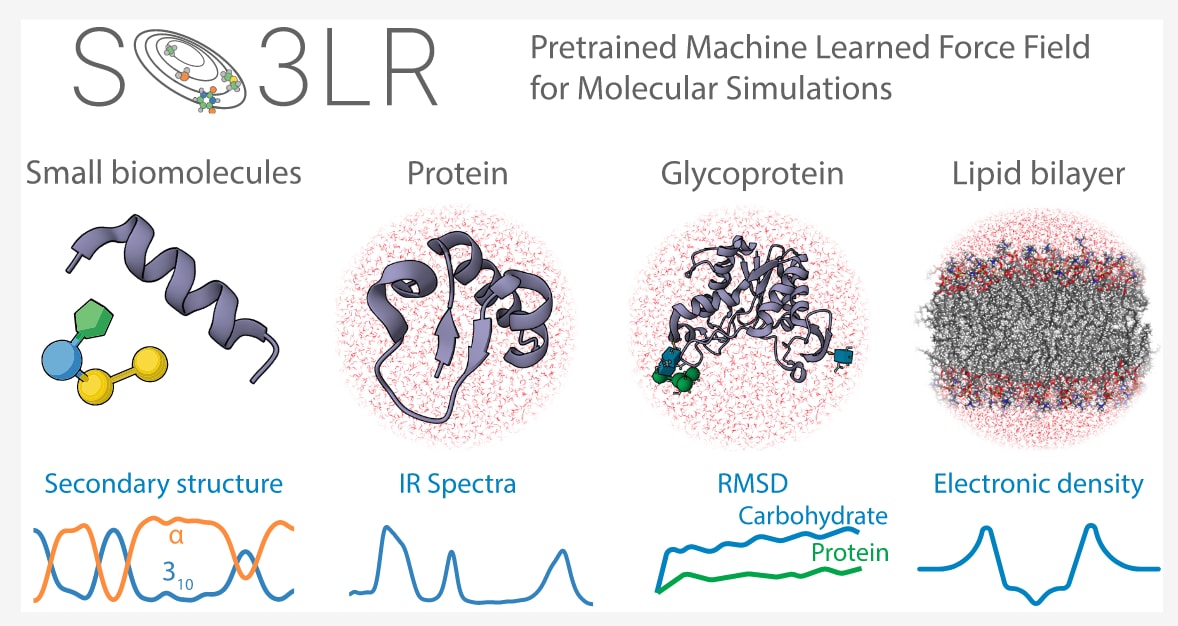
今天介绍的是发表在 J. Am. Chem. Soc. 上的一项工作,作者提出了一种结合预训练神经网络与通用成对力场的新方法——SO3LR,用于实现量子精度的分子模拟。长期以来,分子动力学的目标是构建既高效又可扩展,同时能够准确刻画不同化学空间(如蛋白质、核酸、糖类、脂质与溶剂)的统一力场。SO3LR通过整合SO3krates等变图神经网络、物理启发的短程与长程相互作用模块,以及大规模量子力学数据集训练,成功在效率与准确性之间取得平衡。该方法不仅在小分子和肽类折叠模拟中展现出化学精度,还能扩展到蛋白质、糖蛋白和脂质双层等复杂体系的显式溶剂纳秒级动力学模拟,规模可达数十万原子。值得注意的是,SO3LR在丙氨酸多肽基准上相较传统力场实现了8倍精度提升,同时在单GPU上保持了可接受的计算性能。作为一种开放透明的框架,它展示了机器学习力场与物理模型相结合的潜力,为实现真正的通用分子模拟奠定了重要基础。

获取详情及资源:
- 📄 论文: https://doi.org/10.1021/jacs.5c09558
- 💻 代码: https://github.com/general-molecular-simulations/so3lr
0 引言
机器学习力场(MLFFs)有望推动通用分子模拟的发展,在多样化的分子、材料以及杂化界面体系中,同时实现高效性、准确性、可迁移性与可扩展性。在这一方向上,GEMS方法已为生物分子动力学迈出了重要一步。该研究提出了SO3LR方法,该方法将用于半局域相互作用的快速且稳定的SO3krates神经网络,与针对短程排斥、长程静电及色散相互作用设计的通用成对力场相结合。SO3LR基于400万个中性与带电分子复合物进行训练,这些复合物由量子力学PBE0+MBD水平计算得到,涵盖了广泛的共价与非共价相互作用。该方法兼具计算与数据效率,可在单张GPU上扩展至20万个原子规模,并在有机(生物)分子的化学空间中展现出中高精度。SO3LR被应用于研究四类主要生物分子单元、多肽折叠,以及更大体系(如蛋白质、糖蛋白与脂质双层)在显式溶剂中的纳秒动力学。最后,文章还讨论了通过结合MLFFs与传统原子模型,以实现真正通用分子模拟的未来挑战。
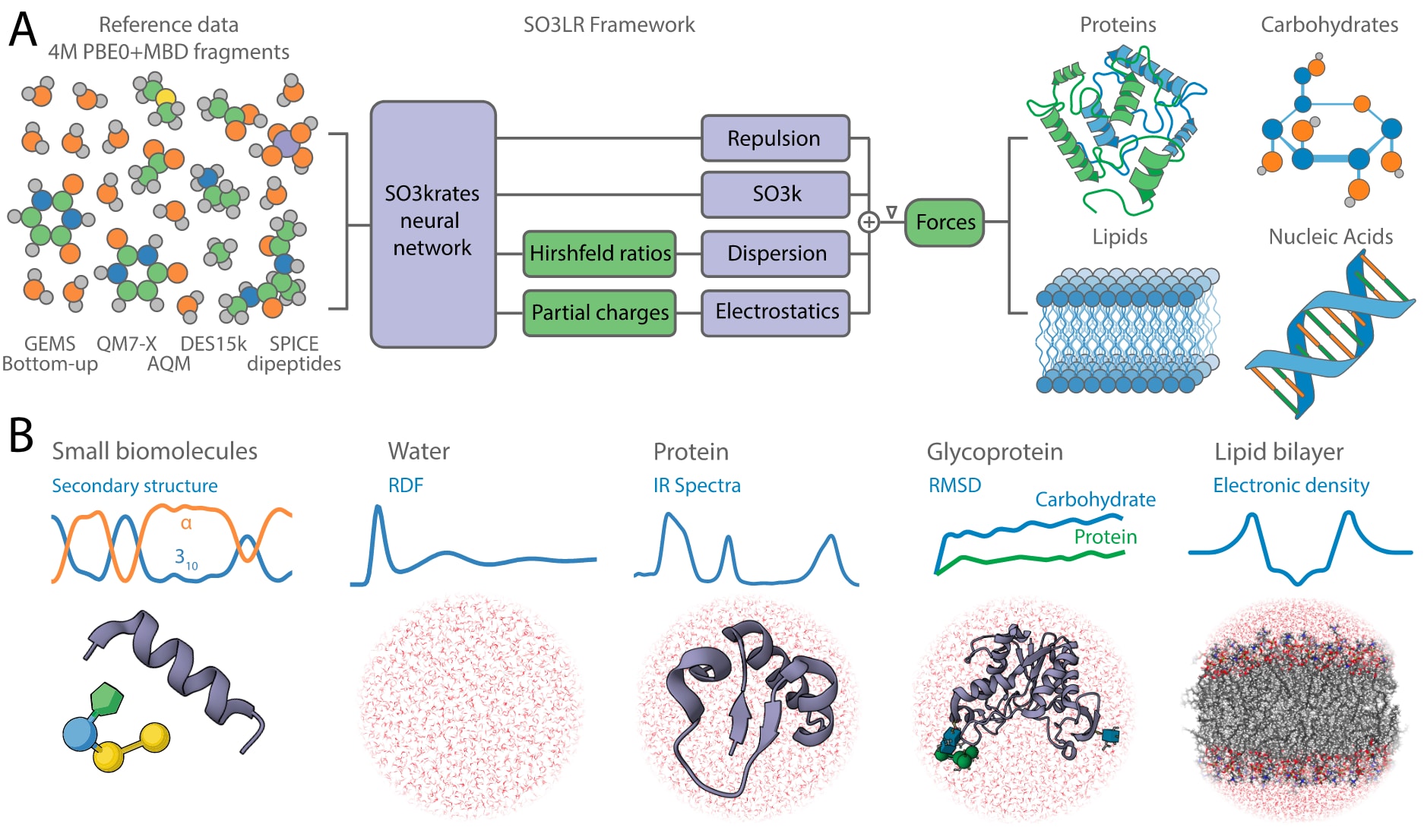
图 1 | SO3LR模型与模拟结果概览。 (A) SO3LR将SO3krates神经网络与物理启发的相互作用相结合,包括ZBL排斥、静电作用以及用于色散的通用成对范德华势,这些作用直接与神经网络模型交互。所有构件在一个精心整理的数据集上进行联合训练,该数据集覆盖了广泛的化学空间与相互作用类型。SO3LR能够用于四大类生物分子单元的小尺度模拟,并支持三类大尺度体系的模拟。(B) 包括液态水、蛋白质、糖蛋白和脂质双层的大规模模拟。
1 引言
早在上世纪,Schrödinger、Dirac和Feynman等科学家就提出了一个愿景,即仅基于核电荷与电子数来进行定量的分子动力学(MD)模拟。然而,尽管过去百年取得了诸多突破性进展,这一设想在分子模拟领域仍未完全实现。现有方法往往需要在效率、准确性、可扩展性与可迁移性(EAST)之间作出重大取舍。在此背景下,原子级建模的多项方法学进展逐渐汇聚,使得实现真正量子精度的分子模拟成为可能。虽然这一目标之路仍然漫长且复杂,但其价值不容置疑,并需要科研共同体的持续努力。
构建满足EAST标准的原子力场模型是分子模拟的核心挑战。传统力场要么源于近似但高效的机制表达式,要么依赖精确却极度耗时的从头算电子结构方法,两者在准确性与效率间均存在折中。近年来,机器学习力场(MLFFs)开始弥合这一鸿沟,它们具备高度灵活的统计建模能力,展现出前所未有的化学空间可迁移性,但在系统规模扩展方面仍面临挑战。要推动MLFF驱动的通用分子模拟走向真正EAST合规,还需解决多项难题,包括:数据与计算高效的半局域原子间相互作用模型、对多体长程相互作用的显式处理、覆盖全面的化学空间数据集的构建,以及GPU加速的分子模拟框架的发展。
该研究针对有机(生物)分子体系提出了解决思路,将化学与计算物理、机器学习及传统力场方法结合。半局域相互作用通过SO3krates机器学习模型描述,采用多体非简谐处理;成对物理项包括Ziegler-Biersack-Littmark短程排斥、长程静电作用及新推导出的通用原子间色散势。通过在400万个分子结构的高质量数据集上进行精心参数化(基于几乎无经验且广泛适用的PBE0+MBD泛函),最终得到SO3LR模型(建议发音为“solar”)。研究表明,SO3LR能够在纳秒尺度上稳定模拟小分子单元、多聚丙氨酸体系、块体水、crambin蛋白、N-连接糖蛋白以及脂质双层。该方法在单张H100 GPU上可扩展至约20万个原子的体系,延迟约为3 μs/原子/步,已接近真实生物分子相关的规模与时间尺度。
2 结果
2.1 SO3LR的组成
普适的分子模拟依赖于对跨体系、跨尺度相互作用的精确刻画。为实现这一目标,SO3LR将势能分解为四部分(图1A):
其中,
2.1.1 EA−SO3krates
该方法的核心在于SO3krates模型,它是一种基于等变图神经网络(详见补充信息中关于不变性与等变性的介绍)的机器学习力场。以原子位置
其中
预测得到的原子能量不仅包含局域邻域信息,还通过平均场更新扩展到更远范围,因此被称为半局域预测。然而,平均场的本质决定了其无法捕捉所有类型的相互作用,并受限于有效截断半径(SO3LR中的有效截断为13.5 Å)。
2.1.2 S−长程色散与静电
为改进长程效应的描述并超越半局域环境,模型显式引入了静电与通用原子间色散势。部分电荷与色散参数均由SO3krates网络预测,且随原子环境而变。以AcAla15NMe为例,Hirshfeld比在氢原子上可分布在0.55至0.8之间,碳与氮的部分电荷则在−0.4至−0.3之间波动。这些变化在原子位移下通过自动微分完整考虑,从而保证了长程项的物理一致性。这些长程作用均满足正确的渐近衰减规律,这对于在训练数据尺度之外实现EAST的可扩展性至关重要。
色散作用通过量子Drude振子(QDO)导出的通用原子间势来计算:
其中
式中
静电相互作用由阻尼库仑势建模:
其中
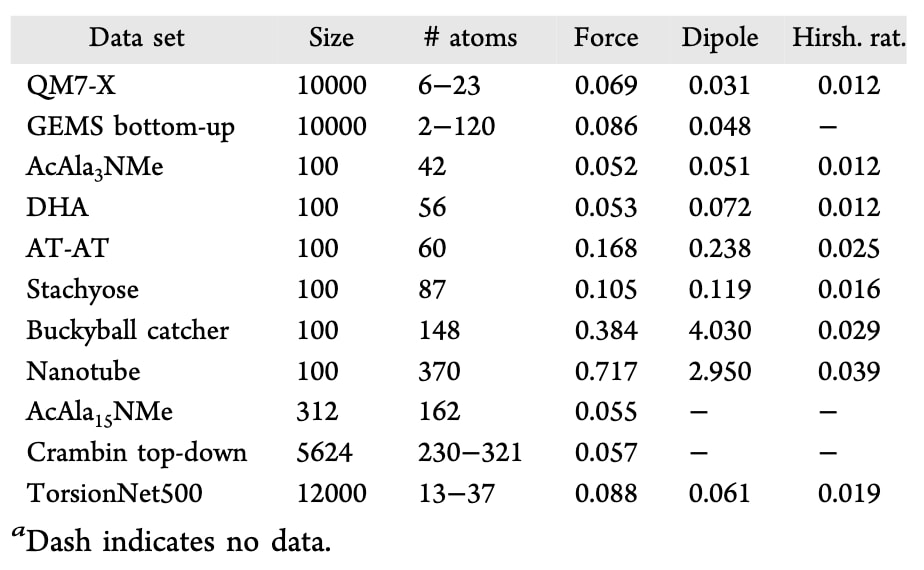
表 1 | 模型在不同测试集上的均方根误差(RMSE),包括力(eV/Å)、偶极矩向量(e×Å)和Hirshfeld比值。
半局域与长程能量的耦合 长程能量与半局域能量的耦合源于势能预测的结构(式1)。长程模块在所有原子对上都有非零贡献,即便在机器学习力场的局域截断范围内也同样如此。因此,长程势的函数形式会改变SO3krates模型所学习到的势能分布。阻尼超参数γ与σ决定了半局域、静电与色散相互作用之间的精细平衡。原则上,SO3krates能够在局域截断范围内修正任意选择的γ与σ,但阻尼对分子动力学模拟中的动力学行为影响显著,尽管整体模型性能几乎不受影响。这是因为半局域与长程相互作用通过参数与超参数的联合优化实现了非线性耦合。基于此,研究者在S66x8基准数据集上对阻尼参数进行了精调。
2.1.3 多样化训练数据上的优化
所有SO3LR模块在覆盖广泛化学空间与多种相互作用类型的多样化数据集上联合优化,从而实现了EAST框架下的可迁移性,涵盖四类主要生物分子。该综合数据集是MLFF发展的关键,包含来自小分子、大分子以及有无溶剂体系的丰富量子化学数据。具体而言,数据集由五部分组成:270万条GEMS片段、100万条QM7-X分子、6万条AQM气相分子、3.3万条SPICE二肽以及1.5万条DES分子二聚体。前三类数据最初在PBE0+MBD水平计算,后两类为保持一致性重新计算。PBE0+MBD方法结合了非经验混合泛函与多体长程色散的显式处理,在多肽、超分子复合物与分子晶体等体系中已展现出与高水平量子化学和实验数据的优异一致性。
这些数据集在构象空间与化学多样性上互补,共覆盖生物体系中常见的8种元素(H, C, N, O, F, P, S, Cl)。例如,QM7-X涵盖小有机分子空间,AQM包含中等规模的类药分子,DES二聚体用于改进非共价作用的描述,SPICE二肽则增强蛋白相关体系的精度,而GEMS则包含气相与显式微溶剂化的蛋白片段及水簇结构。
化学空间覆盖性分析 一个关键问题是:用于训练SO3LR的约400万分子构象是否足以覆盖相关(bio)分子体系的化学空间。尽管全面评估仍待未来探索,但已有的近似估计提供了有价值的洞见。例如,利用SO3krates作为基础MLFF,对中等规模的肽类(如丙氨酸四肽)进行化学精确模拟时,仅需不足1000个构象。若将其外推至所有由20种天然氨基酸组成的四肽组合空间,天真估计约需1.6亿个构象,但这显然是严重高估,主要由于局部化学环境的高冗余性,而这种冗余正是MLFF跨分子与材料体系可迁移性的基础。
在更合理的度量下,化学多样性可通过**轨道数(orbits)**来捕捉,即在不同分子构象中拥有相同局部环境的原子类群。轨道数取决于构建分子子图时的有效距离截断。例如,在丙氨酸四肽中,二阶邻居范围内共有10个轨道,若每个轨道采样100个构象,则训练规模已足够。基于SO3LR、SPICE与GDB-13等公开几何数据集进行统计,含8–10种元素的分子体系轨道数在1万–5万之间。这一初步分析表明,100万至500万条分子构象足以覆盖广泛(bio)分子体系的化学空间。
值得强调的是,这一结论成立的前提是SO3LR结合了消息传递机制与显式的长程物理建模,因此只需图神经网络准确捕捉短程轨道。
联合损失函数 模型优化通过最小化组合损失实现:
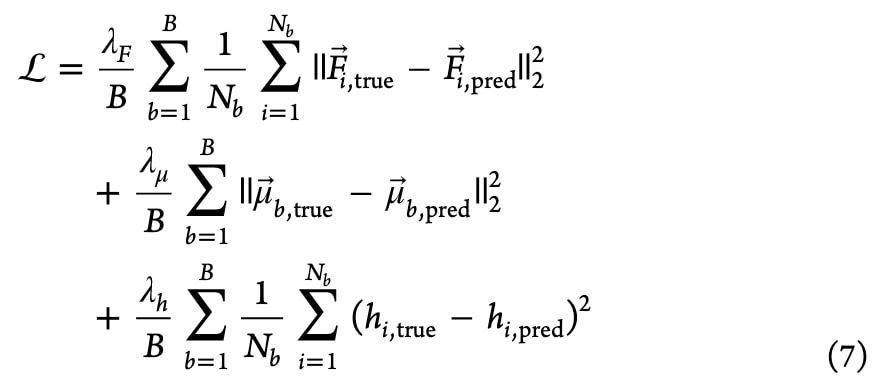
其中
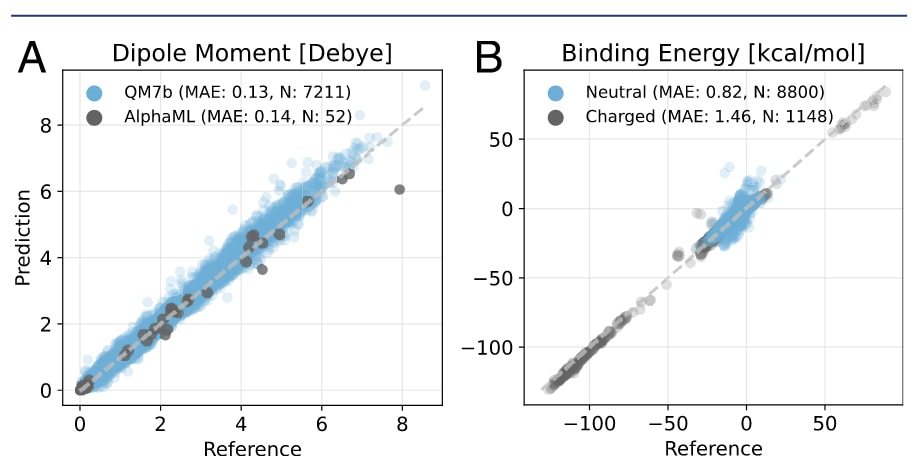
图2 | SO3LR长程模块性能评估。 (A) 在7k QM7b分子与AlphaML数据库上的偶极矩预测表现。(B) 在未见过的SAPT10k数据集上的表现,结果区分为中性与带电子集。
2.2 SO3LR的评估
一个真正符合EAST标准的力场,应当能够准确模拟不同性质与尺度的体系。为展示SO3LR的能力与局限性,研究者依次在测试集与基准集上考察其对原子力、结合能、偶极矩和Hirshfeld比的预测精度,随后分析了来自MD22基准的生物小分子单元的动力学特征,并进一步探讨了真空中聚丙氨酸体系的折叠与稳定性,这些过程依赖于多种相互作用的精妙平衡。在进入更大生物体系模拟之前,还对水的动力学进行了详细分析。最后,将评估范围扩展至含显式水的复杂体系大规模分子动力学模拟,包括蛋白质、糖蛋白和脂质双层(图1B)。
2.2.1 测试集与基准误差
模型首先在量子化学参考数据上进行了精度验证(表1)。测试集包含来自QM7-X与GEMS片段的各1万条随机构象(其余训练集已全部用于训练)。此外,还在PBE0+MBD/tight水平下重新计算了6个MD22参考分子的100条随机结构,并评估了约300条AcAla15NMe结构和约5600条crambin自顶向下片段。结果显示,模型在原子力、偶极矩与Hirshfeld比预测方面表现良好。进一步分析揭示两个关键点:其一,训练集中缺少弯曲碳基体系(如富勒烯捕手、双壁纳米管)片段,导致该类体系误差升高,表明若要实现完整的化学空间可迁移性,需要进一步扩充数据集;其二,AcAla3NMe、AcAla15NMe与crambin片段的误差几乎一致,说明SO3LR具备良好的可扩展性,长程模块能够有效描述分子间相互作用,即便训练主要依赖小分子片段。
为考察构象能量学的准确性,研究者利用在PBE0+MBD水平重新计算的TorsionNet500基准评估了扭转能剖面(图S3)。SO3LR在不同扭转构型上的平均绝对误差(MAE)为1.03 kcal/mol,表明其在生物模拟中常见的多种扭转模式下均具有较高精度。但需要注意的是,缺乏某些官能团(如三唑与三氟甲硫基)的训练样本会显著增加平均误差。
在静电相互作用方面,研究者基于QM7b与AlphaML数据集检验了部分电荷与偶极矩的预测精度,这些数据集在LR-CCSD/d-aug-cc-pVDZ水平下计算得到。SO3LR在偶极矩预测上达到了0.13 D的MAE和5.1°的角度误差(图2A),与B3LYP/d-aug-cc-pVDZ水平(0.09 D)的混合DFT结果相当。在更复杂的AlphaML基准上,B3LYP/d-aug-cc-pVDZ与PBE0+MBD/tight均给出0.10 D的MAE,而SO3LR取得了0.14 D(图2A)。这些结果表明SO3LR能够实现可迁移且精确的偶极矩预测,这对于获得可靠的静电作用能至关重要。
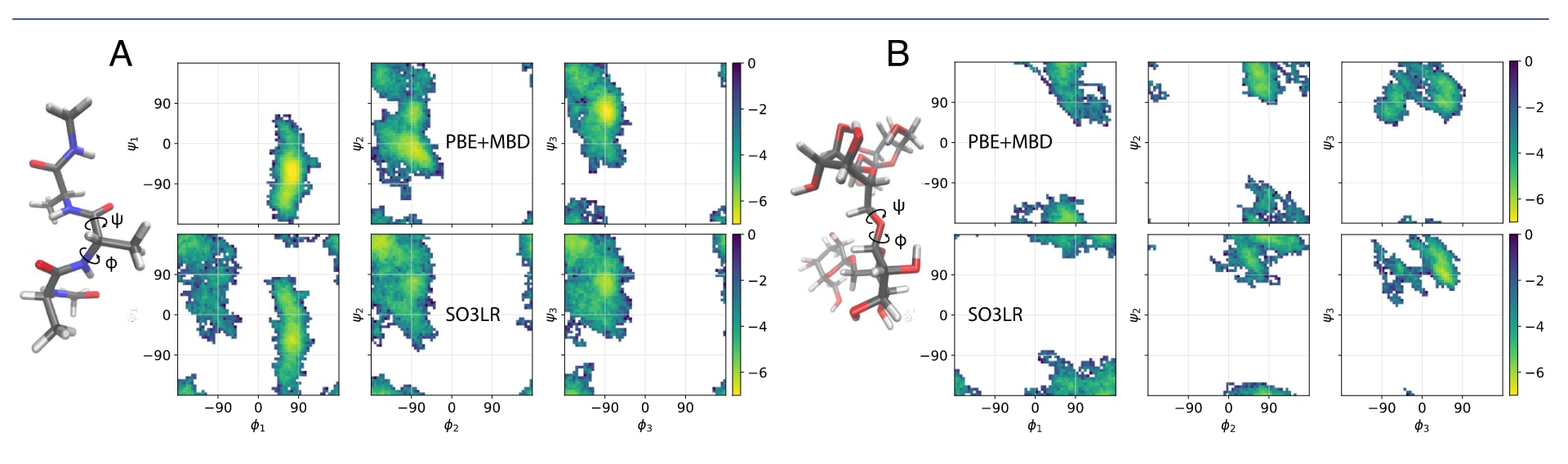
图 3 | 小分子片段模拟。 Ramachandran图(φ/ψ二面角)分别展示(A) AcAla3NMe和(B) stachyose的构象,取自MD22数据集。PBE+MBD与SO3LR在500 K下的模拟,AcAla3NMe为85 ps,stachyose为27 ps;SO3LR额外的500 ps模拟见图S5。轨迹每1 fs采样一次,结果以Boltzmann反演刻度(kcal/mol)表示。PBE+MBD(MD22)与SO3LR(PBE0+MBD训练)的比较仅作参考。
研究者在SAPT2+(3)(CCD)/aug-cc-pVTZ理论水平下计算了大规模SAPT10k基准,其包含70个子集,覆盖以静电或色散作用为主导的复杂结合模式,并在化学空间中展现了广泛的多样性。由于SO3LR仅在8种元素(H, C, N, O, F, P, S, Cl)上训练,9982个复合物中有34个因包含超出训练范围的元素被排除。总体而言,模型表现良好,达到了亚化学精度,平均绝对误差(MAE)为0.90 kcal/mol(图2B)。少数误差高达40 kcal/mol的离群值主要来自训练集中缺失的特殊分子(如ClF、P(CNO)₃、PH₂NO₂)。在PBE0+MBD水平下对这些体系的重新计算确认,误差来源于缺乏训练数据,而非参考方法本身(图S4)。值得注意的是,部分误差还来自CCD与PBE0+MBD参考水平间的差异,因此这一整体性能相当突出。
小分子生物单元的分子动力学模拟 分子动力学模拟是评估力场最直接的检验。研究者选取MD22基准中的6个体系,涵盖四类主要生物分子与两个超分子复合物:AcAla₃NMe四肽、刺槐糖四糖、AT-AT DNA碱基对、二十二碳六烯酸(DHA)、富勒烯捕手以及双壁纳米管。前两个体系在500 K下模拟500 ps,以便与相同条件下的PBE+MBD参考结果比较,其余体系在300 K下进行模拟。
结果显示,SO3LR在所有分子体系中均能实现稳定的构象采样。尤其是四丙氨酸与刺槐糖的自由能面探索,与PBE+MBD的ab initio结果高度一致,Ramachandran图(图3)清晰展示了这种符合性。需要说明的是,图中展示的均为短时间MD轨迹,仅用于与DFT模拟对比,完整500 ps轨迹见图S5。四肽在模拟中遍历了实验蛋白质结构中已知的所有允许(φ/ψ)区间。富勒烯捕手与双壁纳米管复合物尽管在测试集上误差较大,但在动力学模拟中依旧保持稳定(图S6)。这些结果表明,SO3LR即便应用于超出训练域的体系,也能维持稳定性并可靠探索小分子的构象空间。
聚丙氨酸体系的模拟 进一步,研究者考察了聚丙氨酸体系,重点分析真空中延展构象的AcAla₁₅NMe折叠,以及在高温下折叠态AcAla₁₅LysH⁺的稳定性。这类体系极具挑战性,因为其行为依赖氢键、极化与色散相互作用的微妙平衡。此前的尝试若不引入自顶向下片段,往往无法正确折叠AcAla₁₅NMe,或是过度稳定α-螺旋,甚至低估AcAla₁₅LysH⁺的稳定性。
在该研究中,每个体系均进行了4次500 ps模拟。结果显示,延展态的AcAla₁₅NMe在所有模拟中均成功折叠(图4A与图S7A)。其折叠时间尺度与机制与文献48的结果相符:初始阶段以转角构象为主,随后经过“波浪状”中间态,最终折叠为螺旋态,并在α-螺旋与3₁₀-螺旋之间发生动态转换。后者尤为值得注意,因为经验力场往往会高估α-螺旋的稳定性。
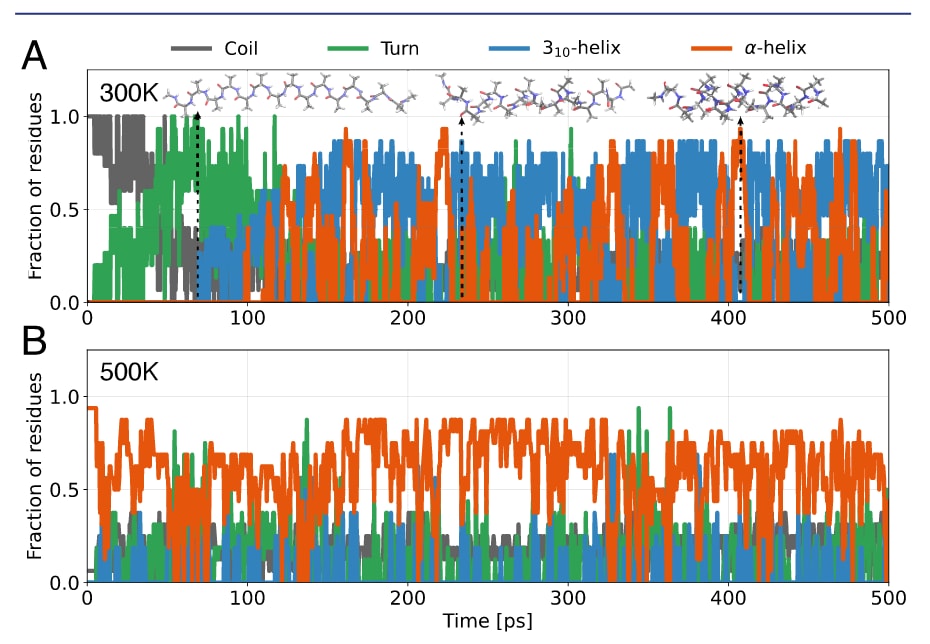
图 4 | 聚丙氨酸模拟。 (A) AcAla15NMe在300 K气相下折叠轨迹中的二级结构。(B) AcAla15LysH+在500 K气相下的折叠轨迹,起始于α-螺旋构象。
折叠态AcAla₁₅LysH⁺ 在折叠态的AcAla₁₅LysH⁺中,α-螺旋结构在500–600 K下依然得以保持(图4B与图S7B)。这一结果与实验观测相符:实验发现,当与氦缓冲气相互作用时,AcAla₁₅LysH⁺在约725 K仍展现出与α-螺旋一致的散射截面。需要指出的是,若要与气相实验进行直接对比,模拟需显式包含氦环境与核量子效应。整体来看,这两个聚丙氨酸体系为SO3LR在中等规模体系的动力学可扩展性提供了良好验证,补充了其在测试误差层面展现的可扩展性。
液态水模拟 液态水在生物体系中至关重要,因此是SO3LR评估的重要对象。研究者在NPT系综下模拟了一个包含4096个水分子的水盒,先进行200 ps平衡化,再在随后300 ps中计算物理量,重点考察径向分布函数、密度与自扩散系数。
结果显示,氧–氧径向分布函数展现出预期的层状结构(图S8),但液相略显过度有序。温度升高至330 K可在一定程度上补偿缺失的核量子效应,从而改善与实验的一致性。水的密度在10–20 Å长程截断范围内介于0.97–1.04 g/cm³(图S9),在后续含显式水的生物模拟中选用12 Å作为折中方案。自扩散系数在300 K下为0.079 Ų/ps,330 K下为0.224 Ų/ps,而室温实验值为0.23 Ų/ps。SO3LR的结果与基于PBE0+vdW泛函的显式ab initio分子动力学模拟高度一致。
值得强调的是,训练数据中仅包含至多40个分子的气相水簇(约1万簇,占数据集总量的0.26%)。已知基于PBE0+vdW的ab initio模拟在水的实验性质再现方面存在局限,主要源于四面体氢键网络对单个氢键轻微高估的放大作用。因此,机器学习力场的表现不会、也不应超越其所依赖的ab initio参考精度。若要提升对水的刻画,可引入更高水平的量子化学数据(如耦合簇或量子蒙特卡洛方法),并在分子动力学中显式纳入核量子效应。对于水溶液中的生物分子而言,氢键只是众多相互作用之一,而结果表明:只要能正确再现实验水密度,基于PBE0+MBD训练的MLFF即可进行可靠的生物分子动力学模拟。
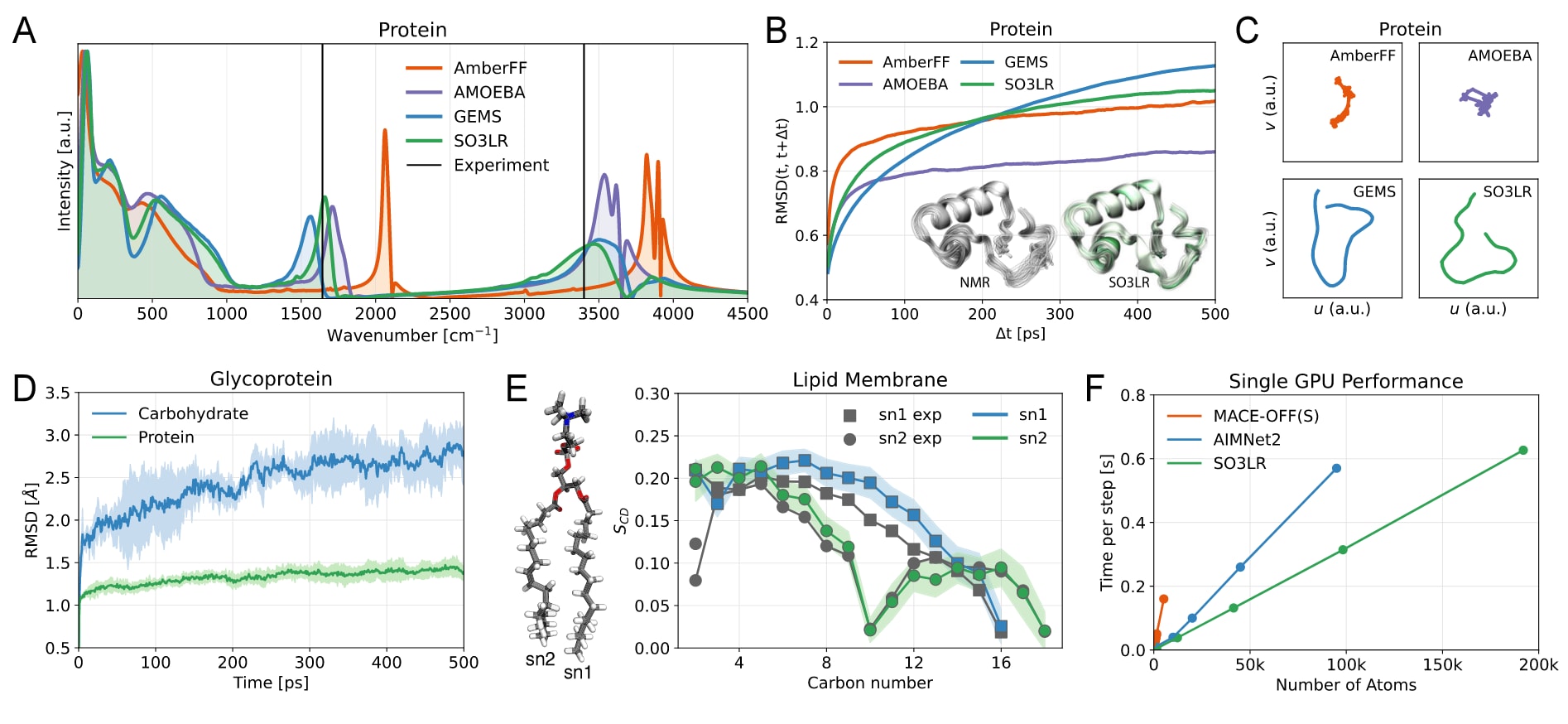
图 5 | 显式溶剂环境下的生物分子模拟。 (A) crambin在水中125 ps动力学得到的功率谱,AmberFF与GEMS结果引自文献。(B) crambin(不含氢)构象在时间t与t+Δt的均方根偏差(RMSD),平均于三个3 ns轨迹;插图显示SO3LR轨迹帧与NMR蛋白结构的叠合。(C) crambin模拟轨迹的二维UMAP嵌入,所有子图采用相同的潜在空间投影。(D) 糖蛋白中蛋白与碳水部分的RMSD,平均于三个500 ps轨迹。(E) 128个POPC脂质双层的尾部基团NMR序参数,比较SO3LR模拟与实验,背景色表示标准差。(F) 单GPU性能。基于液态水分子动力学测得SO3LR延迟,使用JAX-MD在H100 80 GB GPU的NVT系综下运行,斜率为3.25×10⁻⁶ s/atom/step。MACE-OFF(S)与AIMNet2的延迟数据分别在A100与H100上测得,引自相关文献。
大型生物分子模拟 最后,研究者展示了SO3LR在显式水环境下对大型生物分子的模拟潜力。所选体系涵盖多类生物分子组分,每一类都有独特的结构与功能特征,可与已有模拟或实验数据进行验证,包括crambin蛋白、糖蛋白(PDB: 1K7C)以及POPC脂质双层。
对于crambin(含水共2.5万个原子),在1 ns平衡后进行125 ps动力学模拟,并以2.5 fs时间分辨率计算功率谱。结果显示,SO3LR对水的振动峰(1640 cm⁻¹与3200–3600 cm⁻¹)的再现优于GEMS、AMOEBA与AmberFF(图5A)。在三个3 ns模拟的均方根偏差RMSD(t, t+Δt)分析中,SO3LR在长时间尺度下表现出略高的蛋白质流动性,与GEMS结果一致(图5B),整体结构保持折叠状态,无解折或断键迹象(图S10)。进一步通过UMAP投影可视化构象空间,结果显示SO3LR与GEMS在构象空间的采样范围比AMBER与AMOEBA更广(图5C),与NMR实验揭示的高构象多样性相符。
在糖蛋白模拟(含水共4.8万个原子)中,尽管训练集中缺乏碳水化合物,SO3LR依然捕捉到碳水片段更高的柔性,表现为碳水段的RMSD大于蛋白段(图5D),与针对糖类优化的CHARMM力场结果一致。然而,在N-连接区的二面角Cγ−Cβ−Cα−N采样中,模拟仅覆盖了反式(180°)构象,未探索到g⁺(60°)与g⁻(300°),这表明若要获得完整采样,仍需更长时间模拟。
在POPC脂质双层模拟(3.3万个原子,128个脂质+5120个水分子,303 K,500 ps)中,SO3LR对关键结构性质的预测(脂质面积、双层厚度、脂肪链有序参数)与实验和经验力场吻合良好(表2)。其中脂质面积低估约10%,可能源于SO3LR采用各向同性NPT,而经验力场使用半各向同性NPT。基于NMR的脂肪链有序参数也显示SO3LR预测的双层结构与实验一致(图5E)。
在计算性能方面,研究者利用JAX-MD在单GPU上对含1536–192,000原子的水盒进行了NVT模拟(图5F)。结果表明,其计算效率为3.25×10⁻⁶ s/atom/step,可在单GPU上实现约2.6 ns/天(1 fs步长,1万个原子体系),足以在常规硬件上完成纳秒级大体系模拟。但对于毫秒尺度的复杂构象转变(如溶液中蛋白质折叠),仍超出目前能力范围。当前SO3LR模型设定为128个特征、3层交互、4.5 Å局域截断,以平衡精度与效率;通过调整超参数,可在化学保真度不丧失的前提下进一步加速。值得一提的是,该模型仅用单GPU、耗时86 GPU小时完成训练,显示了其在计算成本上的优势。

表 2 | POPC脂质双层结构性质:双层厚度DHH(Å)与每脂质分子面积(Ų)。
3 讨论与结论
原子级模拟领域的长期愿景是发展出统一的力场函数形式,能够跨越多样化的化学空间(如溶剂、蛋白质、DNA、RNA、糖类和脂质)进行应用。这类力场不仅需要尽可能逼近量子力学行为,还必须具备高效与可扩展性,从而在不同条件(如压力、温度及外部环境)下模拟真实的生物分子复合物。
该研究通过SO3LR模型在这一方向上取得了重要进展。SO3LR构建在一个开放透明的框架中,融合了高质量且多样化的量子化学数据集、快速稳定的SO3krates机器学习架构、通用长程相互作用模块、JAX-MD模拟引擎以及完备的分析工具。得益于这些组件的结合,SO3LR实现了跨越更广化学空间的量子精度分子模拟。这一发展与经验力场领域数十年来追求的目标相呼应。
在性能方面,SO3LR在丙氨酸多肽上的力预测精度达到化学精度,相较AmberFF实现了8倍提升(力MAE为0.9 vs 7.6 kcal/mol/Å),而在单GPU上的计算速度仅比GROMOS慢约40倍。其在能量、力、偶极矩与极化率上的评估,以及纳秒级分子动力学轨迹的分析,均显示出SO3LR在生化空间中的高度可迁移性与可扩展性,可处理数十万原子体系。这种能力的实现不依赖于预定义原子类型、谐振约束或为特定体系设计的函数形式,而是通过自下而上的量子数据训练,使模拟能够推广至更广泛的条件范围。相关结果在聚丙氨酸300–800 K的模拟、溶液中crambin的结构与光谱、糖蛋白1K7C以及POPC脂质双层的结构性质中均得到了验证。
SO3LR的独特之处在于显式物理知识与机器学习势能的结合:短程与长程力学模块由物理规律硬编码,而复杂的多体贡献及不同能量项的平衡则通过数据驱动学习获得。这种方式使模型能够在保持物理约束的同时,释放非线性表达力来处理多体效应与能量权衡。
尽管取得了进展,要实现真正通用的分子模拟仍面临诸多挑战。SO3LR的预测精度依赖训练数据,对于曲面碳体系或特定官能团等样本稀缺环境表现欠佳。因此,发展稳健的不确定性量化以检测模型处于外推区尤为重要。未来的改进方向包括:引入涵盖80余种元素的大规模量子化学数据(QCML、MPTrj、OMol25),扩展DFT+MBD训练集至离子、糖类、脂质、DNA、超分子和溶剂体系;在JAX-MD中集成PME求和以改进长程静电;为小片段生成更高水平的参考数据(如耦合簇或量子蒙特卡洛);优化长程相互作用模块以捕捉各向异性多体效应;提升至多GPU架构以扩展规模;以及在经典MD之外引入核量子效应。这些方向均是当前社区积极探索的前沿。
最后,鉴于原子级模拟高度依赖底层力场与模拟参数的细节,亟需建立一套标准化的量子精度MLFF基准,以确保结果可复现,并支持在现实的时间与空间尺度下对实验相关现象的稳健建模。