Nat. Commun. 2025 | DLFea4AMPGen:结合深度学习模型特征的抗菌肽从头设计方法
人工智能正深刻重塑生物药物设计的范式,尤其在抗菌肽(AMP)研究领域展现出巨大潜力。传统AMP筛选依赖实验或数据库过滤,不仅周期长、成本高,还面临序列空间过大与筛选成功率低等问题。近期发表于 Nature Communications 的研究提出了创新性的 DLFea4AMPGen 框架,首次将深度学习模型的特征可解释性与肽分子设计相结合。该方法通过 SHAP(Shapley Additive Explanations) 分析提取与抗菌、抗真菌及抗氧化活性最相关的氨基酸特征,构建“合理序列子空间”,实现从特征驱动的多功能肽生成。实验结果显示,所设计的 16 条候选肽中有 12 条具备生物活性,其中 D1 与 D2 对多重耐药菌表现出强效广谱抗菌作用,并在小鼠感染模型中显著降低炎症反应。这项研究不仅验证了深度学习在生物活性肽设计中的可解释性应用,也为未来基于人工智能的抗感染药物开发开辟了全新方向。

获取详情及资源:
0 摘要
深度学习模型在加速抗菌肽(AMPs)的设计与优化方面展现出巨大潜力,但现有方法仍面临成功率低或虚拟文库规模过大的挑战。在该研究中,研究者提出了一种名为 DLFea4AMPGen 的生物活性肽设计策略,该策略利用深度学习模型识别并提取与抗菌肽活性相关的关键特征,从而生成具有潜在生物活性的肽序列。
研究者采用 SHapley Additive exPlanations (SHAP) 方法,对具有潜在抗菌、抗真菌和抗氧化活性的多功能肽中每种氨基酸的贡献进行定量分析。通过分析,提取出平均贡献最高的关键特征片段(KFFs),并根据氨基酸频率将其划分为四个亚类。这些高频氨基酸被系统排列,用以构建候选肽的合理序列子空间,进而从中筛选出 16 条代表性序列进行实验验证。
实验结果表明,75%(12/16) 的序列表现出至少两种生物活性。值得注意的是,其中的 D1 展现出广谱抗菌活性,对多重耐药临床致病菌在体外和体内均表现出显著的抑制作用。
这项概念验证性研究表明,DLFea4AMPGen 平台 在高效设计与筛选生物活性肽方面具有重要潜力,展示了其在抗菌肽研究领域的应用价值。
1 引言
抗菌肽(AMPs)已成为应对抗生素耐药菌的有前景候选物,因此受到越来越多的研究关注。其独特的作用机制通常通过物理性破坏细菌膜来实现抗菌,为规避现有耐药机制提供了潜在途径。以往的抗菌肽设计主要集中在天然肽的发现,或在天然肽基础上进行修饰以增强其生物活性。近年来,人工智能的快速发展,特别是大模型技术的进步,正在深刻改变肽类药物设计的方式。借助深度学习模型,如今可以快速设计出大量自然界中尚未发现或可能并不存在的抗菌肽。
目前,从头设计抗菌肽主要有两种途径:
第一,开发生成式深度学习模型以生成蛋白质或肽序列。诸如变分自编码器、Chroma 和 ProGen 等生成模型,通过在无标签数据上训练,能够基于对天然蛋白质序列空间的理解生成新的序列。
第二,利用深度学习模型探索整个虚拟肽库,通过预测模型扫描虚拟文库,可以准确识别出具有潜在抗菌活性的序列。
这两种方法都已成功生成了针对耐药病原体的高效抗菌肽,拓宽了可用生物活性肽的范围,并加速了超越天然来源的功能肽发现。然而,尽管这些方法取得了显著成果,仍存在一些限制影响效率,亟需改进。尤其需要更有效的策略,从生成式模型产生的大规模虚拟肽库中筛选出高质量活性肽,以提高实验验证的成功率。
虽然结合不同深度学习模型的策略在一定程度上提高了设计效率,但现有的遍历式设计通常需要列举所有可能的肽序列组合,导致生成数量庞大。例如,一个13个氨基酸的肽片段可能包含超过 20¹³ 种序列组合,计算负担极为沉重。数据库筛选技术可以通过限制氨基酸种类来减少虚拟文库规模,但会降低序列的多样性。
此外,细菌感染会在宿主中引发氧化应激,导致细胞损伤并加重组织破坏,而这一因素在早期肽药物筛选中往往被忽视。因此,抗菌肽的设计还应考虑具备缓解氧化应激和减轻炎症反应的活性。
为应对这些挑战,研究者提出了 DLFea4AMPGen,一种用于生成高生物活性从头抗菌肽的新方法。研究者利用多个模型分别识别出具有潜在抗菌、抗真菌和抗氧化活性的肽序列,并通过 SHAP(SHapley Additive exPlanations) 方法提取在这些肽中起关键作用的 关键特征片段(KFFs)。随后,根据系统分析将这些片段划分为四个亚类,每个亚类展现出独特的氨基酸频率特征。通过在各亚类中重新排列高频氨基酸,研究者构建了四个完整的序列空间,并从中筛选出 16 条具有代表性的候选抗菌肽(c_AMPs)进行实验验证。
结果显示,75%(12/16) 的序列表现出抗菌活性,其中部分肽同时具有抗菌、抗真菌和抗氧化三种活性。值得注意的是,D1 与 D2 展示出针对多种细菌(包括耐药菌株)的广谱抗菌作用。进一步的体内实验表明,D1 能够在败血症小鼠模型中降低细菌负荷并减轻炎症反应。
该研究展示了基于深度学习的生物活性预测如何实现精确的肽分子设计,为获得具有增强生物活性的肽类药物提供了一种高效而可行的新途径。
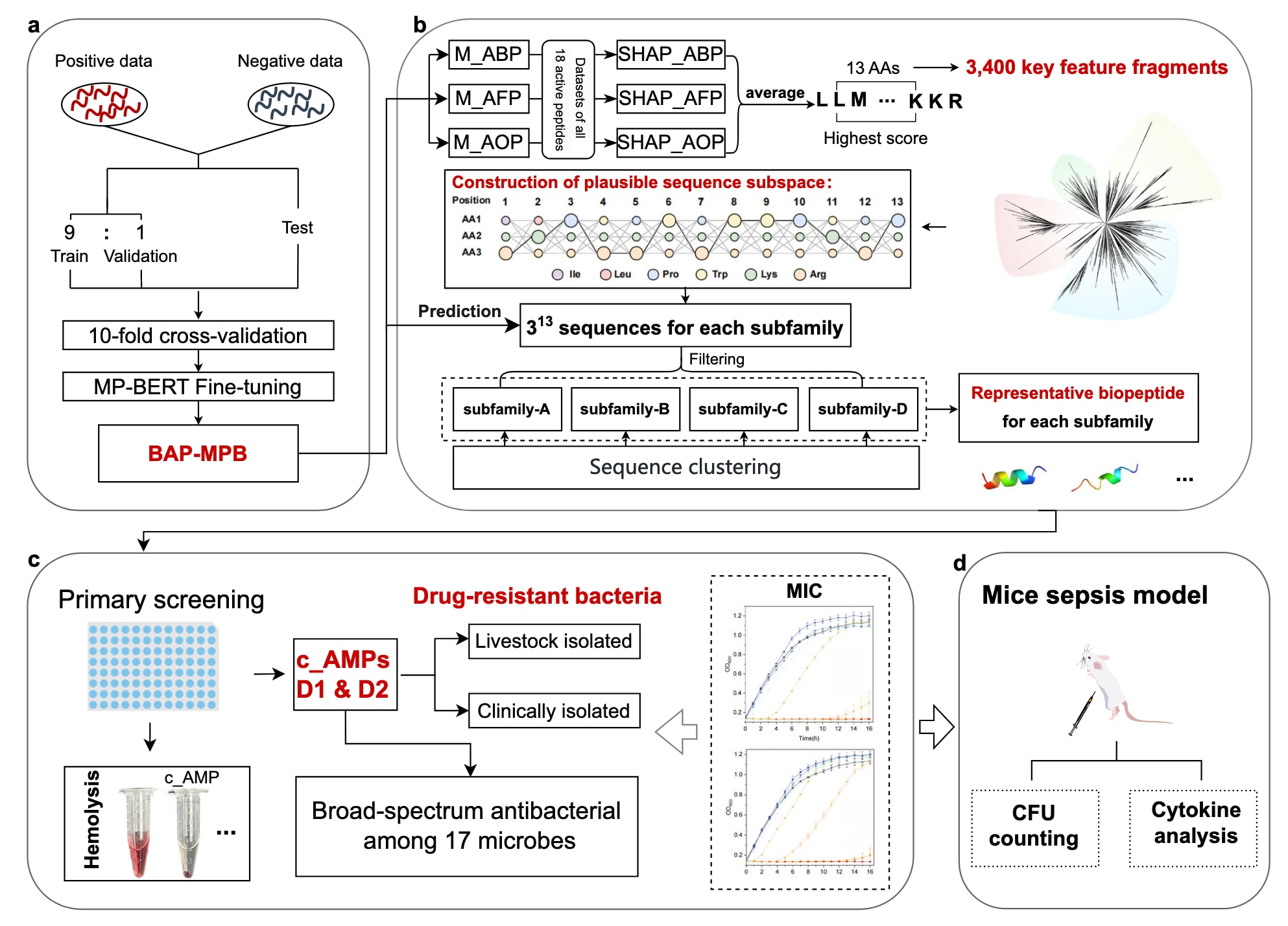
图 1 | 研究工作流程示意图。 a 模型构建阶段:研究者首先基于前期研究中的生物活性肽数据集,对预训练的 MP-BERT 模型进行了微调,建立了三个主要模型,分别为 ABP-MPB(抗菌肽模型)、AFP-MPB(抗真菌肽模型) 和 AOP-MPB(抗氧化肽模型),统称为 BAP-MPB(Bioactive peptide for MP-BERT)。随后,这三个模型被用于预测具有潜在三重活性的肽序列。b 基于这三个模型的 SHAP 解释结果,研究者对每条被三个模型均预测为具有生物活性的肽序列应用 13个氨基酸滑动窗口(13-AA sliding window),以识别平均 SHAP 值最高的 关键特征片段(KFFs)。接着,根据氨基酸特征的差异,将这些 KFFs 分为 四个亚类,并在每个位点上选取出现频率最高的前三种氨基酸,系统性地组合形成所有可能的序列,从而构建出 合理的序列子空间(plausible sequence subspaces)。随后,从每个序列子空间中选取代表性序列作为 候选抗菌肽(c_AMPs) 进行化学合成。c, d 最后,研究者对这些候选肽进行了进一步的 体外抗菌与抗氧化活性验证(最小抑菌浓度 MIC 测试中,n = 3 个独立生物学重复,结果以平均值 ± 标准差表示),并通过 小鼠败血症模型 开展了 体内实验,以评估其抗感染效果和作用机制。
2 结果
2.1 基于特征的多功能抗菌肽设计策略
为建立一个能够预测肽类潜在抗菌、抗真菌及抗氧化活性的深度学习模型框架,研究者基于前人发表的生物活性肽数据集,对预训练的 MindSpore proteinBERT (MP-BERT) 模型进行了微调(图1a,补充图1)。最终获得了三个生物活性肽预测模型,分别为抗菌肽模型(ABP-MPB)、抗真菌肽模型(AFP-MPB)和抗氧化肽模型(AOP-MPB)。此外,该研究还利用前期研究中包含 18种不同生物活性 的 20个生物活性肽数据集(补充图2与补充表1),作为具有潜在三重活性的候选肽集合。
在这一策略中,研究者设计了一个多阶段流程,用于生成同时具备抗菌、抗真菌和抗氧化活性的肽序列(图1b)。首先,使用 ABP-MPB、AFP-MPB 和 AOP-MPB 模型对上述20个生物活性肽数据集中的所有肽序列进行预测,仅保留在三个模型中均被预测为阳性的肽。
随后,研究者应用 SHAP(SHapley Additive exPlanations) 方法提取这些深度学习模型识别到的关键特征,为每个氨基酸分配不同的 SHAP 值。为减少背景氨基酸的干扰,研究者在每条肽序列中,仅提取一个 13个氨基酸(13-AA)片段,其在三个模型中平均 SHAP 值之和最高。基于这一原则,所提取的片段被定义为 关键特征片段(KFFs),即对肽活性影响最大的序列部分。
接着,研究者构建系统发育树,将这些同源的 KFFs 按照氨基酸特征进一步划分为四个亚类。对每个亚类,研究者识别出每个位点上最常出现的氨基酸残基,并将这些高频残基系统性地组合排列,构建出一个 合理的序列子空间(plausible sequence subspace)。最后,从每个子空间中选取代表性序列进行体外和体内实验验证(图1c,d)。
2.2 生物活性肽识别的深度学习模型开发
为构建一个高精度的生物活性肽预测模型,研究者采用了迁移学习(transfer learning)方法,对此前开发的预训练 MP-BERT 蛋白模型进行了微调。该模型的整体架构主要包括多头注意力机制(multi-head attention)、归一化层(normalization)以及前馈神经网络(feed-forward network),这些模块在**六个编码层(encoding layers)**中重复实现(见补充图1)。
在模型的六个隐层中,从第1层到第6层,正负样本的区分度逐渐增强(补充图3),表明模型能够逐步提取出用于样本分类的高信息量特征。
如图2a、b所示,在独立测试集上,ABP-MPB、AFP-MPB 和 AOP-MPB 三个模型均表现出最佳预测性能。此外,即便在完全未用于微调阶段的新数据集上进行评估,模型的预测准确率仍保持在 84% 以上,显示出较强的泛化能力(补充图4)。
在多项性能指标上,包括准确率(Accuracy)、精确率(Precision)、召回率(Recall)、F1 值以及曲线下面积(AUC),该模型的表现均显著优于传统的机器学习方法(如支持向量机 SVM与极端梯度提升 XGBoost),同时也优于其他深度学习模型(如卷积神经网络 CNN)及两种现有的先进抗菌肽预测方法。
此外,在全部20个生物活性肽数据集上,基于 MP-BERT 微调的模型整体性能超过了 UniDL4BioPep(补充图5),而后者此前被认为是表现最优的生物活性肽预测模型之一。
为了识别同时具有抗菌、抗真菌和抗氧化特性的多功能肽,研究者利用 ABP-MPB、AFP-MPB 和 AOP-MPB 三个模型,对来自20个不同生物活性肽数据集的 共23,346条肽序列 进行了预测。结果显示,其中有 4,760 条肽序列 被预测为同时具备三种生物活性(图2c)。
如预期所示,这些具有“三重活性”的肽多数来源于原本与抗菌功能相关的数据集,如 ABP、AMP、ACP 等(图2d)。在此基础上,研究者将重点聚焦于这 4,760 条潜在多功能肽序列,并对其进行后续的特征分析与筛选。
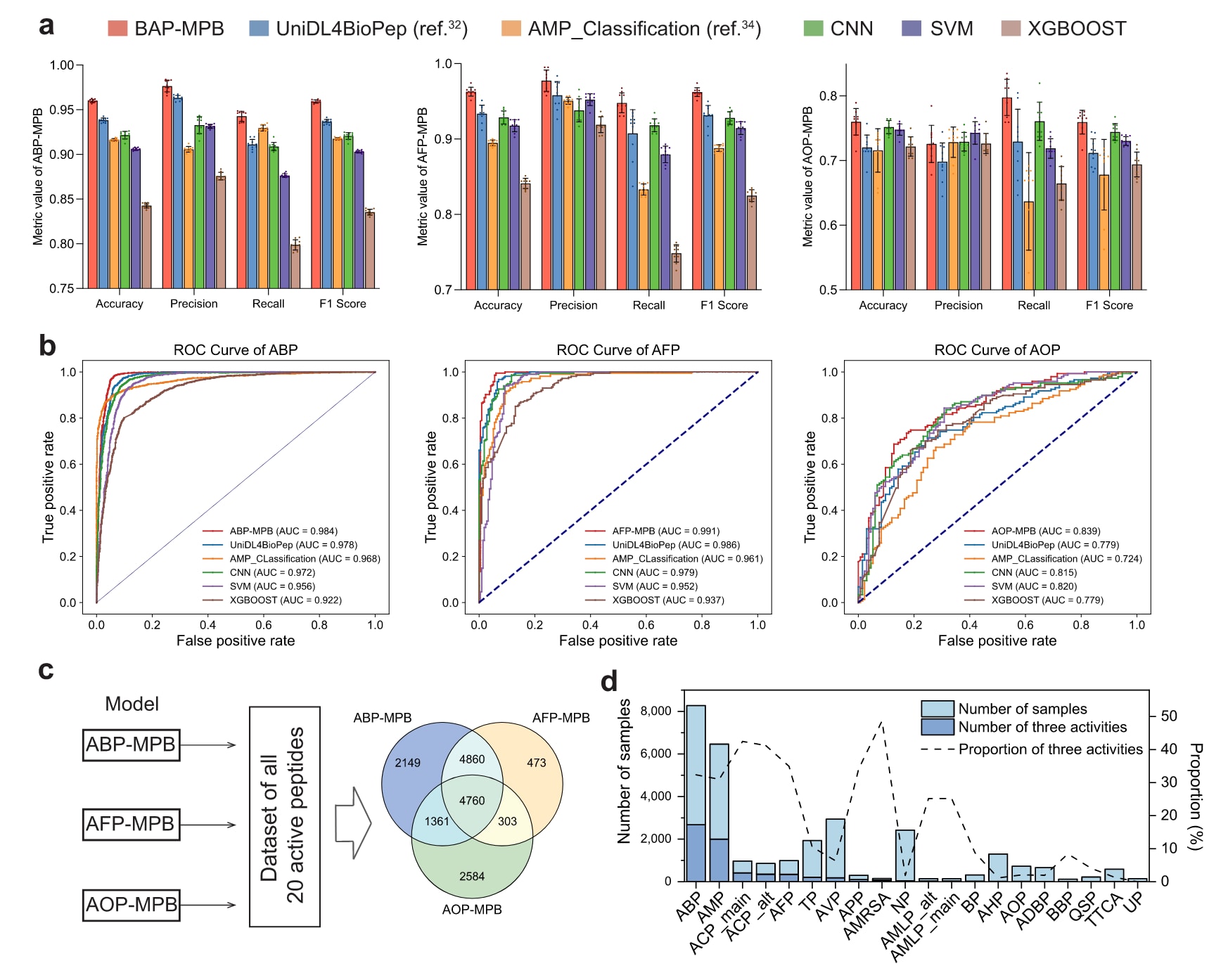
图 2 | 不同模型的性能比较与多功能活性预测分析。 a 测试集上的预测性能。基于 10 折交叉验证(10-fold cross-validation) 的结果(10 次重复的平均值 ± 标准差,n = 10),展示了训练后的 BAP-MPB 模型(即 ABP-MPB、AFP-MPB 和 AOP-MPB)及其他模型的性能表现。在同一数据集上对所有模型进行训练与评估,分别比较了 准确率(Accuracy)、精确率(Precision)、召回率(Recall) 和 F1 值(F1 Score) 四项指标。四个指标的数值越高,模型性能越优。b 六种模型在 ABP、AFP 和 AOP 数据集上的 ROC 曲线,曲线下面积(AUC)用于衡量模型的整体分类能力。c 利用 ABP-MPB、AFP-MPB 和 AOP-MPB 三个模型对全部 20 个生物活性肽数据集进行预测,仅保留被预测为阳性的样本。图中展示了三个模型阳性预测结果的交集,中心的 4760 表示同时被三个模型预测为具有生物活性的肽序列数量。d 每个原始生物活性肽数据集中被预测为阳性的样本数量,以及具有抗菌、抗真菌和抗氧化活性的样本数量(柱状图,左 Y 轴);同时绘制了各数据集中具有三重活性样本所占比例(折线图,右 Y 轴),以反映多功能肽在不同数据集中的分布情况。
2.3 基于 SHAP 方法的关键氨基酸与特征片段提取
为提升模型的可解释性,并深入理解不同生物活性预测背后的分子机制,研究者采用了 SHAP(SHapley Additive exPlanations) 方法,对每个氨基酸(AA)在各个位置上的贡献进行了定量评估。每个氨基酸被视为一个独立特征,并被赋予一个独特的 SHAP 值。不同类型生物活性肽的氨基酸特征存在差异,而与抗菌相关的氨基酸特征具有一定相似性(补充图6)。
统计分析结果显示:
- 在抗菌肽(ABP)中,Cys、Trp、Lys、Pro 和 Arg 对活性贡献最大;
- 在抗真菌肽(AFP)中,Cys、Arg、Lys、Trp 和 His 最为关键;
- 在抗氧化肽(AOP)中,His、Trp、Tyr、Cys 和 Pro 的贡献最为显著(补充图7)。
为了整合三种活性特征,研究者对 ABP-MPB、AFP-MPB 和 AOP-MPB 三个模型的 SHAP 值进行了归一化处理,并计算每种氨基酸的平均贡献值。结果表明(图3a),13个氨基酸长度(13-AA) 是最常被预测为同时具有三重活性的序列长度。
因此,在每条肽序列中,研究者利用 13-AA 滑动窗口 提取出累计平均 SHAP 值最高的片段,以消除背景氨基酸干扰,并将其定义为 关键特征片段(KFF, Key Feature Fragment)(图3b)。去除长度小于13的肽及 SHAP 值总和为负的片段后,共提取出 3400 个 KFFs。
通过成分分析发现,在这些 KFFs 中,Lys、Cys、Gly、Trp、Arg、Pro、Ala、His 和 Leu 的比例均高于原始的20个生物活性肽数据集,提升幅度在 0.14%–4.11% 之间(图3c)。这表明 KFFs 有效整合并突出了与三重生物活性相关的关键氨基酸特征。
随后,研究者基于序列相似性对 3400 个 KFFs 进行系统发育聚类,结果揭示出 四个具有独特氨基酸分布特征的亚类(subfamilies)(图3d)。在每个亚类中,研究者对所有 KFFs 进行序列比对,选取每个位点上出现频率最高的前三种氨基酸,这些氨基酸共同占据了 31.31%–54.38% 的比例,被视为该亚类的基础组成元素。通过系统组合这些氨基酸,构建出各自的 合理序列子空间(plausible sequence subspace),每个亚类包含 313 种可能的组合序列。
如图3e所示:
- 亚类A 的主要氨基酸为 Gly、Ala、Leu、Cys 和 Lys(其中 Ala 与 Cys 各仅出现在一个位置);
- 亚类B 的主要氨基酸为 Gly、Ala、Leu 和 Lys;
- 亚类C 主要包含 Gly、Leu、Cys 和 Lys(其中 Leu 出现在两个位置);
- 亚类D 的主要氨基酸为 Leu、Ile、Trp、Pro、Lys 和 Arg(其中 Leu 与 Ile 各仅出现在一个位置)。
通过利用 SHAP 值定义核心氨基酸特征,研究者成功构建了每个亚类包含313条高置信度序列的序列空间,为后续的候选肽设计提供了坚实基础。
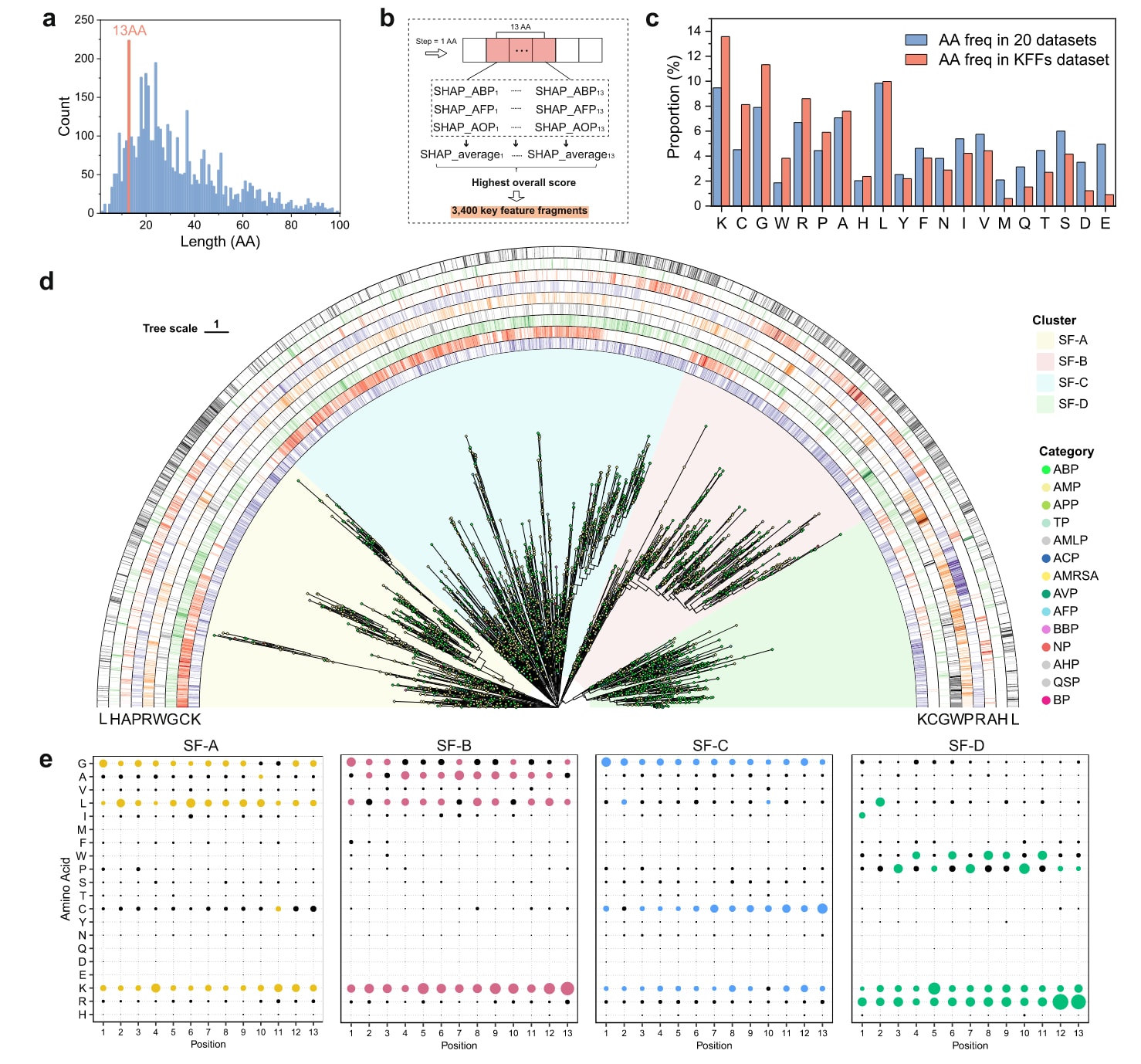
图 3 | 关键氨基酸特征与关键特征片段(KFFs)。 a 具有潜在抗菌、抗真菌和抗氧化活性的 4760 条肽序列 的长度分布。其中,长度为 13 个氨基酸(13 AAs) 的序列出现最为频繁。b **关键特征片段(KFFs)提取示意图。**在整个肽序列上设置一个 13-AA 滑动窗口,步长为 1 个氨基酸。计算每个片段的平均 SHAP 值之和,并选择得分最高且总和大于 0 的片段作为候选。最终,筛选出 满足所有条件的 3400 个片段,定义为 KFFs。c KFFs 与 20 个生物活性肽数据集中所有阳性样本之间的 氨基酸比例比较。结果显示,在 KFFs 中某些关键氨基酸(如 Lys、Cys、Trp、Arg、Pro 等)的占比更高,表明这些氨基酸在多重活性肽中具有重要作用。d 基于系统发育树分析的 3400 个 KFFs 的分布情况,显示其可分为四个亚类:亚类A(SF-A)、亚类B(SF-B)、亚类C(SF-C) 和 亚类D(SF-D)。系统发育树分支末端的彩色圆点表示各 KFFs 所来源的原始生物活性肽类别。e 每个 KFF 亚类在 13 个氨基酸位点上的分布特征。图中高亮显示的氨基酸为各位置上出现频率最高的前三种残基,反映了各亚类在氨基酸组成上的代表性特征。
2.4 构建合理序列子空间与代表性抗菌肽(AMPs)的筛选
在为每个 KFF 亚类生成 313 条基础序列组合(共 1,594,323 条序列) 的合理序列子空间后,研究者通过进一步筛选,剔除质量较低的序列,仅保留在三个模型(ABP-MPB、AFP-MPB、AOP-MPB)中均具有 >0.99 预测概率 的高可信度候选肽。根据既往研究,为确保活性和稳定性,带正电荷数量少于 2 的序列被排除。
筛选后,四个亚类的合理序列子空间分别缩减为:
- 亚类A:893,093 条序列
- 亚类B:273,615 条序列
- 亚类C:127,427 条序列
- 亚类D:666,165 条序列(见图4a)
由于传统基序提取算法在处理短肽序列时存在局限(补充图8),研究者采用了一种改进的策略,以最大化保留序列的多样性。首先,在每个亚类的合理序列子空间内,依据序列相似性进行聚类,将相似序列归为一组。然后,利用 k-means 聚类算法 并结合 肘部法(Elbow method) 确定最佳聚类数(每个子空间分为四个簇,补充图9),最终选取每个簇中心附近的序列作为代表性序列进行后续分析。
分析结果表明:
- 亚类A(图4b) 和 亚类B(图4c) 的代表性肽主要由 α-螺旋(α-helix)、无规卷曲(random coil) 或两者混合结构组成;
- 亚类C(图4d) 的代表性肽主要包含 β-折叠(β-sheet) 与无规卷曲结构;
- 亚类D(图4e) 的代表性肽则主要表现为无规卷曲结构。
通过从头设计(de novo design),研究者共生成了 16 条代表性序列,这些序列在现有数据库中均未被收录(补充图10)。随后,这些序列被用于进一步的实验验证。
2.5 16 条候选抗菌肽(c_AMPs)的体外初步评估
为验证这些候选肽的潜在 抗菌、抗真菌及抗氧化活性,研究者合成并测试了全部 16 条 c_AMPs(图5a与补充表2)。
抗菌实验 在 128 μM 的肽浓度下进行,测试了三种革兰氏阴性菌(Escherichia coli O157:H7、Klebsiella pneumoniae ATCC700603、Salmonella typhimurium ATCC14028)和三种革兰氏阳性菌(Micrococcus luteus ATCC4698、Bacillus subtilis WB600、Staphylococcus aureus ATCC6538)。
结果显示:
-
革兰氏阴性菌 中,B4、D1、D2、D3 和 D4 五组肽表现出显著抑菌活性,其中 D1、D2、D3 的抑菌率最高,达 75%–100%。
-
革兰氏阳性菌 中,10 条 c_AMPs 对至少一种菌株显示出抑制作用,其中 9 条肽的抑制率达到 100%。
具体而言:
- A1、A3、B2、C1、C2 及 D1–D4 可完全抑制 M. luteus ATCC4698 生长;
- C1、C2 及 D1–D4 对 B. subtilis WB600 有明显抑制;
- D1 与 D2 对 S. aureus ATCC6538 亦表现出抑制活性。
抗真菌实验 中,在 128 μM 浓度下,B2、C2 及 D1–D4 对 Candida albicans ATCC10231 的生长实现了 100% 抑制。
抗氧化实验 采用 ABTS⁺ 自由基清除实验(1 mg/mL 肽浓度)进行。结果显示:
- A3、C1–C4 的自由基清除率 ≥ 90%;
- D1、D2、D3、D4 的清除率分别为 72%、54%、62% 和 72%。
综上,D1 与 D2 展现出最强的广谱抗菌作用,同时具备抗真菌和抗氧化三重活性。
此外,生长曲线分析显示部分肽在处理 16 小时后未表现抑制效应,但在细菌或真菌对数生长期早期阶段能够有效抑制其生长(补充图11)。
例如:
- A3 与 B2 能在处理后 3–6 小时内显著抑制 B. subtilis WB600 的生长;
- C4 可在处理后 2 小时内抑制 C. albicans ATCC10231 的生长。
为评估候选肽的生物安全性,研究者在 128 μM 浓度下检测了其对 兔红细胞溶血活性 和 3T3 细胞毒性。结果显示:
- 15 条肽的溶血率低于 4%,表明其具有良好的安全性和临床应用潜力;
- 仅 B2 的溶血率略高,为 6.57%;
- B3 的细胞毒性为 6.97%(图5b)。
总体而言,这些结果证明所设计的 16 条 c_AMPs 具有良好的活性与安全性,其中 D1 与 D2 尤为突出,具备强广谱抗菌能力及多重生物功能。
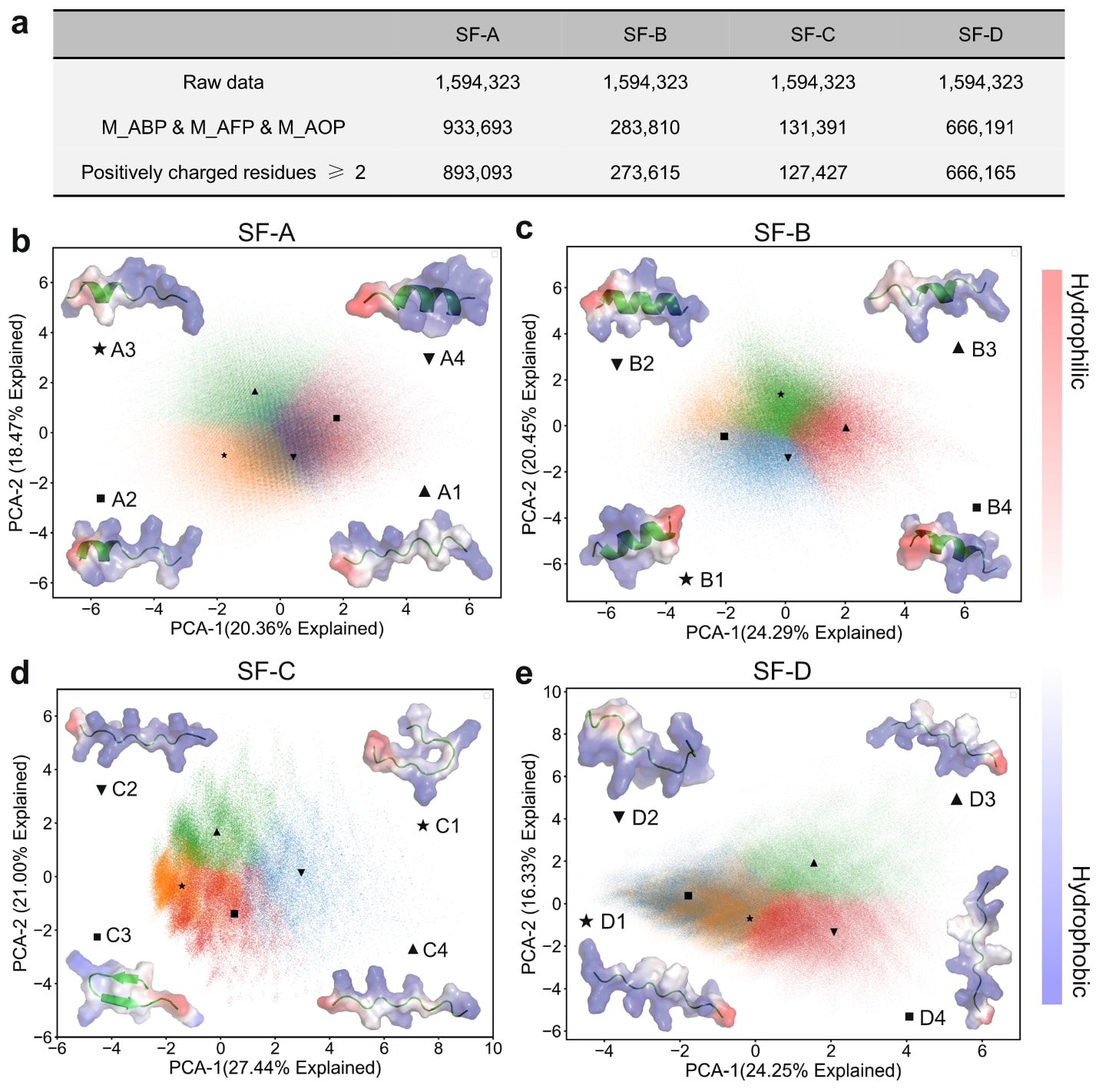
图 4 | 合理序列子空间中的筛选与代表性序列的确定。 a 四个亚类(subfamily-A,SF-A;subfamily-B,SF-B;subfamily-C,SF-C;subfamily-D,SF-D)在逐步应用严格筛选标准后的剩余序列数量。图中展示了每个亚类在筛选流程各阶段(如概率阈值筛选、电荷筛选、冗余去除等)后序列数量的变化。b–e 各亚类内的聚类结果及代表性序列的选取。在 主成分分析(PCA)图 中,不同颜色的点表示不同的聚类簇,而带有黑色标签的点标示出所选取的代表性序列位置。每个代表性序列旁展示其 三维结构模型:
- 内部结构图 显示肽的详细三维构象;
- 外层半透明部分 则展示了肽分子的 疏水性分布(hydrophobicity distribution),以反映其可能的结构–功能特征。
2.6 D1 与 D2 对耐药菌株的进一步评估
鉴于 D1 与 D2 在体外表现出的广谱抗菌活性,研究者进一步评估了它们对来源于畜禽及临床环境的抗生素耐药菌株的抑制效果。
实验结果表明:
-
对于畜禽来源的抗药性菌株 Escherichia coli z44,D1 的最小抑菌浓度(MIC)为 4 μM,而 D2 的 MIC 为 8 μM(图5c)。
值得注意的是,在 2 μM 浓度下,D1 即可在 4 小时内显著抑制细菌生长,与未处理对照组相比表现出明显抑制效果。
-
对于临床分离的抗药性菌株 Staphylococcus aureus 09057,D1 的 MIC 为 16 μM,且在 8 μM 浓度下 能够持续抑制细菌生长达 11 小时;而 D2 的 MIC 为 32 μM(图5d)。
此外,研究者还在多种其他耐药菌株上进行了进一步验证(补充图12与13),结果显示 D1 与 D2 均具备广谱抗菌活性,其中 D1 的抑菌效果略优于 D2。
尤为重要的是,经过 连续 100 代 使用 D1 处理后,在 S. aureus 09057 中未检测到耐药性产生(补充图14),表明 D1 具有较低的诱导耐药风险。
为进一步探究 D1 的作用机制,研究者对经 64 μM D1 处理的 S. aureus 09057 细胞进行了 扫描电子显微镜(SEM) 与 透射电子显微镜(TEM) 观察。
- SEM 图像 显示,随处理时间延长,D1 处理组的细胞膜结构出现明显破裂,与未处理对照组中形态完整的细胞相比,表现出显著的膜损伤(图5e)。
- TEM 图像 同样揭示,D1 处理的细胞出现膜结构破坏与胞质内容物流失,而对照组细胞保持完整形态(图5f 与补充图15)。
这些结果共同表明,D1 通过破坏细胞膜完整性发挥杀菌作用,且对多种耐药菌株具有持久且广谱的抗菌活性。
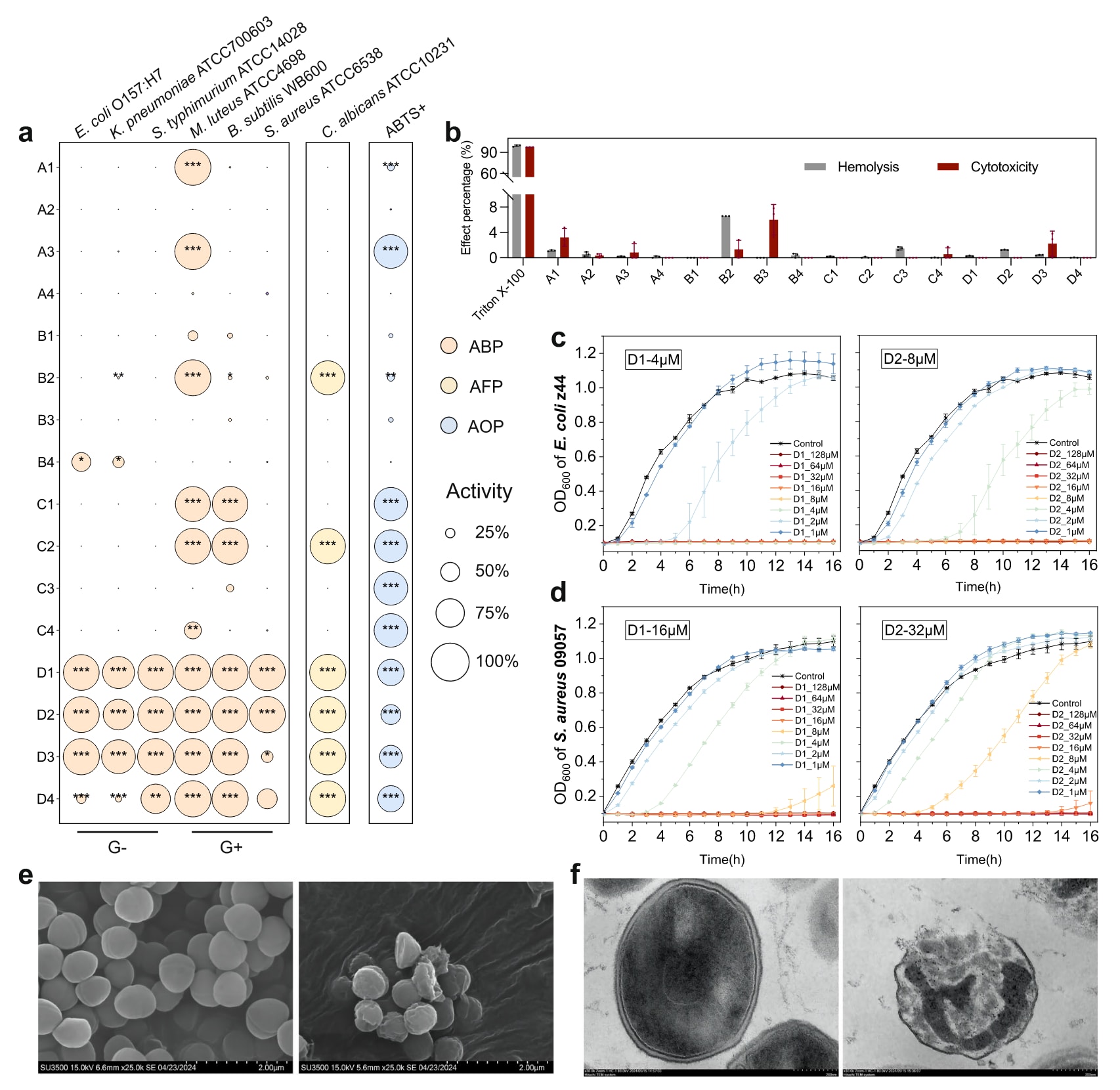
图 5 | c_AMPs 的实验验证与活性检测。
a 在 128 μM 浓度下对 6 种细菌 和 1 种真菌 的抑制率结果,以及在 1 mg/mL 浓度下的 ABTS⁺ 自由基清除率。圆点越大代表活性越强。统计学显著性通过 双侧 Dunnett’s 检验(多重比较校正) 进行评估:
- “*” 表示 0.01 < p < 0.05;
- “**” 表示 0.001 < p ≤ 0.01;
- “***” 表示 p ≤ 0.001。
b 16 条 c_AMPs 在 128 μM 浓度下的 溶血活性与细胞毒性评估。每组包含 3 个独立生物学重复(n = 3),结果以平均值 ± 标准差(mean ± s.d.)表示。
- Triton X-100 作为溶血实验阳性对照;
- PBS 作为溶血实验阴性对照;
- DMEM 为细胞毒性实验对照。
c, d 代表性耐药菌的生长曲线:
-
c 为从家禽中分离得到的 Escherichia coli z44;
-
d 为临床来源的 Staphylococcus aureus 09057。
每组均包含 3 个独立生物学重复(n = 3),结果以平均值 ± 标准差表示。对照组显示正常的细菌生长曲线。
D1 与 D2 的浓度梯度 设置为 128 μM 至 1 μM(两倍递减),不同颜色代表不同浓度。每个面板中标注的浓度为该肽对相应菌株的 最小抑菌浓度(MIC)。
e S. aureus 09057 经过 PBS(对照) 与 D1(64 μM) 处理后的 扫描电子显微镜(SEM)图像,显示低倍放大效果(比例尺:2 μm)。
f 相同条件下 S. aureus 09057 的 透射电子显微镜(TEM)图像,低倍放大观察(比例尺:200 nm)。
所有图像均代表至少 3 次独立实验的典型结果,具有良好重复性。更多图像及不同放大倍数的结果见补充图15,进一步验证了观察结果的可靠性。
2.7 D1 在体内抗菌感染模型中的治疗效果评估
基于前述的体外实验结果,研究者进一步评估了 D1 在小鼠败血症模型中的体内治疗效果(图6a)。
实验中,小鼠首先感染临床分离的高危抗药性菌株 Staphylococcus aureus 09057。感染后 1 小时,通过细菌负荷检测确认败血症模型建立成功(补充图16)。随后,小鼠接受系统性治疗,给予 D1(20 mg/kg,腹腔注射,200 μL)。感染 12 小时后,处死小鼠并取肝脏、肾脏、脾脏和肺等组织器官以定量检测细菌负荷。
结果显示:
- 万古霉素(Vancomycin)治疗组 小鼠的肝、肾、脾和肺中的细菌负荷显著低于未处理感染对照组,验证了感染模型的成功建立。
- 与生理盐水组相比,D1 治疗组 小鼠各脏器中的细菌负荷显著下降,平均降低幅度为 0.44–1.15 log CFU/g(图6b–e)。
此外,与生理盐水对照组相比,D1 治疗组小鼠血清中促炎因子水平显著降低,包括:
- 肿瘤坏死因子 α(TNF-α),
- 白介素-1β(IL-1β),
- 白介素-6(IL-6)(图6f–h)。
这一结果表明,D1 不仅能降低体内细菌负荷,还能有效缓解感染引起的炎症反应。
在 Escherichia coli z44 感染所致的败血症模型中(图6i),在相同治疗条件下,D1 同样显著降低了小鼠脏器中的细菌负荷,平均减少幅度为 0.47–0.67 log CFU/g(图6j–m)。与此同时,与未处理组相比,D1 治疗显著降低了小鼠血清中促炎性细胞因子的水平(图6n–p),包括 TNF-α、IL-1β 和 IL-6。
综上所述,这些结果表明,D1 在体内具有显著的抗菌及抗炎作用,能够有效降低败血症小鼠模型中的细菌负荷和炎症反应,对 S. aureus 与 E. coli 所致感染均展现出良好的治疗潜力。
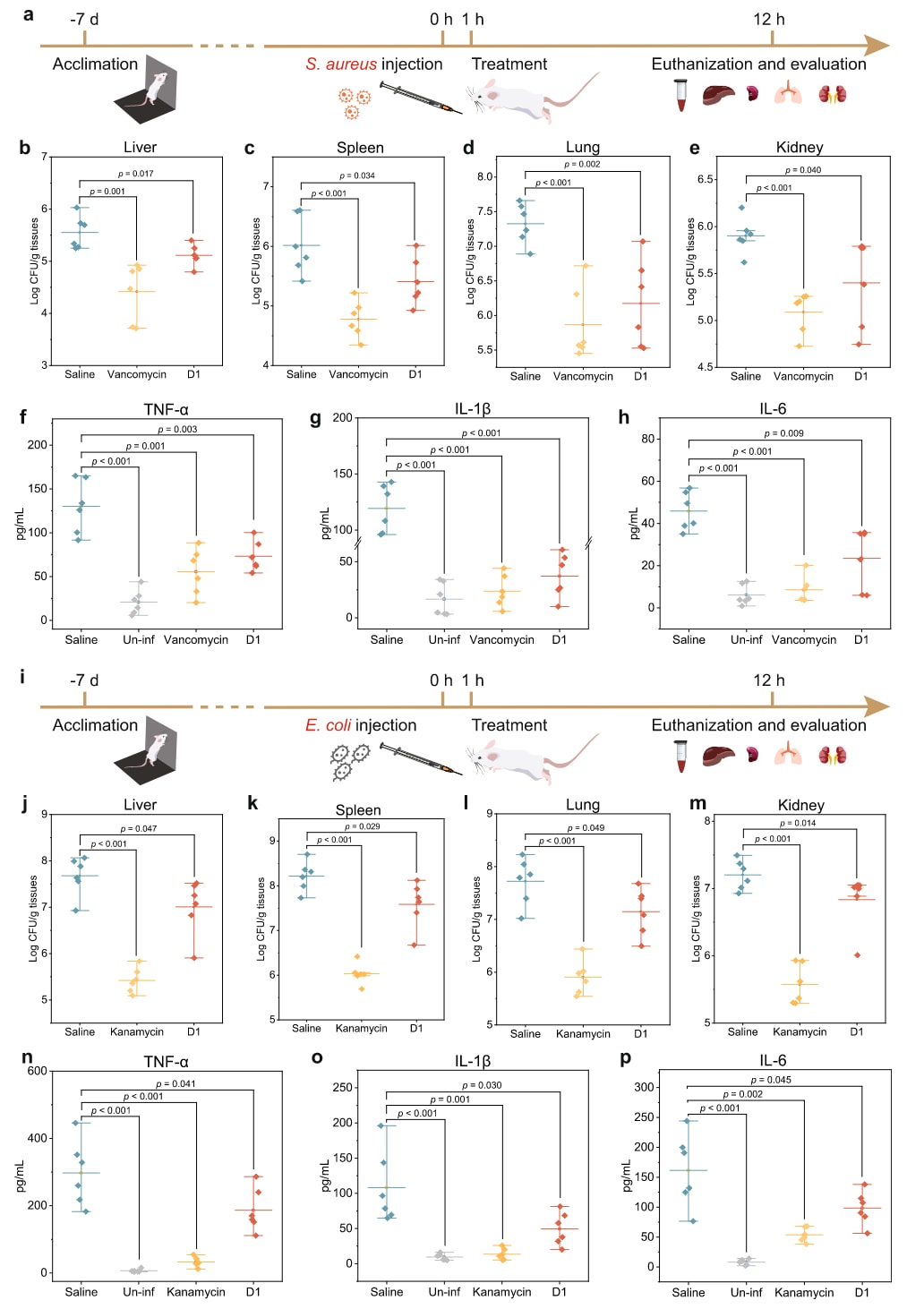
图 6 | D1 在体内的治疗效果。 a S. aureus 09057 系统性感染小鼠模型的实验设计示意图。展示了感染建模、D1 给药及样本采集的实验流程。b–e S. aureus 09057 感染模型中不同处理组小鼠肝脏、脾脏、肺和肾脏中的细菌负荷比较。图中包括未感染组、以及分别接受生理盐水、万古霉素和 D1 治疗的感染组。f–h 健康小鼠及 S. aureus 感染后分别接受生理盐水、万古霉素和 D1 治疗的小鼠血清中 促炎因子 TNF-α、IL-1β 与 IL-6 的水平变化。i E. coli z44 系统性感染小鼠模型的实验设计示意图。展示了感染与药物干预的实验流程。j–m E. coli z44 感染模型中不同处理组小鼠肝脏、脾脏、肺和肾脏的细菌负荷结果。对比了未感染组、以及分别接受生理盐水、卡那霉素和 D1 治疗的小鼠。n–p 健康小鼠及 E. coli 感染后分别接受生理盐水、卡那霉素和 D1 治疗的小鼠血清中 促炎性细胞因子 TNF-α、IL-1β 和 IL-6 的浓度变化。统计分析采用 双侧单因素方差分析(two-sided one-way ANOVA)。数据以 平均值 ± 标准差(mean ± s.d.) 表示,样本量为 n = 6 个独立生物学重复。
3 讨论
深度学习模型在蛋白质功能预测中展现出强大的潜力,但其内部学习到的特征与模型预测结果之间的关系往往难以解释。该研究开发的 DLFea4AMPGen 提供了一种新的从头抗菌肽(AMP)设计策略,利用深度学习模型学习到的氨基酸(AA)特征来指导新序列生成。通过结合 SHAP 方法 对深度学习模型中“黑箱”特征的可解释性分析,该策略能够在综合肽特征空间中同时考虑抗菌、抗真菌和抗氧化三种活性,从而确保新设计的 AMPs 融合与这些目标活性相关的特征,提高总体设计成功率。
实验验证结果显示,在该研究中设计的 16 条候选肽(c_AMPs) 中,有 12 条表现出生物活性,阳性率达 75%,这为 DLFea4AMPGen 的有效性提供了概念验证(proof of concept)。与以往针对所有可能序列进行预测的研究不同,该研究基于深度学习模型的特征提取能力,构建了高阳性率的综合肽序列空间,在保持预测精度的同时显著降低了筛选规模。通过提取对生物活性影响最大的氨基酸并构建合理序列子空间,该策略确保生成的 AMPs 含有关键的抗菌活性特征。
更重要的是,该研究聚焦于 SHAP 分析中最显著的特征,而非随机组合 20 种氨基酸,从而在一定程度上解决了传统肽设计中存在的“全局搜索问题”。相较于常见的数据库筛选方法(通常仅随机组合少量氨基酸,如三种),该研究通过对所有潜在抗菌活性序列的系统分析,并在相似序列中选择代表性肽以最大化序列多样性,从而提高了从头设计 c_AMPs 的有效性。
近年来,AMPs 作为新型抗感染药物候选分子,受到广泛关注。多项研究表明,肽的电荷、二级结构、疏水性及两亲性等因素对其与细胞膜相互作用及抗菌活性具有重要影响。该研究设计的 16 条 c_AMPs 也具备这些典型特征。研究者还尝试使用传统的基序提取方法(MEME)对 ABP、AFP 和 AOP 三个数据集进行分析,但所提取的基序未能呈现出明显模式。这可能是因为抗菌肽与酶类不同,其序列较短,且功能并不依赖于特定活性位点,因此传统基序分析难以揭示其通用功能机制,从而限制了其在 AMP 从头设计中的应用。
DLFea4AMPGen 的优势在于能够在设计过程中同时整合多种活性特征,从而生成兼具抗菌、抗真菌及抗氧化功能的多活性肽。值得注意的是,Trp、Cys 和 Pro 等氨基酸在抗氧化活性中发挥关键作用,其在序列中的富集不仅可降低感染诱发的氧化应激,还能有效减轻宿主炎症反应。
在功能验证中,D1 展现出独特的作用机制——通过破坏细胞膜结构实现杀菌效果,对包括临床耐药菌株在内的 17 种微生物 均表现出广谱抗菌活性。此外,D1 不仅能清除细菌,还能显著降低促炎因子水平,从而改善感染治疗效果并减少并发症的发生。
尽管该研究中 DLFea4AMPGen 生成的 AMPs 显示出优异的效果,但模型仍有进一步优化空间。例如,未来可通过引入 MIC 回归预测模型,实现对抗菌活性的定量评估,而不仅限于定性判断。同时,构建由多模型组成的预测管线有助于提升整体预测精度、降低实验验证成本。结合 MIC 定量预测与经验预筛选,可进一步优化筛选流程,提高最终验证阶段的准确性与效率。
综上所述,DLFea4AMPGen 是一种基于深度学习的抗菌肽生成方法,通过学习与生物活性相关的氨基酸特征,实现了更高效的肽设计。该方法能够降低计算成本、提高设计成功率,并生成具有多样化活性的 AMPs。通过 SHAP 分析识别关键特征并构建合理序列子空间,研究者设计并合成了 16 条新肽,其中 D1 与 D2 表现出强劲且广谱的抗菌活性,包括对耐药菌株的显著抑制作用。
D1 与 D2 的高效表现验证了 DLFea4AMPGen 的可靠性与潜力,为未来开发针对耐药病原体的新型肽类治疗药物提供了有力的依据。DLFea4AMPGen 不仅为 AMP 发现提供了高效的新途径,也为未来多功能肽设计与人工智能驱动的药物研发奠定了重要基础。