JCIM 2025 | FakeRotLib: 在Rosetta中实现非天然氨基酸快速参数化的方法
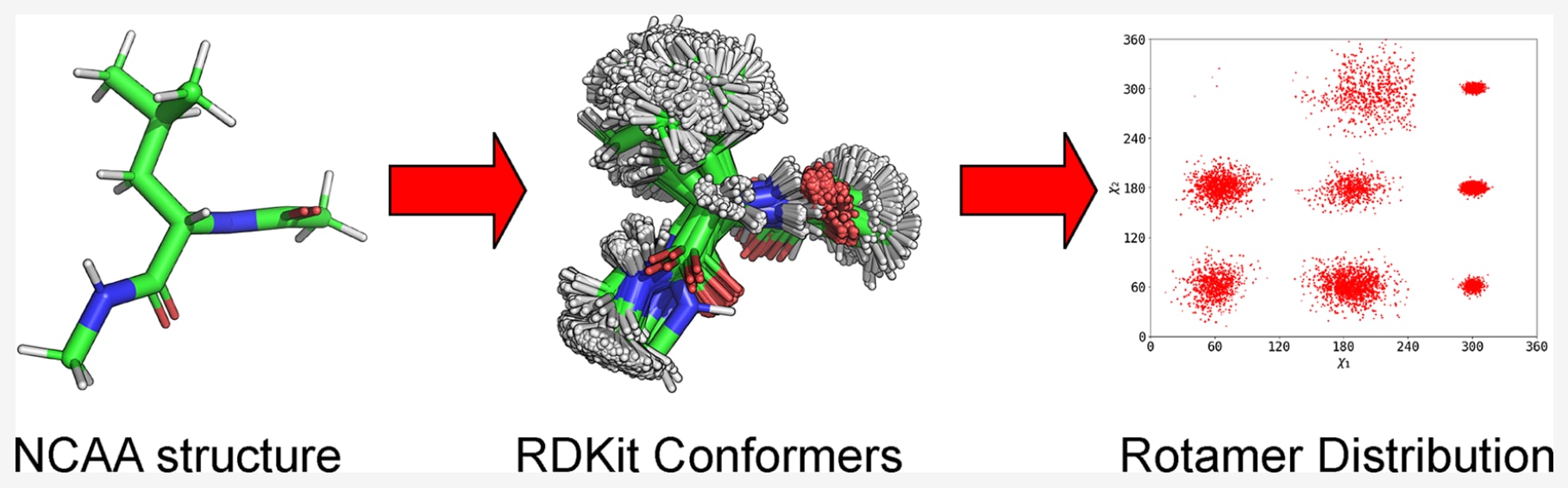
今天介绍的是发表于JCIM的一项研究——FakeRotLib: 在Rosetta中实现非天然氨基酸快速参数化的方法。这项工作针对Rosetta在处理非天然氨基酸(NCAAs)时长期存在的性能与效率瓶颈,提出了一种创新的构象建模方案。传统方法如MakeRotLib虽能生成合理的侧链构象分布,但计算代价高、操作复杂且受限于力场定义。FakeRotLib通过对小分子构象空间的统计拟合,以贝叶斯高斯混合模型(BGMM)快速生成rotamer库,实现了在几秒内完成参数化。结果显示,该方法在rotamer恢复率、序列恢复率与计算效率三方面均优于现有方案,且首次支持脯氨酸衍生物及高柔性侧链等以往难以建模的NCAA类型,为未来Rosetta在新型氨基酸与肽药设计中的应用提供了强有力的工具基础。

获取详情及资源:
0 摘要
非天然氨基酸(Noncanonical Amino Acids, NCAAs)在天然生物体系与合成应用中均占据重要地位。然而,由于缺乏针对这类分子的充分训练数据,目前大多数深度学习方法仍无法有效建模NCAA。相比之下,诸如 Rosetta 等基于生物物理原理的方法在NCAA建模中具有明显优势。该研究系统讨论了在Rosetta中对NCAA进行参数化的各个关键环节,并指出构象异构体(rotamer)分布建模是影响Rosetta性能的最关键因素之一。为此,作者提出了一种名为 FakeRotLib 的新方法,通过对小分子构象的统计拟合来生成合理的rotamer分布。结果显示,FakeRotLib在显著缩短计算时间的同时,其性能优于现有方法,并能够对此前Rosetta无法处理的NCAA类型进行成功参数化。
1 引言
近几年,随着 AlphaFold2、AlphaFold3、ESMFold 等深度学习模型的出现,蛋白质结构建模经历了前所未有的变革。然而,这类模型的训练依赖于大量高质量结构数据,并要求氨基酸序列可由有限的字母表表示。此条件仅适用于20种标准氨基酸,而**修饰型与非天然氨基酸(NCAAs)**在解析结构中稀少,且序列表示通常受限于标准氨基酸字母表。因此,深度学习模型难以有效处理这类氨基酸。
非天然氨基酸在生物体系中具有重要作用,包括翻译后修饰的标准氨基酸、对映异构的D-氨基酸以及诸如γ-氨基丁酸(GABA)等氨基酸代谢物。除天然体系外,NCAAs 在肽设计(尤其是环肽构建)、酶工程以及功能位点解析实验中也极具价值。然而,无法精确建模这些残基的构象,已成为现代蛋白结构预测的显著短板。
虽然深度学习模型正尝试拓展其“可建模氨基酸空间”,如 RoseTTAFold All-Atom 和 AlphaFold3 等“全原子”建模工具开始能够理解任意化学结构,但其在NCAA建模中的表现仍有限。例如,AlphaFold3在糖基化等常见修饰中预测成功率可达72.1%,但对一般修饰残基仅为51.0%。这主要源于公开数据库中含NCAA结构样本稀缺,尤其是罕见类型。
因此,若要准确建模含有NCAA的蛋白,仍需依赖基于生物物理的建模方法。一种常见思路是预先收集NCAA的构象异构体(rotamer) 库以供后续建模使用,但此方法需高昂计算代价,并受限于库中已有的氨基酸类型。分子动力学模拟虽可在原子层面捕捉NCAA的动态行为,但并不适用于需快速筛选大量NCAA的计算肽设计任务。
相较之下,Rosetta 以其可控的粗粒度能量模型成为NCAA建模的理想工具。Rosetta最早通过 MakeRotLib 实现NCAA建模,该方法结合Rosetta与CHARMM势函数,通过能量最小化计算侧链的rotamer分布。然而,MakeRotLib存在显著局限:计算耗时长(对柔性氨基酸需数天)、需人工设定构象分布中心初值、且NCAA扭转角必须存在于CHARMM能量库中。
近年来,仅有少数方法尝试改进NCAA的rotamer生成问题。例如,AutoRotLib 扩展了Rosetta可参数化的化学范围,但依赖商业软件(OpenEye)且计算时间同样较长。
该研究系统评估了当前用于Rosetta的NCAA参数化方法,并分析了不同参数化环节对建模性能的影响。此外,作者提出了一种新的rotamer参数化策略——FakeRotLib,该方法利用开源小分子工具包与笛卡尔坐标空间混合模型,可在更短时间内高效生成NCAA的构象分布,大幅提升了Rosetta在非天然氨基酸建模中的适用性与效率。
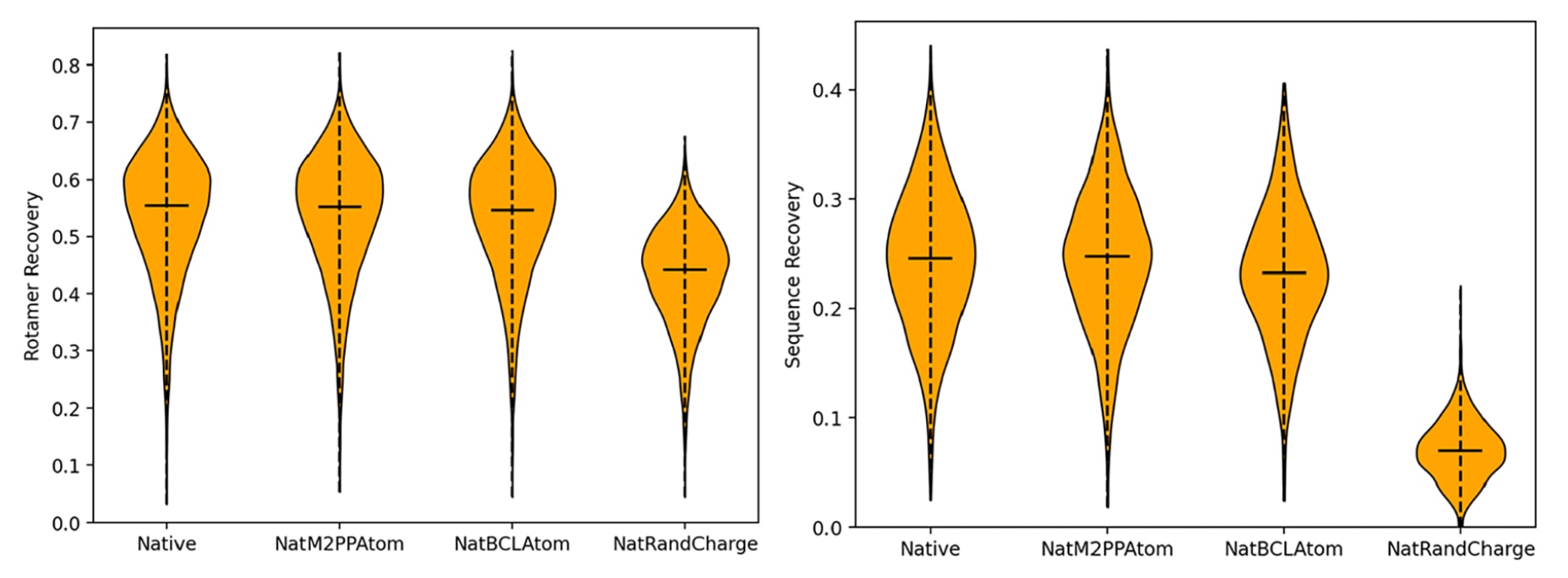
图1|不同参数设置下Rosetta的构象异构体恢复率(左)与序列恢复率(右)性能比较。 对比方法包括:默认Rosetta参数(Native)、由 molfile_to_params_polymer.py 分配原子类型的模型(NatM2PPAtom)、采用 BCL 部分电荷 的模型(NatBCLAtom)以及随机部分电荷**模型(**NatRandCharge)。
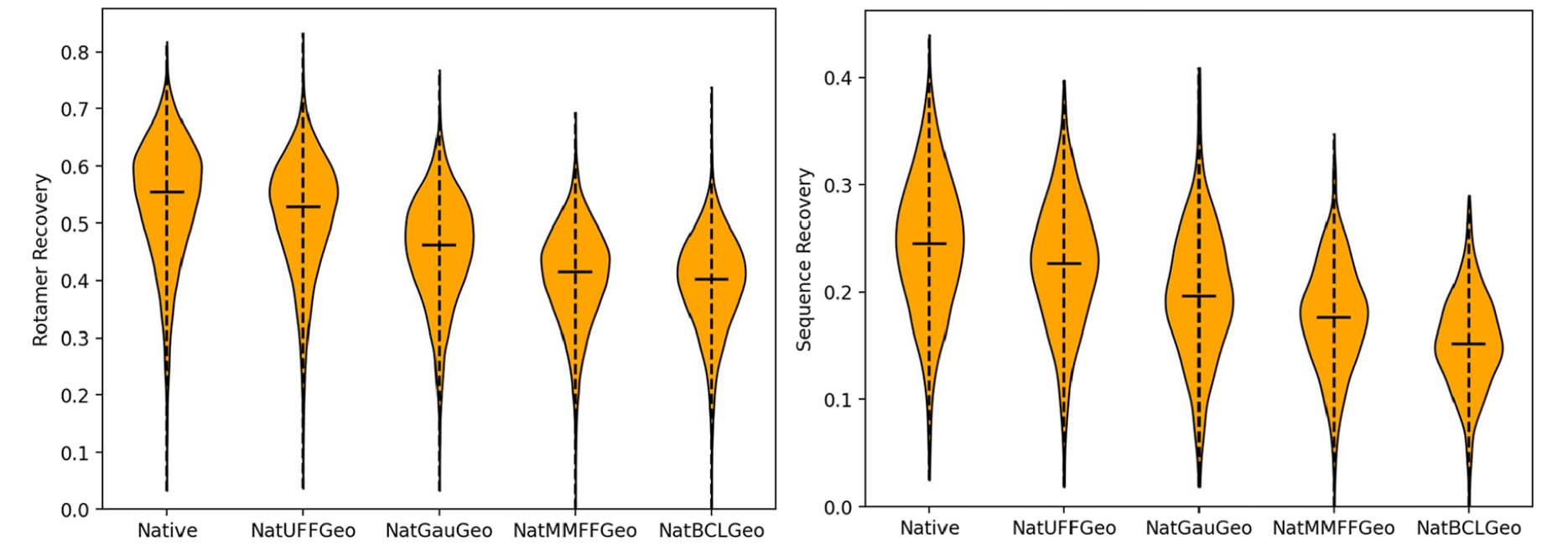
图2|不同几何优化策略下Rosetta的构象异构体恢复率(左)与序列恢复率(右)性能比较。 对比方法包括:默认Rosetta几何(Native)、UFF优化几何(NatUFFGeo)、量子化学(QM)优化几何(NatGauGeo)、MMFF94力场优化几何(NatMMFFGeo)以及BCL优化几何**(**NatBCLGeo)。
2 结果与讨论
2.1 原子类型与部分电荷分配
为了评估原子类型划分对 Rosetta 分子建模性能的影响,研究者为每种标准氨基酸构建了其“非天然(non-canonical)形式”。具体而言,通过替换默认氨基酸参数文件中的“atom”模块,分别采用以下三种方式生成新参数:
- Rosetta自带脚本
molfile_to_params_polymer.py生成的原子块; - 使用 BCL 生成的部分电荷(对二肽中每个原子的σ键与π键电荷求和)并输入至
molfile_to_params_polymer.py; - 采用默认Rosetta原子类型,但从[-1, 1]的均匀分布中随机采样部分电荷值,并随后校正使其总电荷等于氨基酸的形式电荷。
为基准测试Rosetta性能,研究设计了两类任务:
- Rotamer恢复任务:将所有残基转换为其对应的“非天然”形式,并通过重新打包(repacking)来恢复原始的侧链扭转角;
- 序列恢复任务:在给定天然骨架的情况下,使用“非天然氨基酸”重新设计序列,以评估能否恢复原始的氨基酸序列。
结果表明,只要部分电荷分配合理,Rosetta对电荷数值及原子类型的细微差异具有一定鲁棒性(图1)。然而,电荷参数化仍会对性能产生影响——当部分电荷被随机化时,建模精度明显下降。
2.2 侧链几何结构
与原子类型实验类似,研究者通过替换内部坐标模块(internal coordinates block),使用三种不同来源的几何结构生成了“非天然”氨基酸:
- BCL构象生成器生成的几何;
- 基于 RDKit 实现的UFF 与 MMFF94 分子力学优化几何;
- 使用 Gaussian 进行的量子力学优化几何。
采用与前述相同的rotamer恢复与序列恢复任务对其性能进行评估(图2)。结果显示,与原子类型不同,Rosetta对分子几何结构高度敏感。这可能是因为Rosetta的默认运动集仅限于扭转空间(torsional space),即在计算中保持键长与键角固定。而默认的Rosetta几何参数在其能量函数中已被“过度优化(hyper-optimized)”,因此高精度的量子力学几何反而不如分子力学几何表现优异。
综上所述,几何精度越高并不必然提升Rosetta的性能。从结果来看,最佳的参数化策略应是能从头(ab initio)精确复现Rosetta默认几何的方案,其中基于 UFF 力场的优化方法在性能上表现最为接近Rosetta原始几何,可作为最优选择。
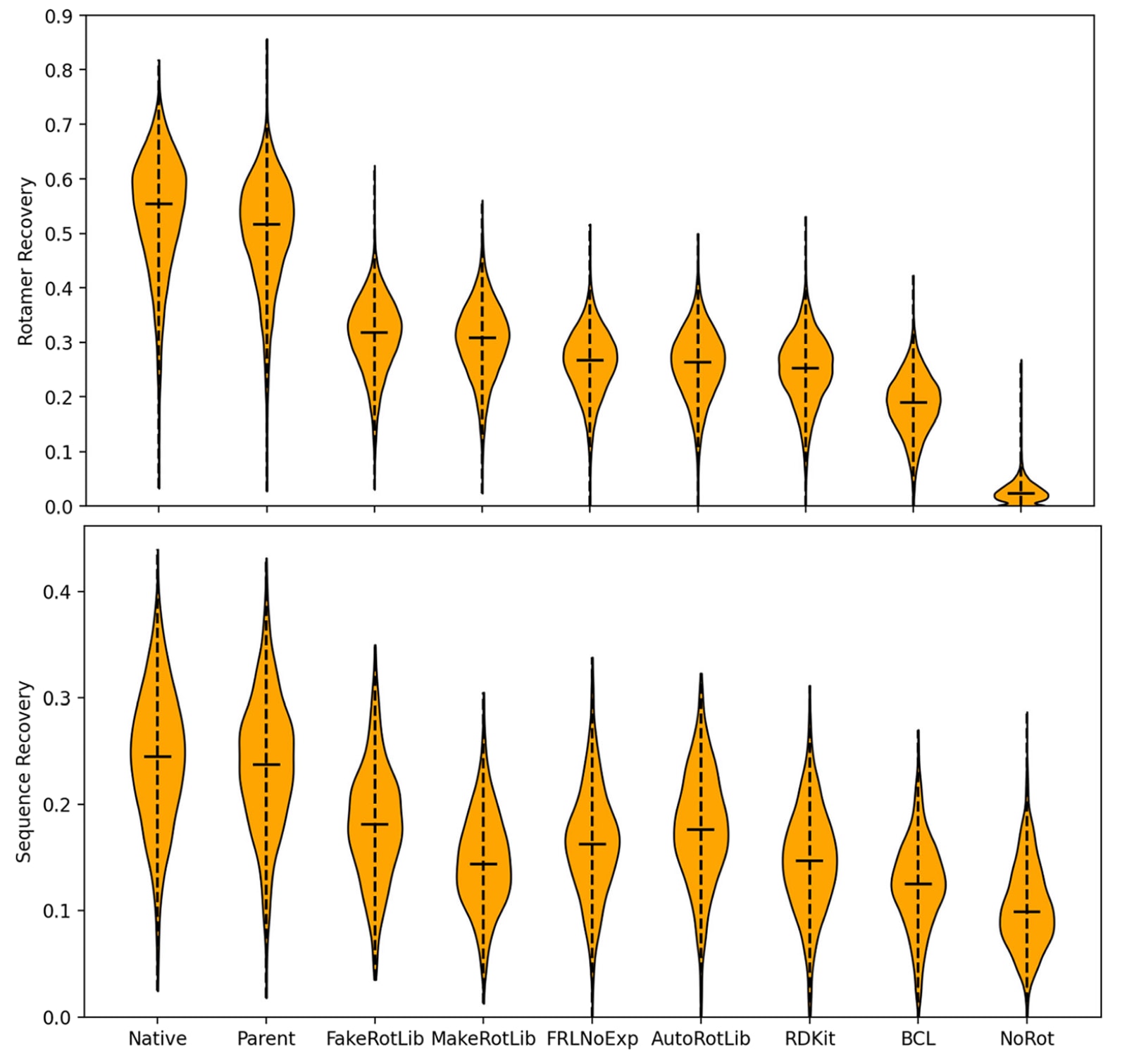
图3|不同构象库(rotamer library)生成方法在 Rosetta 中的性能比较。 上图为 构象异构体恢复率(Rotamer recovery),下图为 序列恢复率(Sequence recovery)。对比的方法包括:
- Native:默认Rosetta自带rotamer库;
- Parent:基于母体氨基酸的rotamer分布;
- FakeRotLib:该研究提出的方法,基于小分子构象统计拟合生成rotamer分布;
- MakeRotLib:Rosetta原有的rotamer生成程序;
- FRLNoExp:FakeRotLib去除实验扭转角约束后的版本;
- AutoRotLib:依赖OpenEye软件的自动rotamer生成方法;
- PDB-RDKit:由RDKit从PDB结构中提取的rotamer分布;
- PDB-BCL:由BCL从PDB中提取的rotamer分布;
- NoRot:无rotamer库情况下的建模。
结果显示,FakeRotLib在rotamer恢复与序列恢复两项任务中均优于传统方法,且计算效率更高。
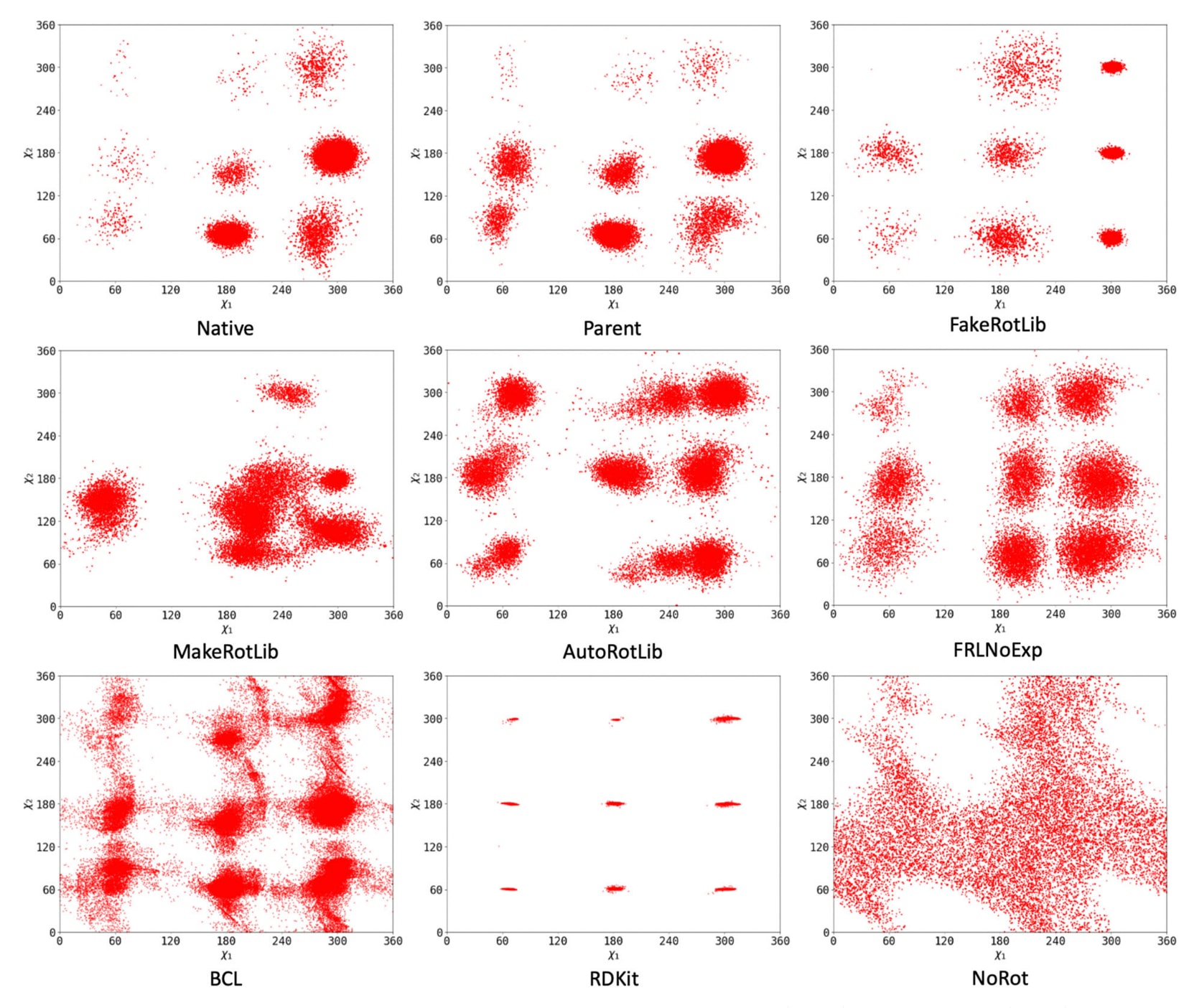
图4 | 各种rotamer生成方法下亮氨酸的构象分布:分别为默认Rosetta(Native)、“parent” rotamer(Parent)、FakeRotLib、MakeRotLib、AutoRotLib、去除实验扭转角的FakeRotLib(FRLNoExp)、基于BCL的PDB rotamer(BCL)、基于RDKit的PDB rotamer(RDKit)以及无rotamer信息(NoRot)。
2.3 侧链构象体(Rotamer)分析
最后,研究者测试了多种为非天然氨基酸生成侧链构象体的方法,并评估其对Rosetta性能的影响。除“native”和“AutoRotLib”两种方法外,其余方案均采用了由molfile_to_params_polymer.py生成的原子类型与部分电荷,并使用UFF进行几何优化。这样设置的目的是在未知非天然氨基酸先验信息的情况下,更真实地再现其在Rosetta中的表现。
共评估了七种不同的构象体生成方法:(1)直接复制天然残基的rotamer,即所谓“parent”方法;(2)Renfrew等人开发的MakeRotLib协议,通过Rosetta/CHARMM混合能量函数最小化侧链构象,从而生成能量谷分布的rotamer;(3)BCL方法,利用BCL构象生成器生成侧链构象并作为PDB rotamer输入;(4)RDKit方法,与前者类似,但使用RDKit替代BCL;(5)FakeRotLib方法,通过RDKit在笛卡尔空间中的构象分布拟合贝叶斯高斯混合模型(BGMM),以生成类似MakeRotLib的rotlib文件;(6)AutoRotLib方法,由Holden与Pavlovicz等人开发,利用OpenEye工具模仿MakeRotLib流程;(7)不提供任何rotamer信息,让Rosetta通过均匀采样模拟侧链(作为负对照)。
结果表明,“parent”方法的表现最接近默认的Rosetta rotamer恢复率,其后依次为FakeRotLib、MakeRotLib、AutoRotLib、RDKit、BCL以及无rotamer方案(图3)。在序列恢复率上趋势相似,仅AutoRotLib与MakeRotLib位置互换。parent方法性能优异并不意外,因为其遵循了Rosetta能量函数原本的参数;其相较默认Rosetta略有下降,可能归因于UFF几何优化带来的偏差。紧随其后的FakeRotLib表现出色,但由于RDKit中包含实验扭转角库,该结果对某些特殊非天然氨基酸可能并不完全适用。为此研究者还测试了排除实验扭转角的FakeRotLib版本,其性能介于MakeRotLib与AutoRotLib之间。
MakeRotLib作为Rosetta的传统工具,在非天然氨基酸rotamer生成上仍有良好表现;AutoRotLib的性能与之相近,但不及FakeRotLib,也未明显劣于PDB rotamer方法。基于PDB的rotamer方法整体表现最差,但仍优于无rotamer信息的对照组。其间,RDKit方法因包含实验扭转角数据而显著优于BCL方法。
为了直观展示这些不同方法生成的rotamer差异,研究者绘制了亮氨酸(leucine)的构象分布(图4)。除BCL与RDKit外,其余方法的分布均通过Metropolis-Hastings采样模拟生成;BCL与RDKit则绘制了等量PDB rotamer的二面角分布。结果显示,“native”分布精确再现了原始Dunbrack rotamer库,而“parent”分布紧密跟随;无rotamer提供时的分布则较为弥散,仅排除了发生内部空间冲突的构象。其余方法中,FakeRotLib最接近天然分布,仅在能量谷标准差上存在轻微偏差。AutoRotLib与不含实验扭转的FakeRotLib虽总体分布准确,但出现谷间模糊或过宽能量谷,可能导致非rotamer型构象的通过。MakeRotLib的分布整体形似天然分布,但显得畸形,原因可能是其初始聚类中心的选择较为随机。
最后,PDB rotamer的九谷分布表现出明显差异:RDKit分布过度依赖实验扭转库,在理想角度附近形成狭窄峰值;BCL分布则形态异常,谷间存在连通,显示出非rotamer构象倾向。需要指出的是,PDB rotamer与rotlib模拟的二面角分布存在一定语境差异——前者基于小分子构象生成,而后者则反映了rotamer在Rosetta环境下的适配情况。此外,图中展示的rotamer数量远超实际应用场景,因为Rosetta对这些rotamer集的实现效率极低,rotamer数量越多,计算时间呈非线性增长。即便采用每个残基100个rotamer的方案,某些目标仍需耗时数日才能完成设计。这种低效性在未来涉及大量非天然氨基酸的Rosetta设计中将成为亟待解决的问题。
3 讨论
基准测试结果表明,在Rosetta中使用非天然氨基酸仍然是一项挑战。其主要原因在于Rosetta的能量函数依赖于基于实验真实数据建立的统计分布与性能优化,而这些实验数据在非天然氨基酸中极其稀缺。由于非天然氨基酸的化学多样性远超数据库可覆盖的范围,因此Rosetta在缺乏可靠统计支持的情况下,其参数化与能量评估均存在明显局限。
基于当前对Rosetta参数化方法的理解,最理想的做法是尽可能遵循默认Rosetta参数体系。对于仅在结构上对天然氨基酸进行小幅修饰的非天然残基,可采用**“parent” rotamer方法**:其与天然残基相同的子结构部分使用默认几何,其余差异部分则通过UFF进行几何优化。
然而,对于结构差异更大的非天然氨基酸,推荐使用该研究提出的FakeRotLib协议。该方法可在文件格式支持的前提下(如单取代氨基酸且χ角不超过四个)生成对应的rotlib文件。提出这一建议有以下三点主要原因:
首先,在前述基准中,该参数化方案的性能优于其他同类方法;
其次,该方法在速度与便利性上均具有显著优势——完整流程仅需一个Python脚本即可实现,只依赖scikit-learn与RDKit两个库,甚至可在数秒内完成高柔性分子的参数化;
最后,FakeRotLib可自动生成PDB rotamer库,使得先前难以被Rosetta支持的非天然氨基酸类型(例如主链结合侧链的脯氨酸衍生物,或具有超过四个χ角的高度柔性侧链)也能顺利完成参数化。
这些推荐基于当前版本的Rosetta能量函数ref2015。该能量模型对非天然氨基酸具有明显排他性:其参考能量难以准确估算,环闭合项默认仅适用于脯氨酸,侧链几何经过强优化且难以复现,Ramachandran项与氨基酸概率项均依赖于氨基酸类型,对非天然残基几乎失效。初步测试显示,在ref2015_cart与beta_nov16能量函数下,所有参数化方案的性能趋势相同(数据未展示),这表明上述问题并非特定能量函数所致,而是普遍存在。
因此,若未来Rosetta的开发仍将继续扩展至非天然氨基酸建模领域,亟需在如何合理生成并评估非天然残基的构象分布方面进行深入的系统性思考与改进。