Nat. Biotechnol. 2025 | GNEprop: 基于深度学习的抗菌化合物虚拟筛选
今天介绍的研究来自 Genentech 团队,发表于 Nature Biotechnology 。该工作提出了一个结合高通量实验与深度学习的抗菌化合物发现框架——GNEprop。研究者首先针对约 200 万种小分子 进行表型高通量筛选,从中获得 5,000 余个对 E. coli ΔtolC 有活性的分子,并据此训练图神经网络模型 GNEprop,用于预测全细胞水平的抗菌活性。随后,模型被用于对 14 亿个可合成分子 进行虚拟筛选,实验验证显示命中率高达 23.8%,比原始 HTS 提高约 90 倍。更重要的是,这些命中化合物在结构上与已知抗生素高度不同,展现出全新的化学骨架和潜在作用机制。进一步的生物学验证确定了部分分子作用于脂多糖和脂肪酸合成通路中的关键酶。该研究展示了深度学习与实验筛选协同加速抗菌药物发现的巨大潜力,为探索新型抗生素开辟了可扩展的计算–实验一体化范式。

获取详情及资源:
0 摘要
多重耐药细菌的不断增加凸显了开发新型抗生素的紧迫需求。该研究将小分子高通量筛选与基于深度学习的虚拟筛选方法相结合,以发现新的抗菌化合物。研究首先针对一株敏感化的大肠杆菌菌株筛选约200万种小分子,获得数千个命中化合物,并利用这些实验数据训练了深度学习模型GNEprop,用于预测抗菌活性。该模型在外部分布泛化与活性突变(activity cliff)预测方面表现出较强的稳健性。进一步对超过14亿种可合成小分子进行虚拟筛选,筛得具有潜在抗菌活性的候选化合物,其中有82种在相同菌株上表现出抗菌活性,命中率相比用于训练的高通量筛选提高了约90倍。值得注意的是,许多新发现的化合物与已知抗生素在结构上差异显著,部分分子对其他细菌株亦显示出活性且具有良好的选择性。生物学表征揭示了部分化合物的特异靶点,为抗生素发现提供了新的研究方向。
1 引言
多重耐药细菌的迅速增加凸显了开发新型抗生素的迫切性。该研究将小分子高通量筛选与基于深度学习的虚拟筛选方法相结合,以发现新的抗菌化合物。研究首先针对一株敏感化的大肠杆菌(Escherichia coli)进行约200万个小分子的筛选,获得数千个命中化合物,并利用这些数据训练深度学习模型GNEprop,用于预测抗菌活性。该模型在外部分布泛化与活性突变(activity cliff)预测中表现出较高的稳健性。通过虚拟筛选超过14亿种可合成小分子,识别出潜在候选化合物,其中82种在相同菌株上表现出抗菌活性,命中率较初始高通量筛选提高约90倍。值得注意的是,许多新发现的化合物与已知抗生素结构差异显著,并对其他菌株表现出良好的活性与选择性。进一步的生物学验证揭示了部分化合物的特异靶点,为抗生素发现提供了新的方向。
当前,抗击不断演化的耐药菌株仍是药物研发的核心挑战。传统方法主要依赖小分子高通量筛选(HTS)、天然产物挖掘、生物制剂(如抗菌肽)的定向进化及后续药物化学优化。然而,这些策略受到化学空间庞大、分子多样性受限及筛选偏倚等问题的制约。尽管已探索约10⁶–10⁷个分子,这一规模在理论上高达10⁶⁰个潜在“类药”化合物的化学空间中仍微不足道,因此多数筛选仅能获得极少数可用先导化合物,甚至无结果。此外,现有化合物库常集中于已知抗生素或旧有药物骨架,难以发掘具有全新作用机制(MoA)的分子结构,这导致抗菌研究长期陷入“发现空窗期”。目前,每年约有127万人因抗生素耐药而直接死亡,其中革兰氏阴性菌尤为严重,其双层膜结构和高效外排系统使得药物难以进入细胞或维持有效浓度。
为突破这一瓶颈,虚拟筛选被引入作为高效替代方案。近年来,诸如Enamine REAL等化合物库已扩展至数十亿分子,为计算筛选提供了前所未有的化学资源。虚拟筛选利用计算算法(基于物理化学模型或机器学习方法)预测化合物活性,并挑选最有潜力的分子进行实验验证。相比传统物理建模,机器学习(ML)尤其擅长预测整体细胞水平的抗菌活性。此前研究表明,基于图神经网络的模型已可从化学结构预测大肠杆菌的抗菌活性,如Stokes等人鉴定的Halicin(原为糖尿病候选药物),以及Liu等人发现的针对鲍曼不动杆菌的窄谱抗菌化合物。尽管这些成果展示了人工智能(AI)在抗生素发现中的潜力,但其应用仍受限于低命中率及训练集规模与多样性的不足。多数模型仅基于几千个样本或已知抗生素构建,难以在广阔化学空间中实现有效泛化。
为此,该研究在化学空间分布广泛的约200万个小分子中进行表型高通量筛选,针对E. coli ΔtolC突变株检测生长抑制效应,获得约5000个抗菌分子(命中率0.26%)。以此为基础训练的GNEprop模型采用图表征学习(graph representation learning)框架,并在外部分布化学空间与活性突变预测中通过系统验证。进一步虚拟筛选超过10亿种可合成分子,预测出约4.4万个潜在抗菌候选物,并选取345种具有不同结构相似度的分子进行实验验证,其中82种表现出抗菌活性,命中率提升至23.8%。其中约三分之一的分子与训练集的Tanimoto相似度低于0.4,几乎所有新化合物均与已知抗生素显著不同。
后续的浓度反应实验(IC₅₀与MIC测定)证实,模型能够识别针对革兰氏阴性菌的活性分子,并可推广至其他菌株(包括野生型大肠杆菌及金黄色葡萄球菌)。通过诱导耐药菌并进行基因组测序,研究确定了若干推测靶点,进一步验证了模型的预测准确性。最后,基于GNEprop的邻域扩展策略成功挖掘出更多活性类似物,拓宽并优化了抗菌活性谱。该研究展示了深度学习结合实验筛选在抗菌药物发现中的巨大潜力,为探索结构新颖、机制独特的抗生素开辟了新的途径。
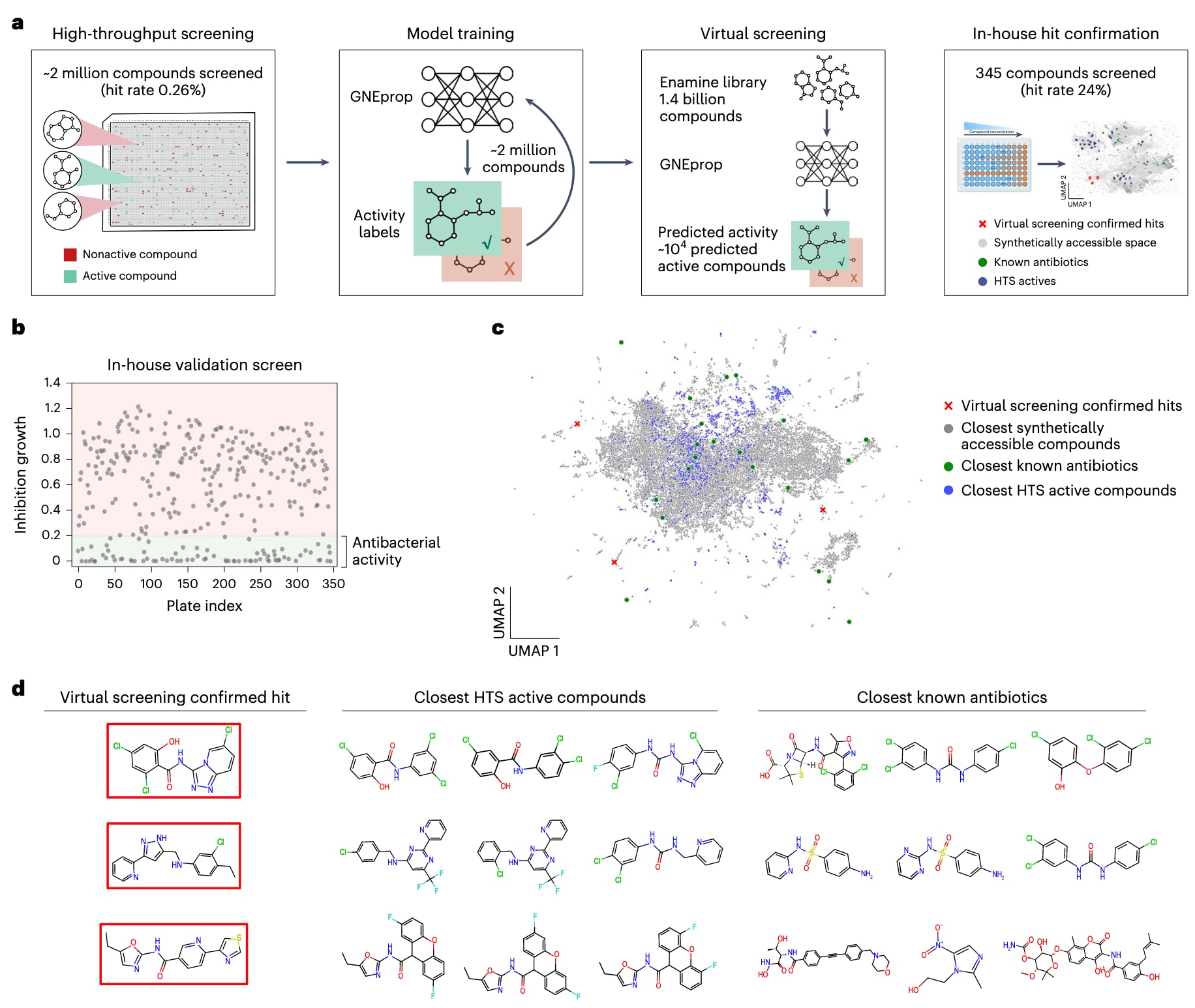
图1|高通量表型筛选结合深度学习虚拟筛选策略以发现多样化的抗菌骨架。 (a) 示意图。该研究通过两步策略将抗菌化合物的搜索空间扩展至已知抗生素之外:首先进行新的表型高通量筛选(HTS),随后采用基于深度学习的超大规模虚拟筛选以探索可合成化学空间。HTS用于鉴定与已知抗生素相似度低、但活性显著的化合物,同时覆盖广阔且多样的化学空间。基于HTS数据训练的深度学习模型 GNEprop 可在虚拟筛选中对庞大的可合成分子空间进行高效预测。虚拟筛选能够在极大化学空间中发现结构新颖、命中率高且与已知抗生素相似度低的候选分子。 (b) 使用GNEprop对超过10亿个可合成小分子进行虚拟筛选,相比HTS数据集显著富集了抗菌活性分子,命中率从 0.26% 提升至 23.8%。 (c, d) 虚拟筛选所得活性分子与HTS活性分子及已知抗生素的结构相似度均较低。 (c) 通过UMAP降维可视化结果显示,虚拟筛选命中分子(红色叉)位于与HTS活性分子(蓝点)及已知抗生素(绿色点)不同的化学空间中。 (d) 展示了虚拟筛选所得代表性分子(对应c中的红叉)与其在训练集和抗生素数据库中最相似的前三个分子的结构对比,可见这些新命中分子均具有显著的结构新颖性。
2 结果
2.1 高通量表型筛选揭示抗菌化合物及精细的构效关系(SAR)
该研究首先开展了一项大规模表型高通量筛选(HTS),使用了一个包含 1,981,993 个小分子化合物 的化合物库(图2a,方法详述)。这一筛选规模显著超过以往用于训练抗菌性质机器学习(ML)模型的已报道数据集。值得注意的是,该化合物库不仅包含与已知抗生素具有结构相似性的分子,还覆盖了更广阔的化学空间,从而能够探索潜在的新型可合成分子领域(图2a)。
筛选采用单点浓度(5 μM)表型实验,针对 E. coli ΔtolC 菌株进行检测。该菌株缺乏多种外排系统的共同通道蛋白,因此对多种抗生素表现出高敏感性,这使其成为发现抗菌化合物的理想模型系统。每个化合物根据其是否能抑制细菌生长被赋予二元标签,最终获得 5,161 个抗菌(活性)化合物,命中率为 0.26%。
研究者进一步分析了HTS数据中抗菌化合物的分布特征。该化合物库包含大量结构相似的分子族群,其中 181,435 个骨架(scaffold) 被两个以上化合物共享(占库中化合物的84%),25,437 个骨架 被10个以上化合物共享。抗菌化合物共覆盖 3,231 个骨架,其中 1,075 个骨架 包含10个以上的活性分子。值得关注的是,即使在同一骨架家族内,微小的结构变化也可引发活性的大幅差异(图2b示例),这一现象被称为 活性悬崖(activity cliffs)。此外,研究者发现化合物活性与其分子量(MW)及计算得出的疏水性(logP)并不存在简单的相关性,这表明模型训练时不会因简单理化性质而产生偏倚。
鉴于数据集的规模与多样性,研究者推断其足以支撑构建能够在新化学空间中外推并解析不同骨架家族中活性悬崖的机器学习模型。基于此,研究者进一步整理并公开了HTS数据的一个高质量子集,命名为 GNEtolC。该数据集包含 115,519 个化合物,覆盖 44,852 个化学骨架,其中包括 2,918 个抗菌分子。
GNEtolC 是迄今公开的最大规模的小分子抗菌活性数据资源之一,覆盖了广泛的化学空间,既包含活性悬崖,也包括与已知抗生素显著不同的化合物(见扩展数据图1与补充表1)。

图2|GNEprop 利用高通量筛选数据在大规模上探索多样化的化学空间。 (a, b) 超大规模高通量表型筛选揭示结构多样的抗菌骨架。 (a) 通过UMAP降维可视化显示,HTS数据集(紫色点)不仅包含与已知抗生素(黄色三角形)结构相似的化合物,还覆盖了更广泛的可合成化学空间(灰色点),从而拓展了可探索的新分子区域。 (b) HTS中存在大量活性悬崖(activity cliffs)——即具有相同骨架但仅有微小结构差异、却导致活性显著不同的化合物。每个矩形代表一个活性悬崖,圈出的化合物为活性分子,其他为无活性分子。(c–g) GNEprop 模型的设计与性能验证:关注外部分布(OOD)泛化、效率与可扩展性。 (c) GNEprop 是一种图神经网络(Graph Neural Network, GNN),用于基于分子图表示预测抗菌活性。模型不仅为每个化合物赋予0至1之间的活性概率,还配备了可解释性模块,用于识别关键活性基团,以及外部分布(OOD)检测模块,用于评估分子结构相对于已知活性分子的独特性。 (d) 左图: 模型经自监督学习获得的表示具有清晰的化学意义,形成有序分区,能捕捉理化性质(如 logP 与类抗生素化学特征;更多性质见扩展数据图2)。 右图: 传统Morgan指纹作为对照,其聚类结构较弱,未能有效区分化学特征。 (e) 表征对比实验(线性评估)。 在三个化学性质数据集(Freesolv、Lipophilicity 与 ESOL)上,比较使用自监督表征(青色)与Morgan指纹(黄色)作为输入时的线性模型预测误差。图中为30次随机划分(80:20训练/测试)的均方根误差(点)及其均值(柱),显示自监督表征在性质预测上具有更高准确性。 (f) GNEprop 预训练可显著提升抗菌预测性能。 柱状图展示模型在不同条件下(颜色区分)的表现。每种条件下进行20次基于骨架划分的独立实验(点),柱为均值,误差线表示标准误。结果显示含预训练的GNEprop表现最佳。 (g) GNEprop 的可扩展性分析。 以推理吞吐量(每秒处理1,000个分子数)衡量,在不同GPU数量下进行8次重复实验,柱为均值,误差线表示标准误。结果表明模型在计算资源扩展时具有良好的线性可扩展性。
2.2 基于深度学习的模型在大规模化学空间中探索新型抗菌区域
研究者利用HTS数据集训练了一个基于深度学习的模型——GNEprop。GNEprop 是一种图神经网络(Graph Neural Network, GNN),通过分子图表示来预测抗菌活性,为每个化合物分配一个介于0到1之间的活性概率(见图2c与方法部分)。该模型设计目标包括:
(1)外部分布(OOD)泛化能力,确保在不同化学空间中保持预测精度;
(2)结构敏感性,能识别导致“活性悬崖(activity cliffs)”的细微结构差异;
(3)可扩展性,能够高效处理数十亿级的可合成化合物。
为实现这些目标,模型结合了图表示学习的最新进展。其编码器基于图同构网络(Graph Isomorphism Network, GIN)**以获得高表达性的原子特征表示,并结合**Jumping Knowledge Network框架构建局部结构的多尺度层级表征。此外,GNEprop通过**对比学习(contrastive learning)**在来自ZINC15数据库的 1.22亿个未标注化合物 上进行自监督预训练,从而学习通用的、可迁移的化学表示,再在带标签的HTS数据集上进行微调。已有研究表明,大规模预训练不仅能增强GNN模型的OOD泛化性能,还能提供高效的化学空间导航与度量方式。
研究者首先评估了预训练对模型泛化与可扩展性的影响。对比学习所获得的自监督表示无需依赖人工选择的分子性质,即可形成具有化学意义的结构空间。结果显示,模型学习到的表征空间呈现出清晰有序的分区,化学性质相似或满足相同药物化学特征的化合物会自动聚集(图2d与扩展数据图2)。在线性评估实验中(图2e,方法详述),自监督表征在多个化学性质预测任务中表现出更高的表达能力。
随后,研究者利用一个包含 2,335 个化合物 的公开抗菌数据集(E. coli生长抑制实验数据)验证了该表示的实际效果。先前的研究曾基于深度学习模型并结合额外的分子描述符进行预测,而研究者的实验表明,自监督表征在不依赖这些描述符的情况下即可获得更优的性能,同时使传统分子描述符变得冗余(图2f,补充表2)。
最后,研究者分析了模型在大规模虚拟筛选中的计算可扩展性。GNEprop通过一次性预训练替代逐分子计算描述符的高成本过程,并采用高效的分子图特征化方法、数据并行及高性能计算库,使其能够在推理阶段实现每秒数千分子的筛选速率(图2g)。在多GPU环境下,模型可进一步提升吞吐量,例如使用 8块A100 GPU 时,虚拟筛选速度超过 每秒30万个分子,足以在数小时内完成对数十亿可合成分子的筛选。

图3|GNEprop 在高通量筛选(HTS)数据集上的回顾性验证。 (a) 骨架簇划分(scaffold-cluster splitting)分析。 与随机划分(绿色)或传统骨架划分(红色)相比,骨架簇划分(紫色)显著降低了训练集与测试集之间的结构相似性。图示为测试集中活性分子与训练集中活性分子之间最大 Tanimoto 相似度的概率密度分布。 (b) 可视化比较。 展示了在骨架簇划分下,三个测试集中活性分子与其最相似的五个训练集分子的结构对比。 (c) 活性悬崖与活性/非活性分子的表示空间。 通过自监督学习(左)与监督微调(右)获得的分子嵌入空间可视化。上方显示活性悬崖(activity cliffs)的分布,下方展示活性(红)与非活性(灰)分子的分区情况。 (d) 不同划分策略下的模型性能对比。 比较 GNEprop 在骨架划分与骨架簇划分下的多项指标表现。灰色柱为所有测试分子结果,紫色柱为仅含活性悬崖子集的结果。柱为平均值,误差线表示标准误。 (e) 活性悬崖预测示例。 模型正确预测的测试集中活性悬崖实例。每个黑色矩形框表示同一骨架家族的化合物组,红色为活性分子。数字表示模型预测的活性概率。 (f) 模型可解释性分析。 对(e)中预测为活性的化合物进行特征重要性可视化。颜色叠加在化学结构上表示模型关注的关键原子与化学键(颜色条示重要性)。 (g) 训练数据规模对性能的影响。 模型在不同划分条件下(颜色区分),以训练数据占比为横轴、预测性能(纵轴)为指标的变化曲线。点为平均值,阴影表示标准误(8次划分)。 (h) 主动学习策略示意图。 GNEprop 结合不确定性估计,用于选择新的候选分子加入训练集,从而迭代优化模型。 (i) 主动学习效果评估。 左图为模型在不同数据使用比例下保留的 AUPRC(平均精确率–召回率)比例;右图为恢复的阳性样本比例。不同颜色代表不同不确定性估计方法。点为均值,阴影为标准误(基于6次骨架划分实验)。
2.3 GNEprop 在大规模表型筛选数据中刻画外部分布(OOD)活性悬崖特征
研究者在HTS数据集上训练了 GNEprop 模型,并通过回顾性分析评估其在外部分布(OOD)化学空间中的虚拟筛选能力,重点考察预测精度、可解释性及其在未见过的“活性悬崖”(activity cliffs)家族上的表现。最后,研究者还研究了训练数据集规模对OOD泛化能力的影响,以及模型在主动学习(active learning)框架下高效引导新数据采集的潜力。
首先,研究者提出了一种更严格的训练–测试划分策略。传统虚拟筛选常使用“骨架划分(scaffold splitting)”方法,但研究者发现该方法仍可能导致训练集与测试集之间存在较高相似度。为此,研究者使用预训练编码器对分子骨架进行特征化,并基于社区检测算法将结构相似的骨架聚类(扩展数据图3左、方法部分)。然后,研究者以整个“骨架簇(scaffold cluster)”为划分单元进行训练、验证与测试,称为骨架簇划分(scaffold-cluster splitting)。与传统方法相比,该策略显著降低了训练集与测试集之间的相似性(图3a,b),因此能更准确地评估模型在新化学空间中的泛化能力。
研究者对HTS数据进行八折训练–验证–测试划分,并在每一折上对预训练模型进行微调(方法;HTS数据集见补充表3,GNEtolC结果见补充表4)。结果表明,模型在最具挑战性的骨架簇划分下仍保持较高预测性能(AUROC = 0.877 ± 0.007,AUPRC = 0.126 ± 0.019),并在“早期识别”指标(BEDROC20 = 0.488 ± 0.014)上表现稳定。
为理解模型学习到的表示,研究者比较了微调前后的分子嵌入(图3c)。微调前,活性化合物与其对应骨架下的非活性化合物混杂在一起;微调后,活性化合物在嵌入空间中聚集于同一区域,同时保留了结构分层信息。
随后,研究者评估了模型在“活性悬崖”任务上的表现。模型若要在此任务中保持预测能力,必须学习到微小结构变化如何导致活性的显著变化,尤其当这些悬崖分子属于与训练集不相似的家族时(即OOD情境)。为此,研究者将测试集限定为仅包含活性悬崖家族(方法)。尽管总体得分略低,GNEprop 在该子集中仍维持合理性能(图3d,补充表5)。进一步地,研究者可视化了这些悬崖分子家族的预测活性分布(图3e)。
为定量评估每个化学特征(原子与键)对抗菌活性的贡献,研究者为GNEprop 配备了基于 积分梯度(Integrated Gradients, IG) 的可解释性模块(方法)。通过该模块,研究者可以直观地查看模型在预测中是否聚焦于活性与非活性分子间的关键差异。对于每一个被正确预测的活性悬崖,研究者生成对应的**结构热图(heat map)**以显示重要特征(图3e,f)。这些热图虽可能带有噪声或不完全,但在多数情况下与实际决定活性的结构差异一致,从而验证了模型的解释合理性。
在回顾性验证的最后,研究者探讨了训练数据规模对OOD泛化的影响。结果表明,训练数据量对模型性能具有显著作用(图3g),尤其在骨架簇划分下,当训练集低于40%时性能下降明显,显示HTS数据对OOD表现至关重要。值得注意的是,当使用全部HTS数据时模型性能仍未达到饱和,说明体系仍能从新增数据中获益。
因此,研究者进一步测试了GNEprop在**主动学习(Active Learning)**框架下指导新数据采集的能力(图3h,方法)。在该设置中,模型通过不确定性估计选择最有可能提升性能的分子加入训练集。结果显示,与随机采样相比,GNEprop主导的数据采集带来了持续性能提升(图3i,扩展数据图4,补充讨论)。
总体而言,这些结果表明 GNEprop 不仅能在大规模表型筛选数据中刻画复杂的外部分布活性规律,还能有效指导数据集扩展,为抗菌活性泛化预测与抗生素发现提供了有力支持。
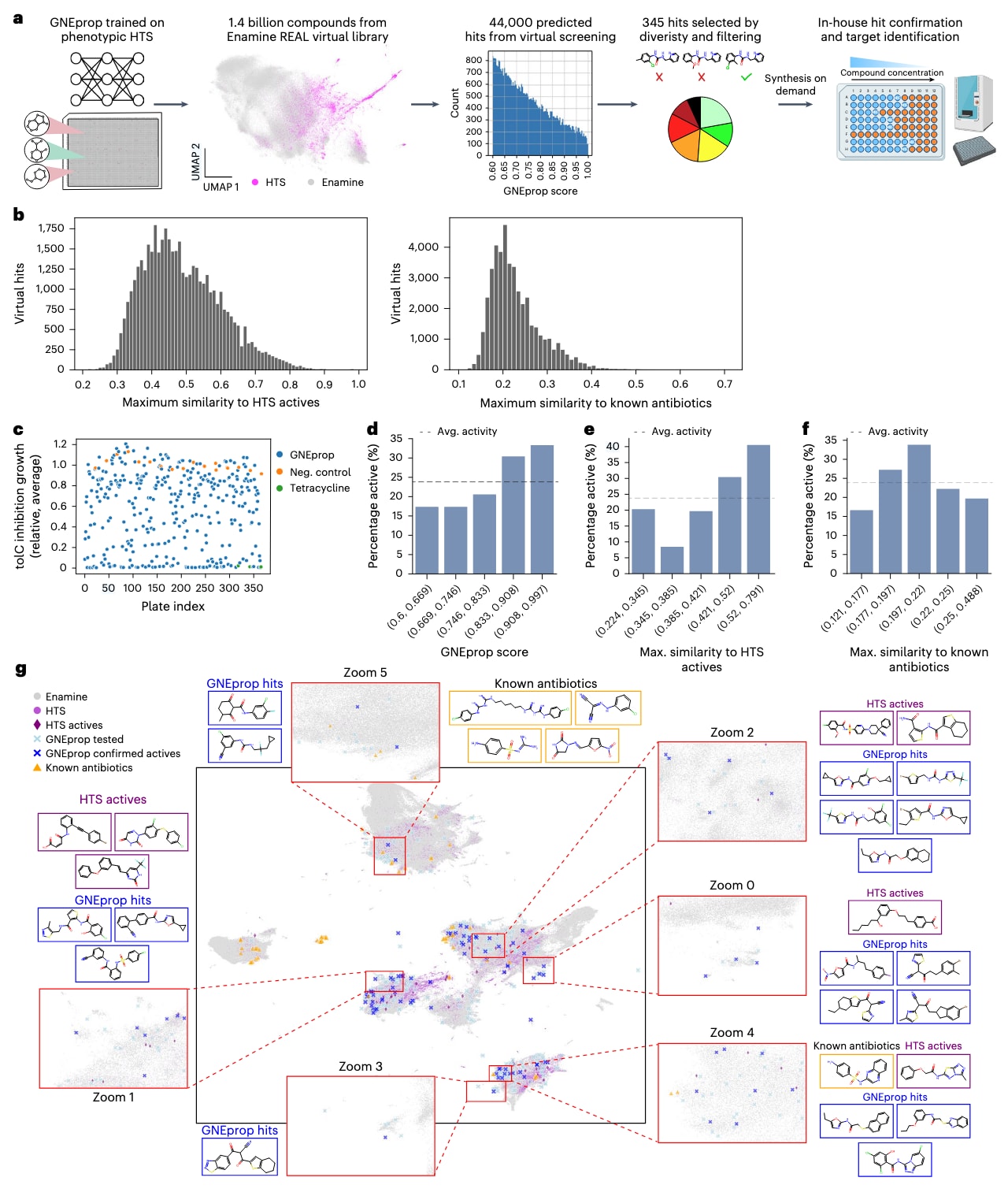
图4|超大规模抗生素虚拟筛选显著提升命中率,发现结构多样且新颖的活性分子。 (a) 虚拟筛选流程示意图。 使用在高通量筛选(HTS)数据集上训练的 GNEprop 模型,对可合成化学空间(Enamine 库)进行预测。模型首先为分子打分(预测活性概率),随后根据多重标准(多样性、与已知抗生素及HTS活性分子的低相似度等)进行筛选与聚类。最终选出的分子被合成,用于命中确认与靶点验证阶段。该图部分由 BioRender.com 绘制。 (b) 相似度分布。 虚拟筛选命中分子与训练集中活性分子(左)及已知抗生素(右)的最大 Tanimoto 相似度分布。 (c) 抑菌活性比较。 实验验证库(蓝点)的相对生长抑制率,与阳性对照四环素(绿点)及阴性对照(橙点)进行对比。 (d–f) 命中率分析。 虚线表示平均命中率。每个柱状区间代表一个分位区间,括号内为该区间的最小与最大值。 (d) 命中率随 GNEprop 预测得分(活性概率)的变化。 (e) 命中率随与 HTS 训练集中活性分子最大 Tanimoto 相似度的变化。 (f) 命中率随与已知抗生素最大 Tanimoto 相似度的变化。 (g) 化学空间可视化(UMAP)。 每个点代表一个分子,颜色表示来源(见图例与方法)。表示基于 GNEprop 自监督学习得到的嵌入向量及欧氏距离。插图为局部放大区域(红框),其中显示对应分子的具体结构,边框颜色与点的颜色一致。为便于可视化,已知抗生素与 GNEprop 命中物经过随机抽样展示。
2.4 超大规模抗生素虚拟筛选显著提升命中率与化学多样性
在利用HTS数据回顾性评估了 GNEprop 模型性能后,研究者将其应用于实际的超大规模虚拟筛选任务(图4a)。模型在约200万个分子的完整HTS数据集上进行训练,并采用95:5比例划分训练与验证数据,结合八次重采样以提高稳定性。
虚拟筛选获得的大量候选分子经过多步后处理,以减少数量、提升多样性并覆盖不同化学假设(方法部分)。具体而言,首先将筛选得到的分子聚类为984个簇,并在每个簇中选择具有最高GNEprop得分的代表性分子。随后根据实验预算进一步筛选,最终选出503个候选化合物,其中49%的结构与训练集中活性化合物的最大相似度(Tanimoto相似度)≤0.4。经过合成可行性评估后,共有345种化合物被成功合成并进行体内实验验证。
对这些化合物在E. coli ΔtolC 菌株上的单点生长抑制实验结果显示,共有82种化合物表现出明显抗菌活性(生长抑制率≥80%),命中率达 23.8%(图4c与补充表6)。相比最初HTS实验的 0.26% 命中率,基于深度学习的虚拟筛选实现了 约90倍的提升。尽管两次筛选的实验设计存在差异,这一结果仍代表着显著的实际改进与应用潜力。
进一步分析发现,预测得分与实际抑制效果之间呈弱正相关(Spearman ρ = 0.22,P = 2.4 × 10⁻⁵),表明模型在仅以二元标签训练的条件下,仍部分学到了反映抑制强度的特征。尤其是高分预测区(得分>0.9)的分子富集了大量活性化合物,命中率高达 36%(图4d)。
当筛选分子与训练集中活性化合物的相似度较低时,模型性能虽有所下降,但仍保持显著效果(图4e)。例如,在最大相似度≤0.4的子集(共170个分子)中,命中率为 14.1%,虽低于总体23.8%的水平,但仍远高于HTS的0.26%。总体来看,约 29.3%(24个) 的验证命中分子与HTS活性分子的最大相似度低于0.4(最低为0.22)。
与已知抗生素相比,大多数新发现的化合物在结构上具有明显差异——其中 98.8% 的最大相似度≤0.4,且 39% 的相似度≤0.2。值得注意的是,命中率与与已知抗生素的相似度之间并无显著相关(图4f),这可能得益于HTS数据集本身已覆盖了更广的化学空间。
综上所述:
- GNEprop 模型在低相似度化合物上仍保持高命中率;
- 大多数命中分子在化学结构上与现有抗生素完全不同。
这一发现通过UMAP低维嵌入可视化进一步得到验证(图4g),其中的典型示例展示了模型如何在新的化学空间中成功识别出抗菌候选分子,为发现具有全新骨架与作用机制的抗生素提供了有力工具。
2.5 已鉴定化合物的生物学表征验证其抗菌活性
为进一步确认并解析所发现化合物的抗菌作用,研究者进行了多层次的生物学实验,包括活性验证、菌株谱测试以及靶标识别。
首先,从所有预测命中物中筛选出 165种化合物 进行初步剂量–反应(dose–response)分析以测定其半数抑制浓度(IC₅₀)(图5a,补充表6)。该集合不仅包括在E. coli ΔtolC中抑制率超过80%的活性化合物(138种),还包括部分抑制率在50–80%之间的分子,以捕获潜在弱活性但有优化潜力的候选物。结果显示,IC₅₀范围从 <1 μM到>40 μM 不等,其中95%的化合物(157种)在E. coli ΔtolC上的IC₅₀低于20 μM(图5b)。
此外,研究者还测定了这些化合物在野生型E. coli MG1655与**金黄色葡萄球菌(S. aureus USA300)**上的IC₅₀,分别处于 1.3–>40 μM 与 0.31–>40 μM 范围(扩展数据图5)。其中,有39种化合物(约23.6%)在野生型E. coli上IC₅₀低于40 μM,表明尽管模型仅基于E. coli ΔtolC数据训练,一部分命中物仍对野生型菌株表现出活性。
为排除广谱裂解性分子,研究者在**酵母(S. cerevisiae)**中进行对照测试,仅6种化合物表现出非特异性毒性并被排除(补充表7)。
接着,研究者采用最低抑菌浓度(MIC)测试,这一方法可更严格评估化合物的抗生素特性。从剂量–反应数据中筛选出26种候选化合物,标准包括:
1️⃣ E. coli ΔtolC IC₅₀ < 5 μM 且对S. aureus具≥3倍选择性(倾向于革兰氏阴性特异性);
2️⃣ E. coli ΔtolC上最高活性(IC₅₀ < 0.35 μM);
3️⃣ 对野生型E. coli 亦具抑制作用(IC₅₀ < 10 μM)。
结果显示,22种化合物 在E. coli ΔtolC上的MIC范围为 0.3–40 μM(图5c,补充表8),说明通过GNEprop预测可快速发现具全细胞活性的候选分子。
然而,这些化合物中无一对具有完整外膜的野生型菌株表现出MIC值,提示仅使用E. coli ΔtolC数据训练的模型存在一定局限,后续需结合**邻近结构筛选(nearest-neighbor screening)**等优化步骤。
为考察抗菌谱,研究者在S. aureus上测试同一批26种化合物的MIC,发现其中15种(58%)同时对E. coli ΔtolC与S. aureus活性(补充表8),表现出潜在的广谱抗菌性。红细胞溶血实验(扩展数据图6)与人肺上皮细胞(A549)通透性实验(扩展数据图7)均未显示毒性,排除了非特异性膜破坏机制。
接着,研究者聚焦于革兰氏阴性菌选择性化合物以确定其分子靶标。两种候选物——N9777 与 N9786——被选中进行深入分析。它们在E. coli ΔtolC中的MIC分别为 0.313 μM 与 10 μM,对S. aureus的MIC均 >40 μM,且在红细胞及膜通透性实验中均无裂解作用(图5d,补充表8)。虽然N9777在野生型E. coli中无MIC值,但显示出可检测的生长抑制(IC₅₀ = 16 μM)。
为确定作用靶点,研究者在E. coli ΔtolC中筛选N9777与N9786的自发抗性突变株(图5e)。
- N9777: 获得两个抗性克隆,MIC较亲本株提升16–128倍。全基因组测序发现它们分别携带 lpxH 基因 的不同突变,该基因编码脂多糖(LPS)生物合成途径的关键酶。进一步分析表明,这些突变位点对应于此前报道的K. pneumoniae中AZ抑制剂结合口袋(图5f,补充表9)。
- N9786: 获得8个抗性克隆,MIC提升4–8倍,测序揭示6个独立的 fabZ 突变与1个 acpP 突变。两者分别编码脂肪酸生物合成中的关键酶——去水酶FabZ与载体蛋白AcpP。虽然脂肪酸合成并非革兰阴性特有,但它是LPS合成的前体步骤,对E. coli至关重要。
综上,这些结果表明,通过GNEprop虚拟筛选发现的抗菌化合物不仅具有革兰氏阴性特异性,还能指向已验证的功能靶标(如LpxH、FabZ),显示出在发现新型抗生素方面的强大潜力。
2.6 邻近结构探索(Nearest-neighbor exploration)揭示更多活性化合物,包括对野生型菌株具MIC的命中物
鉴于 GNEprop 筛选得到的命中化合物具有高度多样性,涵盖不同的骨架与筛选标准,研究者进一步将其作为锚点分子(anchors),通过邻近结构搜索(nearest-neighbor analysis)扩展虚拟筛选空间,以探索并优化其活性谱。
具体而言,研究者在 44,437 个虚拟命中物 中,以两组有前景的锚点为中心进行搜索(方法)。
- 集合1(Set 1) 包含7个对ΔtolC株选择性较高的化合物;
- 集合2(Set 2) 包含6个具有广谱活性的化合物。
最终共获得 67个候选分子 进行合成验证(补充表10、图5g、扩展数据图8)。
集合1的锚点分子分属4种分子骨架,其结构普遍包含两个通过酰胺(amide)、脲(urea)、**酰基磺酰胺(acyl sulfonamide)或硫醚(thioether)**键连接的构件。67个邻近结构中,来自集合1的38个化合物均表现出可测得的 E. coli ΔtolC IC₅₀,其中 66% 的化合物活性与锚点相当或更优。值得注意的是,3个化合物的MIC提升超过 4倍,其中既包括革兰氏阴性选择性分子,也包括广谱型分子。
集合2的锚点分子普遍具有**硝基噻吩(nitro-thiophene)**结构模体。来自集合2的29种化合物中,全部对E. coli ΔtolC展现可测IC₅₀值,其中 93% 的化合物活性保持或提升,并且有相当比例的分子在E. coli ΔtolC与S. aureus上均实现≥2倍的IC₅₀提升(分别为31%与24%)。
尤为重要的是,尽管集合2的锚点分子均未在野生型E. coli中显示MIC活性(均>40 μM),但其邻近结构中出现了3个具野生型MIC值的化合物,其中两者MIC分别为 10 μM 与 20 μM(补充表10、图5g示例)。这些分子在红细胞裂解实验中亦未显示溶血活性(扩展数据图9)。
总体而言,GNEprop引导的邻近结构扩展 证明是一种高效的化合物挖掘策略,可在有限实验资源下进一步提升虚拟筛选命中率与化学多样性。约 15% 的验证化合物在E. coli ΔtolC中的MIC提升超过两倍,并首次发现了具野生型E. coli MIC的分子——这在训练集仅包含单点ΔtolC抑制数据的前提下尤为显著。
这表明,即便初始锚点分子活性有限,它们仍可作为GNEprop驱动的探索节点,通过邻近结构搜索发现具改进和多样化活性的候选物,同时生成更丰富的构效关系(SAR)数据,为后续模型再训练与抗菌剂优化奠定基础。
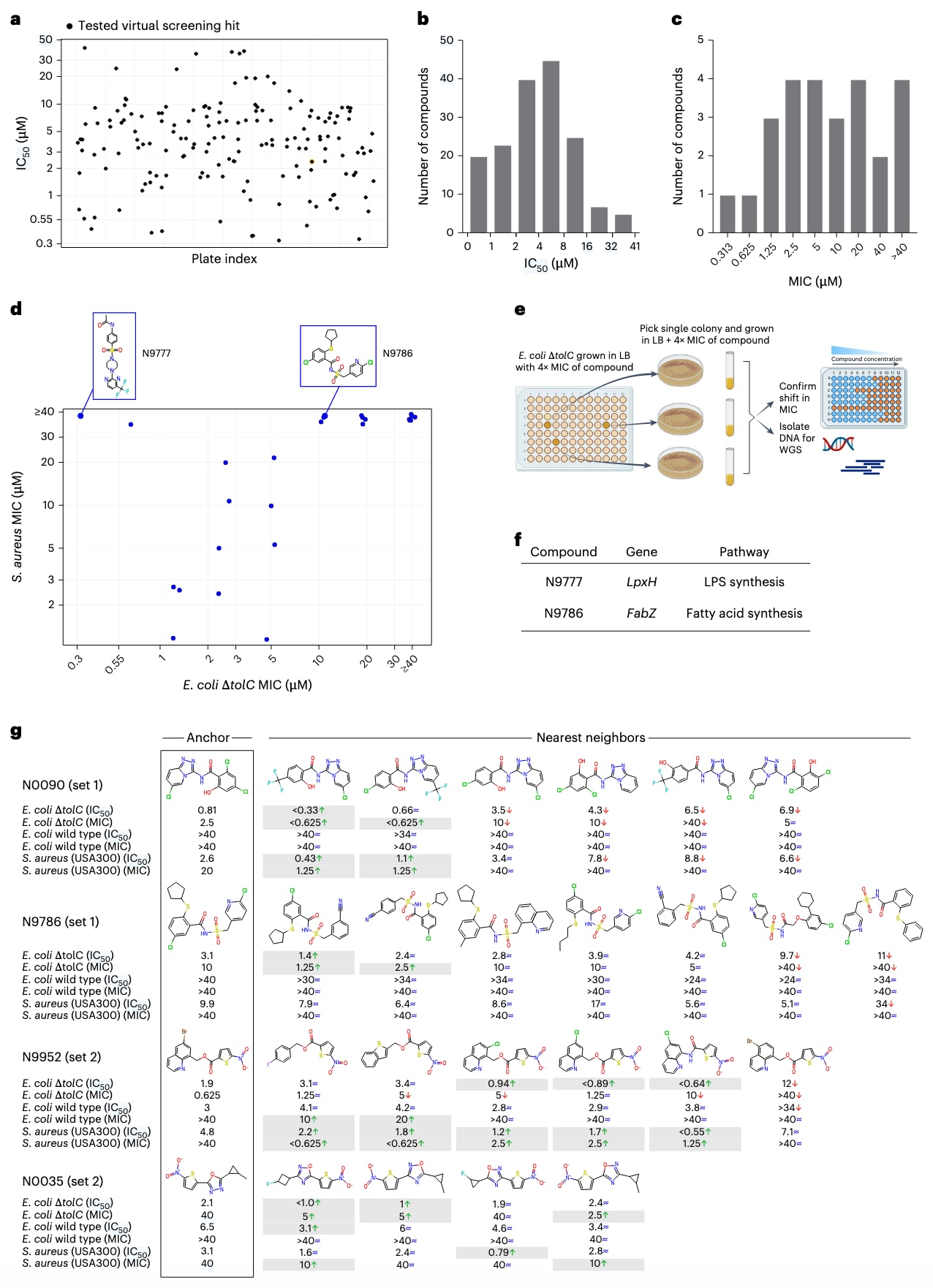
图5|虚拟筛选命中化合物的生物学表征验证其抗菌活性。 (a) 剂量–反应实验(dose–response)IC₅₀ 测定。 展示了针对 E. coli ΔtolC 的化合物在剂量–反应实验中的 IC₅₀ 分布(横轴为化合物编号),显示筛选流程的早期阶段。 (b) IC₅₀ 分布。 所有测试化合物在 E. coli ΔtolC 中的 IC₅₀ 值分布。大部分活性化合物的 IC₅₀ 低于 20 μM。 (c) 最低抑菌浓度(MIC)分布。 展示了对 E. coli ΔtolC 测得的 MIC 值分布范围(0.3–40 μM)。 (d) 不同菌株的活性谱。 将选定化合物在 E. coli ΔtolC(x 轴)与 S. aureus(y 轴)中的 MIC 值进行对比。用于靶标鉴定的代表性化合物以蓝色矩形标出。 (e) 抗性突变株筛选示意图。 展示了通过药物暴露筛选出自发获得抗性的菌株流程示意。 (f) 抗性株全基因组测序结果汇总。 图中总结了对抗性克隆测序后鉴定出的关键突变基因。结果显示:化合物 N9777 的抗性克隆中出现 lpxH 基因突变(脂多糖合成关键酶);化合物 N9786 的抗性克隆中出现 fabZ 与 acpP 突变(脂肪酸合成通路相关酶)。 这些结果揭示了化合物作用于革兰氏阴性菌的特异代谢靶点。 (g) 基于邻近结构的虚拟筛选扩展。 以 GNEprop 预测得分高的“锚点化合物”为中心(首列示例),在模型优先区域内进行局部邻域探索,从而选择额外候选分子(其余列)。每个化合物下方标注了在 E. coli ΔtolC、野生型 E. coli 与 S. aureus 中的 IC₅₀ 与 MIC 值。符号表示与锚点化合物相比的变化:↑ 表示提升超过两倍,↓ 表示降低超过两倍,≈ 表示差异在两倍以内。前两个示例展示了集合1(Set 1)中对 E. coli ΔtolC 活性(IC₅₀ 与 MIC)显著改善的化合物(如 N0090、N9786);第三个示例展示了集合2(Set 2)中两种化合物在野生型 E. coli 上 MIC 改善;第四个示例展示了集合2中在 E. coli ΔtolC 上活性显著增强的化合物。
2.7 改进革兰氏阴性菌细胞包膜屏障与作用机制(MoA)表征的探索
该研究的虚拟筛选策略主要聚焦于早期命中化合物的发现阶段,成功获得了一系列结构多样、骨架新颖的抗菌候选分子。然而,该流程本身并不能直接揭示化合物的分子靶标或作用机制(Mechanisms of Action, MoA),这需要进一步的实验表征。此外,研究者所用的高通量筛选(HTS)基于 E. coli ΔtolC 突变株——该菌株因外排通道缺陷而对毒性分子敏感。虽然此设计增加了可用于训练与验证 GNEprop 的抗菌样本数量,但其局限在于结果未必适用于外膜完整的野生型菌株。
为此,研究者在研究末期开展了两项探索性扩展工作:(1)尝试直接对细胞通透性障碍进行建模;(2)探索如何在早期识别命中物的潜在新型作用机制。尽管这些分析超出了主研究范围,但它们为未来方向奠定了基础。
一、细胞包膜通透性(permeability)的建模探索
为验证模型区分穿透性差异的能力,研究者重新筛选了先前HTS中已鉴定的5,161个ΔtolC活性分子中的3,074个(取决于化合物可得性),并在野生型E. coli 上再次测试。结果获得:
- 306种“双重活性(dual-active)”化合物,即同时对ΔtolC与野生型菌株活性,标记为“cell-penetrating(可穿透)”;
- 2,768种仅在ΔtolC中活性的化合物,标记为“inhibiting(抑制型)”。
虽然该数据集在规模与多样性上不及主HTS集,但研究者仍将其用于测试 GNEprop 区分穿透性差异的潜力。将模型扩展为**三分类任务(非活性 / 抑制型 / 可穿透型)**后,GNEprop 仍保持了良好的整体预测性能(图6a),但对“可穿透”化合物的分类准确率约为50%,反映出样本稀疏与化学空间重叠的影响(扩展数据图10)。
进一步地,研究者利用 模型可解释性管线(explainability pipeline)区分“inhibiting”与“cell-penetrating”分子(图6b)。分析显示,大约25%的正确预测“可穿透”分子在结构上与训练集中的“抑制型”分子最相似,提示存在与通透性相关的结构悬崖(structural cliffs)(图6c)。模型热图分析识别出多种被认为与穿膜能力相关的关键子结构(图6d)。例如,伯胺基(primary amine)**被模型标记为促进穿透性的关键特征,这与先前研究报道的革兰阴性菌药物积累规律一致。虽然目前数据仍不足以归纳通用性规则,但这一方法展示了未来通过深度学习实现**大规模细胞通透性定量建模的潜力。
二、基于模型嵌入的作用机制(MoA)预测探索
研究者进一步探索了 GNEprop 在区分潜在不同作用机制化合物方面的可行性。受近期 深度学习异常检测(out-of-distribution, OOD detection) 研究启发,研究者构建了一条可扩展流程,用于判断某一化合物是否可能具有区别于已知抗生素的作用机制。
基于自监督学习得到的分子表示,研究者观察到已知抗生素可在嵌入空间中自然聚类为不同的靶标区域(图6e,补充表11)。据此,研究者提出:若某一抗菌化合物在嵌入空间中距离所有已知靶标簇较远(即“outlier”),则其更可能具有新型作用机制。
为量化这一“距离”,研究者在每个已知靶标簇上拟合条件高斯分布,并基于 Mahalanobis 距离 计算OOD分数(图6f)。尽管仅使用了不到200个带注释的样本进行回顾性验证,该方法仍能在多数情况下优先排序出具备独特机制的化合物。GNEprop的自监督表示在这一任务上优于或接近等规模的分子指纹(fingerprints),能区分不同靶标的得分分布(图6g)。
需要指出的是,大维度指纹特征在某些场景下表现相似甚至略优,这说明现有分子嵌入在MoA区分上的表达能力仍有限。然而,模型嵌入的优势在于其为GPU可加速的稠密向量表示,具备进一步针对该任务微调与扩展的潜力。
综上,这些探索性研究展示了利用深度学习方法同时表征细胞包膜通透性与潜在作用机制的可行性。随着更丰富的标注数据和更强的分子表示学习技术的加入,这一方向有望实现针对革兰氏阴性细菌的机制导向型虚拟筛选,为未来抗生素发现开辟新路径。
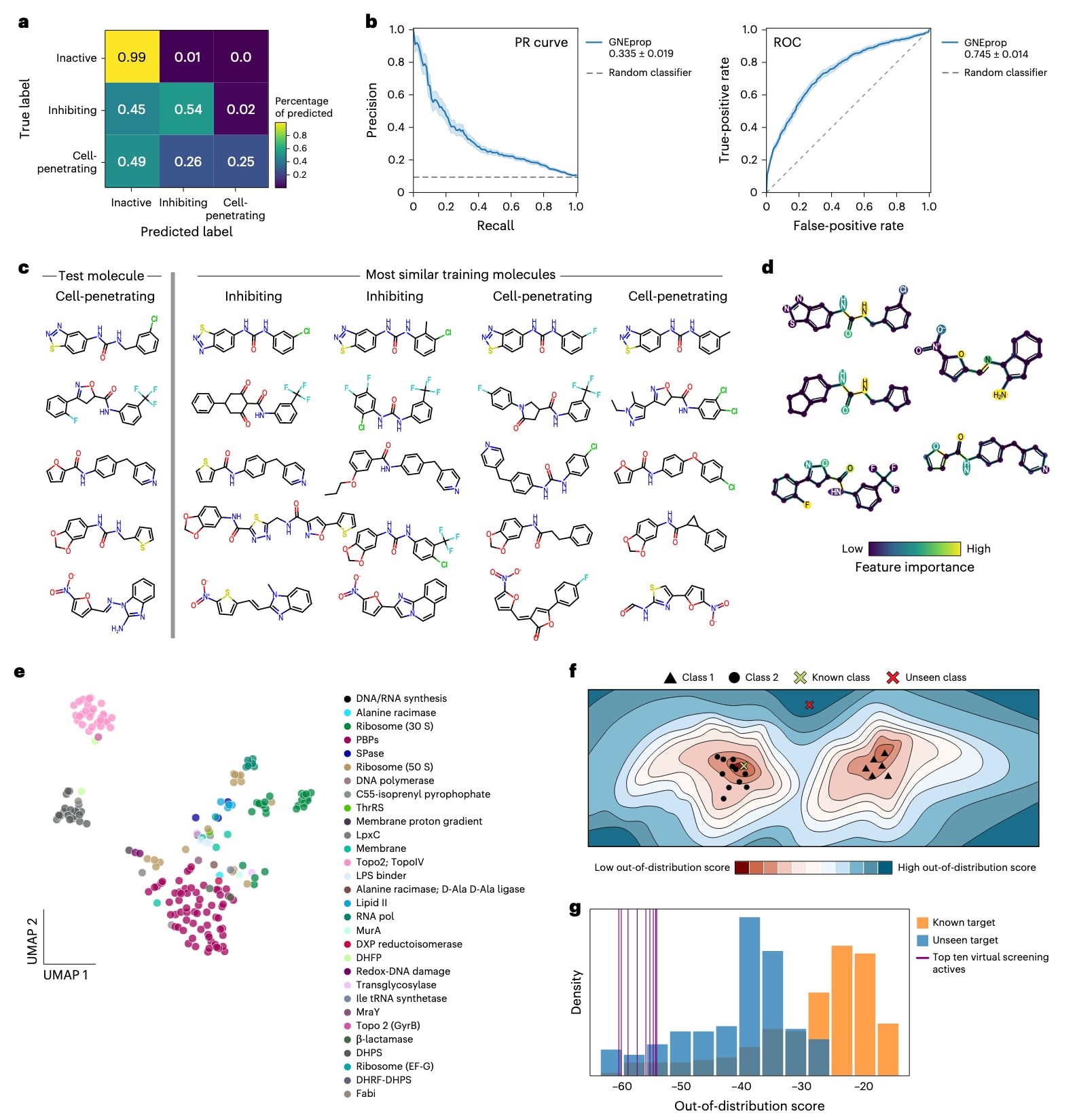
图6|利用 GNEprop 探索革兰氏阴性菌的细胞包膜通透性与作用机制(MoA)特征。 (a–d) 基于双重筛选(double-assayed HTS)的 GNEprop 模型揭示细胞包膜屏障特征。 (a) 混淆矩阵。 模型区分三类化合物:非活性(inactive)、抑制型(inhibiting,仅对 E. coli ΔtolC 有活性)及可穿透型(cell-penetrating,同时对 E. coli ΔtolC 与野生型 E. coli 活性)。 (b) 模型分类性能。 展示 GNEprop 区分“可穿透型”与“抑制型”化合物的 ROC 曲线,图例中的数值表示曲线下面积(AUC),阴影区域为基于20次骨架划分的标准误。 (c) 通透性活性悬崖(activity cliffs in permeability)。 对五种代表性可穿透化合物(左列),展示其最相似的两种“抑制型”和“可穿透型”结构对比,揭示细微结构差异如何影响通透性。 (d) 化学特征重要性可视化。 将模型预测的关键化学特征(原子与键的重要性)叠加于(c)中选定的可穿透化合物结构上。模型识别出的高重要性区域(颜色高亮)与已知促进通透的结构基团(如伯胺)一致。(e–g) 探索性分析:早期识别潜在新型作用机制(MoA)。 (e) 低维表示空间中的抗生素聚类。 基于 GNEprop 自监督表示的二维嵌入,已知抗生素按靶点着色,显示不同靶标在嵌入空间中形成独立簇。 (f) 原理示意图。 在模型嵌入空间中,具新型(未见过)作用机制的分子(红色 X)远离已知靶标簇,而已有作用机制的分子(绿色 X)靠近相应簇。 (g) 分布分析与 OOD(分布外)得分。 展示通过 Mahalanobis 距离计算的 OOD 得分分布。较低得分代表结构与已知靶标差异更大。灰色与蓝色曲线分别代表已知靶标与未见靶标的参考分布,紫色标记显示虚拟筛选中得分最低的前10个候选分子,这些分子最可能具有全新作用机制。
3 讨论
该研究结合了两种优势:一方面是由近 200万种小分子 组成的多样化高通量筛选(HTS)库,另一方面是基于深度学习的虚拟筛选策略,其虚拟化合物库规模超过 14亿个化合物,从而在大尺度上探索了化学空间。通过这种方法,研究者显著提高了命中率,并增强了所鉴定化合物的结构多样性。
模型性能分析表明:训练数据的规模与多样性对模型在分布外(OOD)任务上的表现至关重要。值得注意的是,即便使用了全部训练数据,GNEprop 的性能仍未出现饱和,说明模型可从更大量的数据中继续受益。进一步实验显示,当使用仅 10% 的数据重新训练模型时,仍能恢复约 90% 的原始性能,这表明一种基于主动学习(active learning) 的策略——让深度学习模型基于不确定性引导实验室迭代采样与筛选——可能极具潜力。
在该研究中,研究者还为 GNEprop 配备了可解释性分析管线(explainability pipeline),证明该方法能够揭示化学结构中的微小差异如何导致活性显著变化,包括细胞膜通透性等关键性质。虽然该研究仅将此用于验证模型预测,但类似技术未来有望帮助研究者发现支配不同菌株活性的一般规律,例如通过全局可解释性(global explainability) 方法。该领域的一大挑战在于:现有化合物库规模有限且缺乏多样性,难以提取真正因果性(causal) 的结构特征。未来,结合因果表征学习(causal representation learning) 的方法,有望从结构属性中提取出与活性(如通透性、外排效应)具有因果关联的特征。
研究者的深度学习策略建立在近年来快速发展的分子嵌入(molecular embeddings) 技术基础上,因此无论是无监督还是有监督学习的进一步进步,都将直接推动这一流程的性能提升。特别是在作用机制(MoA) 的解析方面,更具表达力的分子表示能够同时捕捉结构属性与生物效应信息,从而提高对不同作用机制的分辨能力。
该研究的一项局限性在于其目标菌株为 E. coli ΔtolC。该菌株缺乏多个外排系统共用的外膜通道,对多种抗生素更为敏感。虽然这一选择使研究者能在化学空间中更远离已知抗生素与天然产物,并获得更多用于模型训练的活性分子,但也导致了在野生型菌株中的命中率显著降低。具体而言,在约 200万种化合物 的HTS中,只有 0.26% 对 E. coli ΔtolC 表现活性;进一步在野生型 E. coli 上的再筛选中,仅约 10% 的 ΔtolC 活性化合物保留活性,相当于原始筛选中仅 0.03% 的野生型命中率。这一比例远低于 Stokes 等人基于野生型 E. coli 筛选得到的结果。研究者认为这种差异主要源自该研究化合物库的更高多样性与新颖性。
尽管该研究主要聚焦于 E. coli ΔtolC 的活性预测,但该流程可轻松扩展至其他菌株或生物活性任务。例如,在数据可用的情况下,可通过多任务学习(multitask learning) 模型实现跨菌种建模。研究者的框架主要以结构多样性作为生化多样性的代理指标,假设结构差异大的化合物更可能具有不同的作用机制。这一假设在多数情况下成立,但 MoA 的表征仍相对粗略,未来可通过引入高通量、高内容(high-content)数据模态进一步完善,以揭示小分子的多层次功能效应。
此外,该研究仅关注小分子药物发现。然而,寻找新型抗菌化合物类别的工作亦可利用新兴治疗分子类型(如抗菌肽)或其他药物形式,这些方向近来也显示出令人鼓舞的前景。未来,借助人工智能(AI)整合跨模态、多类型治疗剂与多样化实验读出(readouts)的数据,将成为一项充满挑战但极具潜力的研究方向。