Patterns 2025 | LigUnity: 通过学习共享的口袋-配体空间实现分层亲和力景观导航
今天介绍的是发表于《Patterns》2025年的一项研究——“LigUnity:通过学习共享的口袋–配体空间实现分层亲和力景观导航”。药物发现的关键在于精准预测小分子与蛋白靶点之间的结合亲和力,但传统方法常受限于计算成本高、实验数据稀缺及任务割裂等问题。尽管已有数千种人类蛋白,但目前仅约一成能被高效小分子靶向,而多数机器学习方法仍将活性预测简化为“有活性”或“无活性”的二元分类,无法捕捉分子结构差异对亲和力的细微影响。
为此,研究团队提出了LigUnity——一种可同时服务于虚拟筛选(Virtual Screening)与从命中到先导优化(Hit-to-Lead Optimization)的统一基础模型。该模型通过骨架区分(Scaffold Discrimination)与药效团排序(Pharmacophore Ranking)学习蛋白结合口袋与小分子之间的共享表示,从而兼顾广泛化学空间探索与精确亲和力预测。
LigUnity在多个基准上超越现有24种方法,在精度与效率上均表现突出,并能在主动学习框架中高效筛选出最优配体,为计算驱动的药物设计提供了新范式,也展现出成为通用亲和力预测基础模型的巨大潜力。

获取详情及资源:
0 摘要
蛋白质结合口袋的结构通过提供关键的分子间相互作用与空间匹配性,决定了配体的结合亲和力。虽然现有方法已利用这些结构信息推动亲和力预测的发展,但 虚拟筛选与从命中到先导化合物优化(hit-to-lead optimization) 常被分开处理,主要是因为二者在速度与精度上的需求不兼容。然而,这两类任务本质上是互补的,它们的整合能够在保持对决定亲和力关键亚结构关注的同时, 实现更广泛的化学空间探索。
该研究提出了配体统一亲和力模型(LigUnity, ligand unified affinity),这是一种用于亲和力预测的基础模型,能够 将配体与蛋白口袋共同嵌入到一个共享表示空间中。具体而言,LigUnity通过 骨架区分(scaffold discrimination) 学习粗粒度的活性/非活性分类,同时通过 药效团排序(pharmacophore ranking) 捕捉精细的口袋特异性配体偏好。
在八个基准数据集、六种不同任务设置的验证中,LigUnity展现出 显著的准确性与通用性。在虚拟筛选任务中,LigUnity较24种现有方法取得了 超过50%的性能提升,并在新靶点上表现出强大的泛化能力;在hit-to-lead优化中,它在 时间划分、骨架划分与单元划分 等多种设置下均达到了 当前最优性能,成为相较于自由能微扰(free energy perturbation, FEP)更具性价比的替代方案。此外,研究还展示了LigUnity在 酪氨酸激酶2(TYK2) 活性学习框架中的应用,可高效地筛选出最优配体。
总体而言,LigUnity被确立为一种通用且高效的亲和力预测基础模型,在药物发现流程的多个环节中展现出广泛的适用潜力。
1 引言
蛋白质结合口袋(protein binding pocket)是一种具有明确空间结构的配体结合腔体,它通过决定与配体之间的非共价相互作用与立体化学互补性,成为计算机辅助药物设计(CADD)的结构基础。这一结构特征使得能够更准确地预测蛋白质-配体结合亲和力,而结合亲和力又是药物效力与靶点选择性的关键决定因素。
在基于靶点的药物发现流程中,准确预测结合亲和力在两个连续阶段中起着至关重要的作用:其一是高通量虚拟筛选(high-throughput virtual screening),旨在从大规模化学库中发现能够与目标蛋白结合的活性化合物;其二是从命中到先导化合物优化(hit-to-lead optimization),目标是进一步优化这些活性配体的结构,以提高其结合亲和力与类药性质。
在虚拟筛选中,分子对接(docking)方法被广泛应用并取得了一些成功,但它仍受限于化学搜索空间大小与计算成本之间的权衡。而在hit-to-lead优化中,基于物理的计算方法——如自由能微扰(FEP)与分子力学广义Born表面积法(MM/GBSA)——虽然表现出一定潜力,但往往与实验亲和力相关性较弱或需要大量计算资源进行采样。
为应对这些挑战,近年来机器学习(ML)方法被引入虚拟筛选(如DrugCLIP)与hit-to-lead优化(如ActFound与PBCNet)中。这些方法在保持与传统物理计算相当精度的同时,显著提升了计算效率,使其成为大规模应用的高效替代方案。然而,大多数现有ML模型仍将这两个任务分开研究。事实上,二者是相互依赖、互为补充的:
若模型仅关注hit-to-lead优化,则容易受限于局部化学空间,难以推广至新颖骨架的配体;反之,若模型仅用于虚拟筛选,则可能忽视决定蛋白-配体相互作用的关键亚结构,从而难以区分具有微小结构差异的分子。
一些近期研究(如GenScore、PIGNet2、IGModel与EquiScore)尝试通过再对接、交叉对接与虚假分子生成等数据增强手段弥合两者间的差距,但它们仍面临一个核心问题——缺乏结构相似的活性配体对,而这对学习亚结构变化如何影响结合亲和力至关重要。基于以上观察,研究者提出一种假设:若能建立一个同时处理虚拟筛选与hit-to-lead优化的统一基础模型(foundation model),即可利用两者间的协同效应提升整体性能。
因此,该研究提出了LigUnity(ligand unified affinity)——一种用于蛋白-配体亲和力预测的基础模型。LigUnity的核心创新在于同时实现广泛筛选与精确亲和力预测的集成能力,这一目标通过骨架区分(scaffold discrimination)与药效团排序(pharmacophore ranking)的结合实现。模型将配体与蛋白口袋共同嵌入到一个共享结构-化学空间,以捕捉二者之间的互补性。
在骨架区分阶段,LigUnity学习区分活性与非活性配体,从而揭示跨骨架的全局结构-活性关系;在药效团排序阶段,模型进一步在共享空间中学习同一口袋下活性配体的相对排序,刻画微小结构差异对结合亲和力的影响。借助这种联合嵌入表示,LigUnity能够快速筛选大规模虚拟化学库并高效识别活性配体,兼具虚拟筛选与hit-to-lead优化的实用性。
然而,要构建一个同时适用于这两类任务的结构感知(structure-aware)数据集仍具有挑战。现有大型亲和力数据库(如BindingDB与ChEMBL)虽包含丰富结合数据,但缺乏能表征结合口袋的结构信息。为此,研究团队构建了PocketAffDB——一个包含约80万条亲和力数据、50万个独特配体与53,406个口袋结构的综合结构感知结合实验数据库。PocketAffDB以实验测定(assay)为组织单位,同一实验下的亲和力数据采用一致方法测定,具有可比性。基于此,研究者提出了一个基于实验指导的口袋匹配算法(assay-guided pocket matching),通过假设大多数实验针对特定结合位点设计,为每个蛋白-配体对分配合理的口袋结构。PocketAffDB因此成为迄今最大规模的融合生物实验数据与结构信息的亲和力数据库,为模型训练提供了宝贵资源。
为了验证LigUnity的有效性,研究在8个基准任务与6种不同实验设置中进行了系统评估。在三个虚拟筛选数据集(DUD-E、Dekois与LIT-PCBA)上,LigUnity均超越了全部24种现有方法;在未见过的新蛋白靶点上也保持了稳健提升,显示出优异的泛化能力。在hit-to-lead优化任务中,LigUnity在两个FEP基准上实现了**零样本与少样本(zero-shot/few-shot)**下的最优性能,表明其有望成为昂贵FEP计算的高效替代方案。此外,在ChEMBL与BindingDB上的时间划分、骨架划分与单元划分测试中,LigUnity再次优于八种比较方法。
更进一步地,在模拟多轮药物优化过程的主动学习框架中,LigUnity仅通过少数迭代便成功识别出具有最优亲和力的配体,体现出其在降低分子优化实验成本方面的潜在价值。
综上所述,LigUnity作为一种面向虚拟筛选与hit-to-lead优化的统一基础模型,在药物发现流程的多个环节展现出广泛的适用性与高效性。
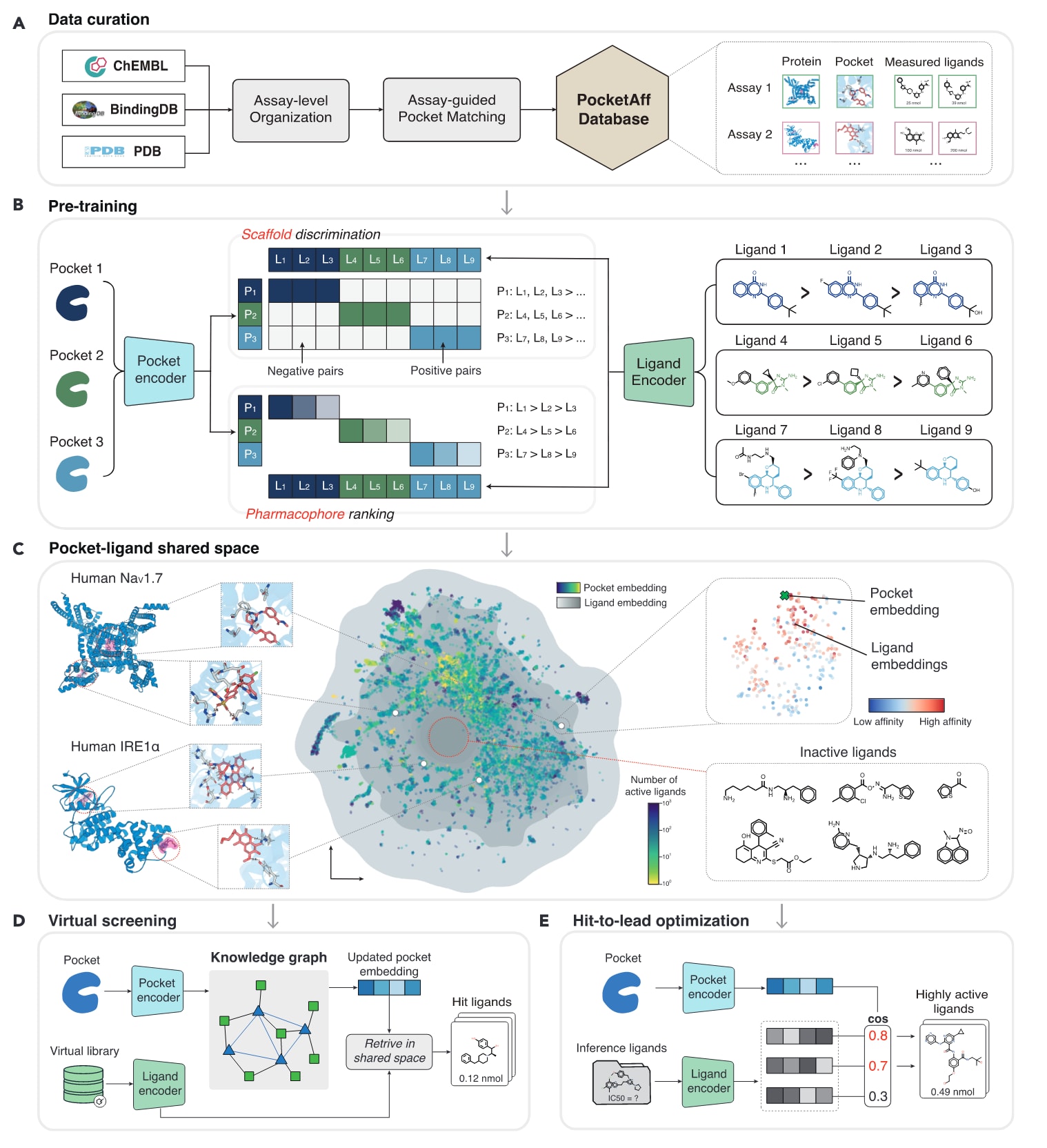
图1|LigUnity整体框架概述 (A) PocketAffDB的数据整理流程。 (B) LigUnity通过分层亲和力景观导航(hierarchical affinity landscape navigation)对口袋与配体编码器进行预训练。模型首先将蛋白口袋与配体映射到共享嵌入空间,然后利用骨架区分(scaffold discrimination)捕捉活性/非活性配体的粗粒度特征差异,并通过药效团排序(pharmacophore ranking)进一步对嵌入空间进行精细化调整,使其能够对齐亲和力的微小差异。 (C) LigUnity生成的口袋与配体嵌入表示的UMAP可视化图,展示模型学习到的分层结构模式。 (D) 在虚拟筛选(virtual screening)任务中,LigUnity首先通过图神经网络(graph neural network)对查询口袋的嵌入表示进行优化,随后利用更新后的口袋嵌入在共享口袋–配体空间中检索潜在命中配体。 (E) 在先导化合物优化(lead optimization)任务中,LigUnity直接计算口袋与配体嵌入之间的余弦相似度(cosine similarity),对未测定配体进行排序,从中筛选出高活性候选配体。
2 结果
2.1 LigUnity 概述
LigUnity是一种用于虚拟筛选(virtual screening)与从命中到先导化合物优化(hit-to-lead optimization)的蛋白–配体亲和力基础模型,利用三维结合口袋结构预测蛋白–配体的结合亲和力。为训练该模型,研究团队构建了一个结构感知的综合数据库——PocketAffDB,该数据库由大型实验亲和力数据库(BindingDB与ChEMBL)及蛋白质结构数据库PDB整理而成(见图1A)。
研究首先将亲和力数据按实验测定(assay)进行收集,并利用基于实验指导的口袋匹配方法(assay-guided pocket matching),为每个蛋白–配体对分配相应的结合口袋结构(方法详见原文)。据目前所知,PocketAffDB是迄今为止最大规模的结合生物实验数据与结构口袋信息的整合亲和力数据库,共包含约80万条亲和力数据,覆盖50万个独特配体、53,406个口袋与26,748个实验测定。
在预训练阶段,LigUnity通过学习口袋–配体共享空间(pocket-ligand shared space),捕捉两者间的结构与化学互补性。该过程通过**骨架区分(scaffold discrimination)与药效团排序(pharmacophore ranking)**两个模块的集成框架实现(图1B)。
- 骨架区分模块侧重于化学骨架的结构差异,学习区分活性与非活性化合物,从而在嵌入空间中让正确的口袋–配体对相互吸引,而让结构差异较大的负样本对相互排斥。
- 药效团排序模块进一步在嵌入空间中进行精细对齐,关注配体药效团层面的差异,通过比较口袋与配体嵌入间的相似度排序,预测其相对结合亲和力。
这两个模块在预训练阶段协同工作、互为补充,共同塑造LigUnity的高效嵌入表示空间。
经过预训练后,LigUnity的嵌入空间呈现出有意义的分层结构模式(hierarchical patterns)(图1C):
- 在粗粒度层面,结合同一口袋的配体聚集成明显簇群,而结合同一蛋白不同口袋的配体则相互分离,说明LigUnity的嵌入表示能够捕捉结合位点层面的差异信息,展现出用于虚拟筛选的潜力;
- 在精细层面,嵌入空间中具有更高结合亲和力的配体更接近其靶口袋,表明LigUnity能够学习到影响亲和力的药效团级微结构差异,具备执行hit-to-lead优化的能力。
在推理阶段(inference),LigUnity针对不同任务分别进行调整,以充分利用特定任务数据:
- 对于虚拟筛选,LigUnity采用基于图的策略来整合已有数据库中的大规模蛋白–配体相互作用信息(图1D)。具体而言,首先构建一个大型的知识图谱(knowledge pocket-ligand graph),其中包含约83万条口袋–配体边(已知相互作用)与1600万条基于结构相似性的口袋–口袋边。随后,利用**图神经网络(GNN)**聚合邻域信息,优化查询口袋的嵌入表示。
- 对于hit-to-lead优化,LigUnity可直接通过**计算口袋与配体嵌入间的余弦相似度(cosine similarity)来预测未测量配体的结合亲和力(图1E)。若实验中已有少量测得配体数据,还可进一步进行小样本微调(fine-tuning)**以提升预测精度。
通过在嵌入空间中建模亲和力景观(affinity landscape),LigUnity在推理阶段显著降低计算成本,实现了相较传统分子对接方法高达六个数量级的加速,从而在保持准确性的同时,大幅提升了药物筛选与优化的效率。

图2|虚拟筛选性能评估 (A, B) LigUnity与其他对照方法在DUD-E与Dekois-2.0基准上的表现对比,指标为富集因子(Enrichment Factor, EF)1%。白色圆点表示平均值,分别基于n = 102(DUD-E)与n = 81(Dekois-2.0)个靶点计算。 (C, D) LigUnity与无结构方法(structure-free methods)在不同训练蛋白集下的表现对比,同样以EF 1%为指标。横轴表示训练集与测试集之间的最大序列相似度。白色圆点为平均值,分别基于n = 102(DUD-E)与n = 81(Dekois-2.0)个靶点计算。 (E, F) LigUnity与其他对照方法在LIT-PCBA基准上的性能对比,评估指标包括EF 1%与Boltzmann增强型ROC区分度(BEDROC, α = 80.5)。白色圆点为平均值,基于n = 15个靶点计算。 (A–F) 图中p值表示LigUnity在单侧Wilcoxon检验中显著优于最佳对照方法的统计显著性水平。
2.2 LigUnity 提升虚拟筛选性能
研究首先在DUD-E基准集的102个蛋白靶点与DEKOIS 2.0基准集的81个蛋白靶点上评估了LigUnity的虚拟筛选性能。这两个基准覆盖了多种类型的靶点,包括酶、离子通道、G蛋白偶联受体(GPCRs)与转录因子(见图S1与S2),具有广泛的代表性。
在比较实验中,LigUnity与24种主流方法进行了系统对比,这些方法包括分子对接法、基于结构的预测法与无结构的机器学习方法。结果显示,LigUnity在两个基准测试中均优于所有对照方法(图2A、2B与S3–S6;表S1)。尤其与性能最优的结构方法Denvis-G与RTMScore相比,LigUnity在富集因子(EF)1%指标上仍提升超过50%(p < 10⁻⁹)。同时,一旦嵌入表示计算完成,LigUnity在筛选速度上比商业对接软件Glide-SP快10⁶倍,因为它不需要生成分子对接构象(图S7)。
为验证其对新靶点的泛化能力,研究在训练集中过滤掉与测试蛋白序列相似的样本(图2C、2D、S8、S9)。即使在严格的30%序列相似性阈值下,LigUnity依然显著优于DrugCLIP与Glide-SP(p < 0.05),证明其具备对未知蛋白的强泛化能力。
接着,研究在LIT-PCBA基准集上进一步评估了LigUnity的表现。该基准包含15个靶点的实验测得配体亲和力数据,并同时收录活性与非活性配体。与DUD-E与DEKOIS相比,LIT-PCBA更具挑战性,因为其中包含大量结构上与活性配体相似的非活性配体。相比之下,DUD-E与DEKOIS的“干扰分子(decoys)”多来自未经实验验证的ZINC数据库,并刻意排除了与活性配体结构相似的分子,以减少假阴性,这使得它们对机器学习模型相对“更友好”。此外,LIT-PCBA维持1:1000的活性-非活性比例,更贴近真实药物发现中的筛选场景。尽管如此,LigUnity在该更具挑战性的基准上依然表现出卓越性能(图2E、2F),即便在排除序列相似蛋白的严格条件下,性能提升依然显著(图S10),进一步验证了模型的有效性与广泛适用性。
为分析各模块对整体性能的贡献,研究进行了消融实验(ablation study)(图S11–S13)。结果表明:
- 当移除骨架区分模块后,EF 1%指标下降超过60%,说明其在区分活性配体与虚假分子时具有关键作用;
- 去除异质图神经网络(H-GNN)后,性能持续下降,证明该模块能有效利用口袋–配体异质图信息提升预测能力;
- 药效团排序模块在LIT-PCBA基准上表现尤为突出(图S13),能够帮助LigUnity识别结构极为相似的配体间的微小差异,而这些差异往往决定其亲和力强弱。
综上所述,消融研究验证了LigUnity提出的三项核心技术思路——骨架区分、异质图推理与药效团排序——均不可或缺,其联合优化对于同时实现虚拟筛选与hit-to-lead优化具有决定性意义。

图3|从命中到先导化合物优化的性能评估 (A) 在Merck基准上的结合亲和力预测结果,指标为Spearman秩相关系数(ρₛ),评估设置为零样本(zero-shot)。白色圆点表示平均值,基于n = 8个靶点计算。 (B, C) LigUnity在不同微调条件下的结合亲和力预测表现。横轴为用于微调的实验结合亲和力数据比例(20%、40%、60%、80%),纵轴为预测结果与实验值之间的相关系数
2.3 LigUnity 提升从命中到先导化合物优化性能(Hit-to-Lead Optimization)
从命中到先导化合物优化的核心目标在于通过对初始活性配体的结构进行优化,以提升结合亲和力。这通常涉及在一组结构相似的候选配体中进行排序,从而找出活性最强的分子。鉴于LigUnity在虚拟筛选中已展现出显著的性能提升,特别是其捕捉配体细微结构差异的能力,研究进一步考察了LigUnity在hit-to-lead优化任务中的适用性。
研究在两个自由能微扰(FEP)基准上——JACS与Merck基准——评估了LigUnity在预测结合自由能方面的表现,这是hit-to-lead优化中最常用的性能指标。这两个基准共涵盖16个靶点,每个靶点平均包含约29个实验测得的配体。为避免潜在的数据泄漏,预训练阶段排除了与FEP基准相关的实验数据,同时移除了基准中的所有配体(详见方法部分)。
在零样本(zero-shot)设置下,即模型在训练与微调阶段均未使用任何测试蛋白的实验配体数据,LigUnity在Merck FEP基准的8个靶点上超越了所有现有计算方法(如Glide-SP与MM/GBSA)、基于结构的方法(如GenScore)以及无结构的机器学习方法(如DrugCLIP)(图3A),验证了其在亲和力预测中的有效性。
为了进一步评估模型在更复杂情境下的表现,研究设定了三种更具挑战性的条件:
-
无相似配体设置:训练集中排除所有与测试配体的Tanimoto相似度超过50%的配体;
-
无相似蛋白设置:排除所有与测试蛋白序列相似度超过30%的训练蛋白;
-
无相似配体与蛋白联合设置:同时应用上述两种排除标准。
在这三种严格设定下,LigUnity依然稳定优于所有对照方法(图S14),展现出极强的泛化能力。
在真实的药物研发场景中,研究人员通常仅能获得少量配体的实验亲和力数据。一个优秀的hit-to-lead优化模型应能利用这些有限数据进行小样本学习与预测增强。为验证这一点,研究在不同比例的实验数据下对LigUnity与其他方法进行微调比较。结果显示,LigUnity在所有四种实验条件下均持续领先(图3B、3C与S15;表S2)。在最严格的“双排除条件”下,当使用80%的实验配体数据进行微调(每个靶点约23个配体)时,LigUnity在Merck基准上取得了r² = 0.472的表现,已接近商业计算化学软件FEP+ (OPLS4)的性能(r² = 0.528)。这证明LigUnity能够在大幅降低计算资源消耗的同时,提供精确且高效的亲和力预测。
与虚拟筛选部分类似,研究通过**消融实验(ablation study)**分析了各模块的贡献(图S16)。结果显示:
- 移除骨架区分模块或药效团排序模块均会显著降低性能;
- 尤其在零样本与少样本条件下,去除药效团排序会使性能下降超过50%,凸显其在捕捉配体间相对活性差异方面的关键作用。
为验证LigUnity是否真正学习到了有意义的蛋白–配体相互作用模式,研究进一步在FEP基准上进行了关键残基屏蔽实验。利用ProteinPlus识别关键结合残基后发现,屏蔽强氢键或离子键残基会导致性能显著下降(Δρₛ = 6.34%),而随机屏蔽几乎无影响(Δρₛ = 0.46%)(表S3),说明LigUnity确实依赖真实的结构相互作用来进行有效预测。
此外,研究还通过原子与残基重要性分析,评估模型对结构特征的解释性(图3D–3G与S17)。以SHP2靶点为例,LigUnity正确识别出两个氨基(-NH₂)为关键活性基团,与晶体结构中观察到的氢键与离子相互作用高度一致(图3E、3F)。进一步地,模型还准确识别了Glu250E在氢键中的核心作用,以及Phe113A、Arg249A与Arg111A等极性残基的重要性(图3G)。虽然模型未显式使用配体构象(pose-free)导致其部分低估疏水腔体残基(如Thr253A与Pro491A)的贡献,但总体上仍能准确捕捉关键的相互作用模式。
综上所述,LigUnity不仅在命中到先导化合物优化任务中展现出高精度与强泛化能力,还具备结构解释性,可作为一种可解释的计算工具用于探索结构–活性关系(SAR)并指导药物优化设计。

图4|多情境下的模型评估 (A, B) 在按时间划分(split-by-time)的实验设置中,对LigUnity的结合亲和力预测性能进行评估。纵轴为预测与实验值的相关系数
2.4 LigUnity:面向多应用场景的通用亲和力预测基础模型
为进一步验证LigUnity作为亲和力预测基础模型(foundation model)的适用性,研究在两个综合实验数据库——ChEMBL与BindingDB——上评估了其性能。这两者包含大量实验测得的结合亲和力数据,涵盖多种类型的生物测定体系,如蛋白水平、细胞水平、细胞膜水平与亚细胞水平的实验。
首先,研究采用按时间划分(split-by-time)的设定进行测试:模型在2019年3月前发布的18,552个实验测定(assays)上进行预训练,并在2019年3月后新增的161个实验上测试(平均每个实验包含约48.6个配体)。为避免数据泄漏,所有测试靶点若在预训练数据中出现则被排除。结果显示,LigUnity在不同微调数据量(4–16个实验配体)下均表现出持续提升(图4A与S18)。与专为小样本亲和力预测设计的最佳对照方法ActFound相比,LigUnity在
在更具挑战性的按骨架划分(split-by-scaffold)测试中,即训练与测试配体骨架完全不同的情况下,LigUnity依然获得最佳表现(图4B),表明其对未知化学骨架具有强泛化能力。
随后,研究以磷酸二酯酶10A(PDE10A)为案例展开应用验证。该靶点是抗精神病药物的潜在治疗靶标,目前尚无获批药物。该数据集由Roche于2022年发布,包含1,162个配体,每个样本均附有实验亲和力与时间戳信息。研究采用时间划分策略:以首批20%的实验配体用于微调,剩余80%用于测试(图4C),并将预训练数据限制为2019年3月前发布的实验。结果表明,LigUnity在测试集上取得Pearson相关系数0.55的成绩,显著优于DrugCLIP的0.37(图4D、4E)。此外,LigUnity在前10个预测结果中成功识别出4个高活性配体(IC₅₀ < 1 nmol),在前50个预测结果中识别出24个高活性分子(DrugCLIP仅为11个)(图S19)。这些结果表明,LigUnity具备高效识别强效配体的能力,可有效降低命中到先导化合物优化阶段的研发成本。
最后,研究评估了LigUnity在按单位划分(split-by-unit)情境下的性能。在实际药物设计中,约24.4%的ChEMBL实验以百分比(%)形式报告活性,而大多数机器学习模型只适用于以摩尔浓度(nmol)或质量密度(μg/mL)为单位的数据。由于两类数据分布差异较大,按单位划分的测试对模型泛化性提出了更高要求。研究者假设LigUnity的药效团排序机制能增强其在不同计量体系下的稳健性,因为该方法学习的是口袋特异性的配体相对排序,而非绝对亲和力数值。基于此,研究以ChEMBL中使用百分比单位的实验作为测试集,以使用摩尔或质量单位的实验作为预训练数据,最终在包含65个实验(平均每个实验约30.5个配体)的测试集中,LigUnity在4–16个配体微调的不同条件下均超越所有对照方法(图4F、4G、S18)。值得注意的是,与回归模型Pocket-DTA相比,LigUnity在该设定下性能提升达40.2%,显著高于在时间划分设定下的13.8%提升,进一步验证了药效团排序策略在不同测量体系中的泛化优势,确立了LigUnity作为药物发现领域高效基础模型的地位。
2.5 LigUnity促进主动学习框架下的药物发现(Active Learning Framework)
在现实的药物研发流程中,由于实验资源有限与结合亲和力预测难度高,分子优化往往需要多轮迭代实验与计算。为提高效率,主动学习(Active Learning)已成为常用策略。基于此,研究进一步探讨了LigUnity在主动学习框架中的应用,以优化酪氨酸激酶2(TYK2)这一自身免疫疾病治疗靶点的配体。
实验采用一个包含10,000个配体的TYK2数据集,其结合自由能由FEP计算获得,总计算耗时约80,000 GPU小时(约9.1 GPU年)。研究目标是在受限计算资源条件下通过主动学习筛选高活性配体。具体流程为:
首先,使用少量随机选取的配体(具有已知结合自由能)训练LigUnity;随后在每一轮迭代中,从未标记配体中选择一部分进行FEP计算以获取新标签,再将这些数据加入训练集并重新训练模型(图5A)。
研究在两种主动学习策略下比较了模型性能:
- 贪心策略(greedy strategy);
- 探索–利用平衡策略(exploration–exploitation strategy)。
结果显示,LigUnity在仅少数迭代后便能准确识别出最强活性配体(图5B),且高活性配体的比例随迭代次数持续增加(图5C),证明了其在主动学习框架中的高效性。无论采用哪种策略,LigUnity均显著优于其他方法,在
综上所述,LigUnity不仅能在传统亲和力预测与优化任务中表现卓越,还能高效融入主动学习流程,在有限实验与计算资源下快速识别高活性配体,展现出在药物设计与分子优化领域的广泛应用潜力。
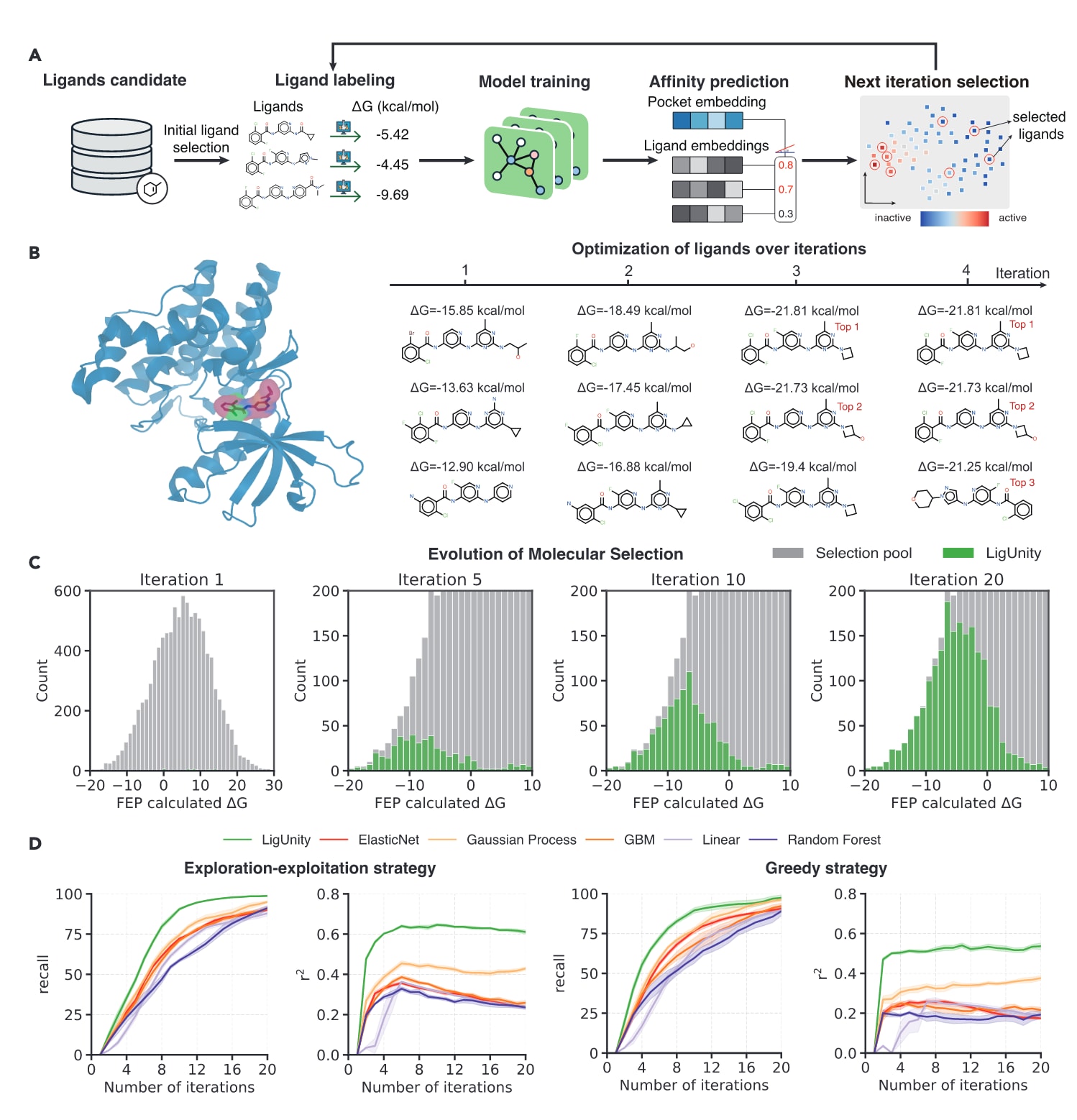
图5|LigUnity在主动学习框架下对TYK2靶点的评估 (A) 基于LigUnity构建的主动学习(Active Learning)框架示意图。模型通过迭代选择与再训练的方式,不断优化配体预测与筛选。 (B) TYK2靶点的三维结构(左,PDB编号:4GIH)及在探索–利用平衡策略(exploration–exploitation strategy)下于第1、2、3、4轮迭代中选出的高活性配体(右)。每个配体旁标注其经FEP计算获得的结合自由能(ΔG)。 (C) 基于贪心策略(greedy selection strategy)的每轮迭代中所选配体的ΔG分布直方图,展示高活性配体比例随迭代的动态变化趋势。 (D) LigUnity与其他对照方法在TYK2数据集上的性能比较图。左图为在探索–利用平衡策略下的结果,右图为在贪心策略下的结果,评估指标包括前2%召回率(top 2% recall)与相关系数
3 讨论
该研究提出了LigUnity——一种面向**虚拟筛选(virtual screening)与从命中到先导化合物优化(hit-to-lead optimization)的蛋白–配体亲和力基础模型。该模型通过学习口袋–配体共享空间(pocket–ligand shared space)来捕捉二者之间的结构与化学互补性,其实现依托于骨架区分(scaffold discrimination)与药效团排序(pharmacophore ranking)**相结合的框架。
在虚拟筛选任务中,LigUnity在三个主流基准(DUD-E、LIT-PCBA与Dekois 2.0)上全面超越24种对照方法;在hit-to-lead优化中,模型在两个FEP基准上取得最佳性能,并在时间划分、骨架划分与单位划分等多种测试条件下展现出优异的精度与稳健性。此外,通过引入主动学习框架(active learning framework)以模拟真实的先导化合物优化流程,LigUnity能在有限计算资源下高效识别最优结合分子。总体而言,这些结果展示了LigUnity在早期药物发现流程中的强泛化能力与实际可用性,确立了其作为计算机辅助药物设计(CADD)通用基础模型的潜力与可信度。
3.1 与现有工作的比较
LigUnity与两类研究方向密切相关:一类为无结构的药物–靶标亲和力预测(DTA)方法,另一类为基于结构的亲和力预测方法。
相比DTA方法(Drug–Target Affinity prediction),LigUnity在以下三个方面具有显著差异:
- 数据层面: DTA方法受限于实验测得亲和力的蛋白数量较少(如BindingDB中仅有2,773个蛋白),而LigUnity整合了53,406个结合口袋、覆盖4,847种蛋白的结构信息,使其在未充分研究的蛋白上依然表现稳健。
- 学习机制: DTA模型通常直接对绝对亲和力数值进行回归预测,而LigUnity通过骨架区分与药效团排序学习口袋–配体共享空间,在实验中表现出更一致的性能提升。此外,药效团排序本身相当于一种归一化策略,聚焦于配体的相对排序而非绝对数值,从而减弱了实验条件差异(如pH、温度)与测定类型差异(如IC₅₀与Kᴅ)的影响。
- 计算效率: DTA模型通常需要在每次预测时进行高强度计算,而LigUnity通过预计算并存储嵌入表示实现了快速大规模筛选,显著提升了应用效率。
相比基于结构的亲和力预测方法(如GenScore、RTMScore、EquiScore等),LigUnity同样展现出三方面改进:
- 数据覆盖: 结构方法依赖共晶复合物的三维构象,但这类结构极为有限,例如在BindingDB的57万种活性化合物中,仅有约7.5%具备晶体结构。而LigUnity基于45万个独特配体的多样化训练集构建,具备更强的泛化能力,可扩展至全新化学骨架。
- 计算成本: 传统结构方法通常需要生成结合构象(pose generation),例如Glide对接每个化合物约需1分钟,而LigUnity无需构象信息即可实现相当精度,极大提升了大规模筛选的可行性。
- 结构灵活性: 现有结构方法多基于静态蛋白构象,而LigUnity通过整合每个口袋的多个轻微变异结构(平均每个实验含10.96个口袋结构)隐式捕捉结合口袋的柔性特征。
此外,LigUnity的框架具有高度可扩展性与灵活性。对于缺乏实验结构的新靶点,LigUnity可利用AlphaFold 3或同源建模预测的口袋结构;若完全缺少结构数据,也可采用基于序列信息的LigUnity变体进行预测。
3.2 局限与展望
尽管LigUnity在结合亲和力预测中展现出优异性能,仍存在两方面限制:
- 适用范围受限: 当前模型仅适用于具有已知蛋白靶点信息的测定实验,因此不适用于缺乏靶标注释的表型实验(phenotype assays)。未来可通过引入**多模态信息(如基因表达特征)**来扩展模型适用性。
- 结构解释性限制: 虽然LigUnity能识别关键亚结构与残基并学习有意义的相互作用模式,但其不依赖构象的特性(pose-free)使其难以提供关于结合姿势的精确机制解释。后续工作计划结合对接姿势信息,发展基于结构的LigUnity变体,以提升预测精度与可解释性,尤其适用于具备充分计算资源、可获取高精度结合构象的场景。
综上,LigUnity不仅为跨任务的亲和力建模提供了统一框架,也为结构驱动的药物发现奠定了通用基础,展现出在虚拟筛选、先导优化与主动学习等多层次应用中的广泛潜力与持续拓展空间。