ACS Chem. Neurosci. 2024 | 机器学习分类人源μ-阿片受体配体的内在活性

今天介绍的是一项发表于 ACS Chemical Neuroscience 2024 的研究,题为《机器学习分类人源 μ-阿片受体配体的内在活性》。阿片类药物是现代医学中极具争议的一类化合物,它们通过与 μ-阿片受体(μOR) 结合来发挥镇痛作用,而像 纳洛酮(naloxone) 这样的拮抗剂则可逆转其致命的呼吸抑制效应。该研究团队以人工精炼的 983 个μOR配体数据集 为基础,结合 SMILES 分子表示与二维结构描述符,利用机器学习算法(包括 极端随机树ET模型 与 消息传递神经网络MPNN)实现了对配体 激动/拮抗活性 的高精度分类。模型在测试集上取得 AUC超91% 的优异表现,并在引入无标签数据的 三重训练(Tritraining with disagreement) 半监督策略后,AUC 进一步提升至 95.7%。这项研究不仅展示了人工智能在药理学预测中的潜力,也为 新型阿片拮抗剂开发与公共安全风险评估 提供了新思路。

0 摘要
阿片类化合物是一类作用于μ-阿片受体(μ-opioid receptor, μOR)的小分子激动剂,而纳洛酮等逆转剂则作为μOR拮抗剂发挥作用。该研究基于SMILES分子表示与二维分子描述符,构建了机器学习(Machine Learning, ML)模型,用于分类配体在人源μOR上的内在活性(intrinsic activity)。
首先,研究团队手动整理了一个包含983种小分子的数据集,这些分子均具有已测定的人源μOR 最大效能值(Emax)。通过化学空间分析,识别出主要的分子骨架以及结构上相似的激动剂与拮抗剂。随后,利用**决策树模型(Decision Tree Models)与定向消息传递神经网络(Directed Message Passing Neural Networks, MPNNs)**训练模型以区分两类配体。
在保留测试集上,Extra-Tree(ET)模型与MPNN模型的AUC(曲线下面积)分别达到91.5 ± 3.9%与91.8 ± 4.4%。为克服小样本带来的限制,研究进一步采用了名为三重训练(tritraining with disagreement)的师生式学习方法,在一个包含15,816个未标记配体(来自人、小鼠与大鼠的μOR、κOR及δOR)的扩展数据集上进行半监督训练。结果显示,该策略可将MPNN模型的测试AUC提升至最高95.7%。
该研究表明,即便在有限数据条件下,机器学习模型仍可准确预测μOR配体的内在活性。该方法未来有望应用于评估未知物质的公共安全风险,以及发现新型阿片过量解毒药物等领域。
1 引言
阿片类药物危机是一场复杂且持续的公共卫生灾难,已导致无数死亡,并造成了巨大的社会与经济损失。尽管已有多种干预手段——如处方药监控项目、药物辅助治疗、纳洛酮分发计划以及公众教育计划等——自2019年以来,阿片类相关死亡率仍在持续上升(来源:https://www.cdc.gov/opioids/basics/epidemic.html),这凸显出开发**新型创新策略**的迫切需求。
借助大数据与机器学习(Machine Learning, ML),研究者能够发现潜在的模式、风险因素及干预方向。近年来,机器学习已被用于多种与阿片危机相关的数据分析,包括电子健康记录、社交媒体数据、以及阿片滥用与过量风险因素等。此外,也有研究利用机器学习预测**药物与阿片受体(Opioid Receptors, ORs)**的结合能力。
其中,Sakamuru 等人的研究具有代表性,他们基于 NCATS药物库(含2805种已批准或在研化合物)通过高通量qHTS cAMP实验测得配体在μOR、κOR与δOR上的内在活性(intrinsic activity),并训练了多种传统机器学习模型(如树模型与支持向量机)来预测激动剂与拮抗剂。然而,他们将激动剂与拮抗剂分开建模,且预测效果差异明显——激动剂AUC为88%、拮抗剂仅76%,平衡准确率(BA)分别为73%与61%。
该研究的目标是开发用于分类人源μ-阿片受体(μOR)配体激动与拮抗活性的机器学习模型。与NCATS使用的cAMP数据不同,该研究手动整理了983种具有实测Emax值的人源μOR配体数据集,其数据来源于 [35S]GTPγS结合实验。相比cAMP检测,该实验在信号传导早期阶段直接量化受体占据率,能更准确反映激动剂活性,避免信号放大带来的偏差。
基于此数据集,研究训练了两类模型:传统树模型与定向消息传递神经网络(Directed Message Passing Neural Networks, MPNNs),输入为配体的SMILES分子表示。图神经网络(Graph Neural Networks, GNNs)作为一类能处理图结构数据的深度学习模型,近年来在药物溶解度、毒性预测、蛋白–配体亲和力预测、抗生素发现等小分子性质研究中展现出巨大潜力。MPNN则是一种针对化学结构优化的GNN变体,通过沿化学键(有向边)传播信息以捕捉分子结构特征。
在100次独立留出测试中,Extra Tree(ET)模型与MPNN模型的平均AUC分别为91.5%与91.8%,平均平衡准确率(BA)为83.3%与85.1%。MPNN模型进一步允许识别与拮抗活性最相关的最小结构片段,从而增强了解释性。
为突破小样本带来的限制,研究者还引入了名为**“三重训练(tritraining with disagreement)”的师生式学习策略,结合包含15,816个未标记的阿片受体配体(涵盖人、鼠与大鼠的μOR、κOR与δOR)**的新数据集进行半监督学习。
该研究为建立高性能机器学习模型以预测阿片受体配体内在活性迈出了关键一步,为未来阿片类药物风险评估与新型解毒剂发现提供了重要的技术基础。
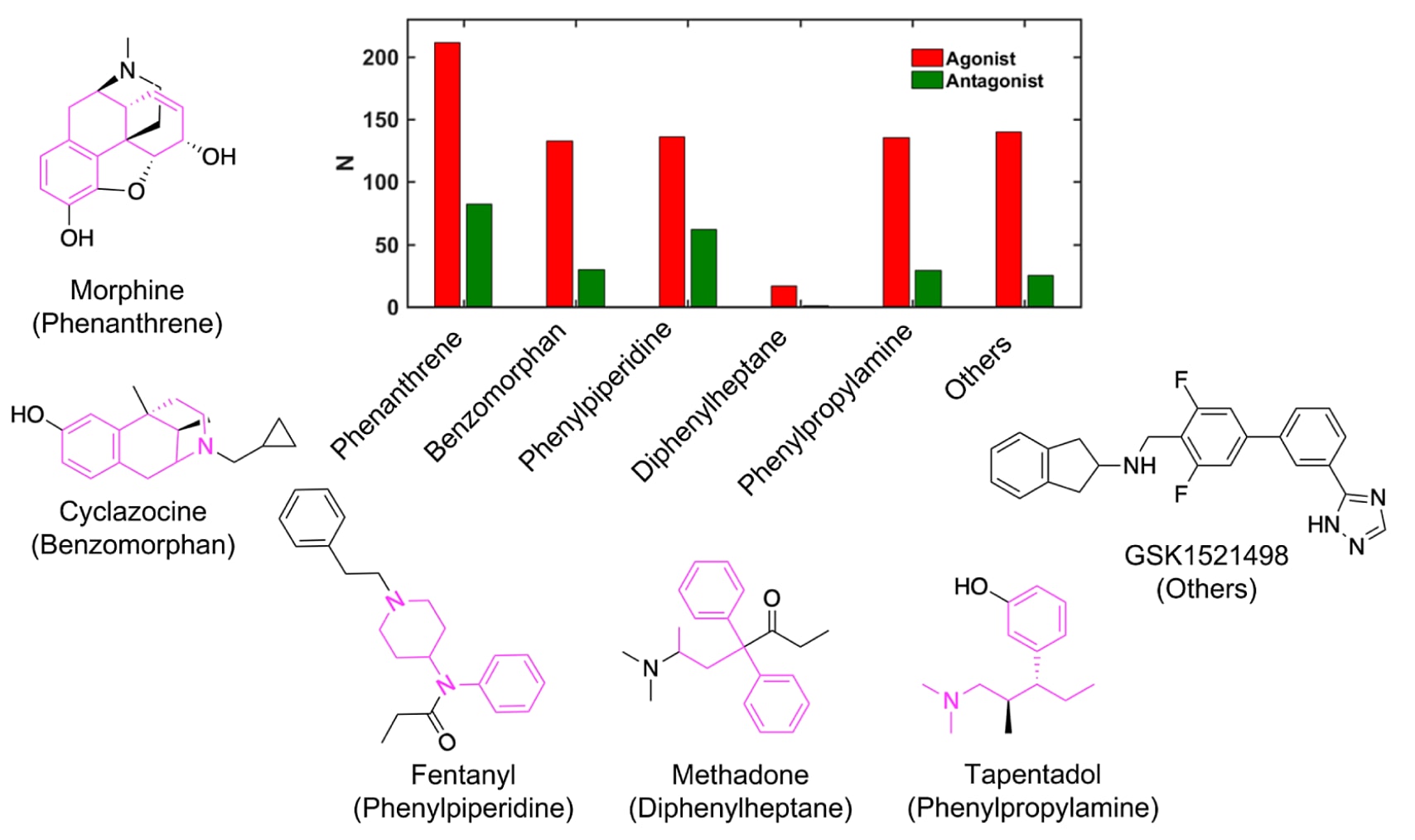
图1|人源μ-阿片受体(μOR)激动剂与拮抗剂训练数据集中代表性的阿片类化合物结构类型。 图中展示了五大主要化学骨架类别下的配体分布,包括苯并菲(phenanthrene)、苯并吗啡烷(benzomorphan)、苯基哌啶(phenylpiperidine)、二苯庚烷(diphenylheptane)与苯丙胺(phenylpropylamine)。其中,红色表示激动剂(agonists),绿色表示拮抗剂(antagonists),对应的数量以N标注。其余不属于上述骨架的分子被归入“others”类别。图中以洋红色标示了来源于这些主要骨架的常见阿片类药物,并展示了一个新型合成阿片化合物作为“others”类的示例。
2 结果与讨论
2.1 数据集的构建
研究团队从四个在线数据库中整合了983种人源μ-阿片受体(μOR)配体,并对文献中关于这些化合物在人源μOR上**[35S]GTPγS功能实验的活性表征进行了人工筛选与整理**。根据文献作者对药理活性的描述,将配体分为μOR激动剂(agonist)和拮抗剂(antagonist)两类;若文献未提供明确分类,则以纳曲酮(naltrexone)Emax = 14%作为阈值进行区分(详见“方法与实验流程”部分)。最终得到755个激动剂与228个拮抗剂。
随后,利用各化合物的SMILES分子式,通过RDKit计算得到其二维分子描述符(2D molecular descriptors),为机器学习建模提供基础数据。
2.2 数据集的化学空间分析
研究者进一步使用RDKit进行子结构分析,结果如图1所示。大部分化合物为阿片类分子,其核心骨架可划分为六大类结构类型:
苯并菲(phenanthrene)、苯并吗啡烷(benzomorphan)、苯基哌啶(phenylpiperidine)、二苯庚烷(diphenylheptane)、苯丙胺(phenylpropylamine)及其他类别(others)。
其中,苯并菲类衍生物数量最多,共288种分子(206个激动剂,82个拮抗剂)。这类化合物多来源于罂粟属植物的天然产物,代表结构为吗啡(morphine),还包括吗啡烷(morphinan)、4,5α-环氧吗啡烷(epoxymorphinan)与奥利帕维因(oripavine)。
苯并吗啡烷类(benzomorphan)共158种分子(128个激动剂,30个拮抗剂),其结构可视为从吗啡分子中去除了“C环”。典型例子包括氯环吗啡酮(ketocyclazocine)、乙基氯环吗啡酮(ethylketocyclazocine)、苯吗啡酮(phenazocine)及喷他佐辛(pentazocine)。
大多数天然与半合成阿片药物均属于苯并菲或苯并吗啡烷类。
此外,苯基哌啶类(phenylpiperidine)包含132个激动剂与62个拮抗剂。这类全合成阿片药物结构较简单,骨架中环系较少,代表化合物包括芬太尼(fentanyl)、阿芬太尼(alfentanil)、**舒芬太尼(sufentanil)等,以及其衍生的4-苯胺基哌啶(4-anilidopiperidines)**化合物。
二苯庚烷类(diphenylheptane)数量最少,仅含17个激动剂与9个拮抗剂,代表分子为美沙酮(methadone)与丙氧芬(propoxyphene)。
苯丙胺类(phenylpropylamine)中有132个激动剂与29个拮抗剂,主要包括曲马多(tramadol)与他喷他多(tapentadol)。
此外,还有137个激动剂与25个拮抗剂不属于以上任一类别,被归入“others”,显示出更为多样化的新型结构特征。
为进一步理解训练数据集的化学分布,研究者基于分子之间的Tanimoto距离(1 − Tanimoto相似度)对其SMILES结构进行聚类分析。聚类方法采用Scikit-learn库中的层次凝聚算法(hierarchical agglomerative clustering),并将Tanimoto距离阈值设为0.7作为聚类标准。
最终共得到23个聚类(clusters),各类的相对分布与拮抗剂比例如图2所示。其中,最大两个聚类(cluster #1与#2)分别包含约35%与20%的数据,其余每类均不足10%。在这两个最大聚类中,约30%的分子为拮抗剂,与整体数据集约**1:3.3(拮抗剂:激动剂)**的比例一致。
聚类分析还帮助识别出与激动剂结构高度相似的拮抗剂。例如,cluster #1 的代表性分子(中心分子)asalhydromorphone 是强效阿片类药物氢吗啡酮(hydromorphone)的前体药物,其结构与纳美啶托斯酸盐(naldemedine tosylate)——一种用于治疗阿片类便秘的μOR拮抗剂——高度相似(见图2,cluster #1)。
此外,通过分析拮抗剂比例,还发现了一个拮抗剂占优势的聚类cluster #11,共含13个分子,其中10个(77%)为拮抗剂。该类的代表分子为一个典型μOR拮抗剂,其结构及与之最相似的激动剂结构在图2中展示。
这些结果表明,聚类分析不仅揭示了μOR配体在化学空间中的分布规律,也为结构相似的激动剂与拮抗剂关系研究提供了关键线索。
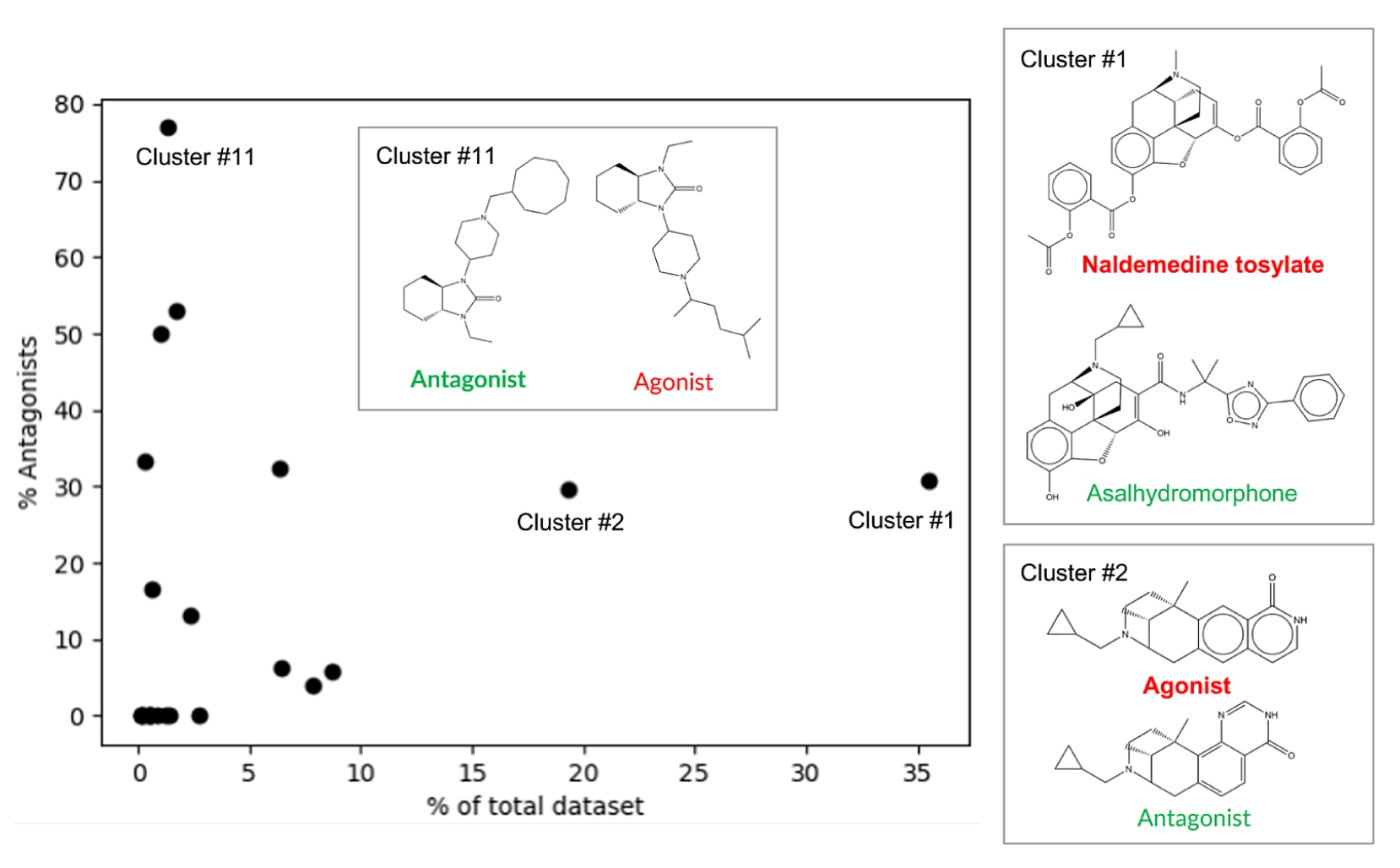
图2|人源μ-阿片受体(μOR)激动剂与拮抗剂训练数据集中化学结构的聚类分析。 横坐标表示聚类编号(按分子数量由大到小排列),纵坐标表示拮抗剂所占百分比。图中展示了各聚类的相对分布情况。对于cluster #1、#2与#11,给出了其代表性分子结构(聚类中心,粗体标示)及其在相反功能类别中结构最相似的化合物(即具有最高Tanimoto相似度的分子)。文中提及的这些具体化合物(如asalhydromorphone与naldemedine tosylate等)在正文部分进行了进一步讨论。
2.3 决策树模型的性能评估
研究团队利用 PyCaret 平台训练了随机森林(Random Forest, RF)与极端随机树(Extra Tree, ET)模型,用于二分类任务:判断化合物是μOR激动剂还是拮抗剂。由于数据集中激动剂数量约为拮抗剂的 3.3 倍,在模型训练中将拮抗剂标记为正类(positive),激动剂标记为负类(negative),以缓解类别不平衡问题。
数据集按 9:1 比例随机划分为训练/交叉验证集(CV)与测试集。每个分子以其 SMILES 分子式为输入,利用 RDKit 计算得到 二维分子描述符。为进一步平衡样本比例,采用了**SMOTE(Synthetic Minority Oversampling Technique)**过采样算法,为拮抗剂(少数类)生成合成样本。经过特征筛选后,去除高相关性与不变性特征,最终保留 91 个分子描述符。
为减少模型过拟合,研究进一步通过调整特征选择阈值 α(0.2–1),构建并测试了 9 组不同规模的特征集(49–91维)。所有实验(数据划分、训练与评估)均重复 100 次以确保统计稳健性。性能评估指标包括:AUC(曲线下面积)、平衡准确率(BA, balanced accuracy)以及针对少数类的 F1 分数(见表 S1)。
结果显示,模型性能对特征维度依赖性较低:
- **RF模型:**AUC为 91.0–91.8%,BA为 81.7–83.1%,F1为 72.6–74.9%;
- **ET模型:**AUC为 90.3–91.5%,BA为 80.7–83.3%,F1为 71.3–75.4%。
最佳模型表现如下:
- **RF模型:**AUC = 91.8 ± 2.7%(90个特征,α=0.8),BA = 83.1 ± 5.2%(70个特征,α=0.4),F1 = 74.9 ± 6.4%(49个特征,α=0.2);
- **ET模型:**AUC = 91.5 ± 3.9%(90个特征,α=0.8),BA = 83.3 ± 5.0%(同上),F1 = 75.4 ± 6.4%(90个特征,α=0.8)。
进一步分析最佳BA模型的召回率(recall)与精确率(precision):
- **激动剂(多数类)**预测表现优异:
- RF模型:召回率 93.3 ± 2.8%,精确率 92.0 ± 2.7%;
- ET模型:召回率 94.1 ± 3.0%,精确率 92.0 ± 2.6%。
- **拮抗剂(少数类)**预测较低但仍可接受:
- RF模型:召回率 72.9 ± 9.9%,精确率 77.0 ± 8.1%;
- ET模型:召回率 72.6 ± 9.9%,精确率 79.5 ± 8.1%。
考虑到激动剂与拮抗剂数量比例超过 3:1,这种性能差异在预期范围内,即使在使用过采样技术的情况下亦然。总体而言,两种模型的平均 AUC均超过91%,表明其对μOR配体内在活性的分类能力较强。
相比之下,Sakamuru 等人基于 NCATS 数据集采用多数类下采样(undersampling)的RF模型,其预测性能为:激动剂模型 AUC = 88%,BA = 73%。尽管该研究的训练数据规模更小,所构建的RF模型仍实现了更高的准确性(AUC = 91%,BA = 83.1%)(见表1),显示出更强的泛化与学习能力。
表1|最佳随机森林(RF)与极端随机树(ET)模型在留出测试集上的性能评估指标汇总。

2.4 模型预测中关键官能团的重要性分析
为了阐明决策树模型的预测依据,研究团队计算了SHAP值(SHapley Additive exPlanations),该方法基于博弈论原理,用于定量评估每个特征对模型输出的影响。
图3展示了按平均绝对SHAP值排序的前10个特征,纵轴为特征名称,横轴为各特征在不同样本中的SHAP值。SHAP值反映了每个特征对模型预测结果相较于平均输出的贡献程度:正值表示使预测结果倾向于“拮抗剂”,而负值则代表倾向于“激动剂”。
根据平均SHAP值分析,模型中最具影响力的七个分子特征为:
- 脂肪族杂环数量(NAlHc)
- 芳香杂环数量(NArHc)
- XCCNR基团数量(即三级胺结构,NXCCNR)
- 芳香环数量(NArRi)
- 脂肪族羟基数量(NAlOH)
- 饱和环数量(NSaRi)
- 咪唑环数量(Nimd)
从SHAP蜂群图(beeswarm plot)可见:
- NAlOH(脂肪族羟基数量) 对模型输出的正向影响最大,即羟基数越多,模型越倾向预测为拮抗剂(图3a);
- NAlHc(脂肪族杂环数量) 同样呈现显著的正向作用,其值增加亦会推动预测结果向拮抗剂方向移动;
- 相反,NXCCNR(三取代胺基)的增加则产生最显著的负向影响,其次是**芳香杂环(NArHc)与咪唑环(Nimd)**数量的增加,这些特征值增大时,模型预测更倾向于激动剂。
为验证SHAP结果的合理性,研究进一步比较了这些特征在激动剂与拮抗剂中的分布差异(见附图S2),并分析了训练集中三个代表性化合物(图3b)。
- NAlHc(脂肪族杂环数量)在数据中呈三种主要模式:0、1、2,其中模式1和2由拮抗剂主导,而模式0主要对应激动剂。这与SHAP分析一致。例如,拮抗剂nor-binaltorphimine的NAlHc值最高(4),而激动剂oliceridine和etonitazene的值分别为1与0。
- NAlOH(脂肪族羟基数量)的分布范围为0–4,其中0和1主要为激动剂,2和3主要为拮抗剂。这与较高NAlOH值预测为拮抗剂的趋势相符。三种示例化合物中,仅nor-binaltorphimine(拮抗剂)的NAlOH=2,而oliceridine与etonitazene均为0。
- 同样地,NXCCNR、NArHc与Nimd等特征的分布模式也与蜂群图分析结果一致:这些特征值的增加会推动模型输出向激动剂方向转移(图3a)。
综上,SHAP分析清晰揭示了模型识别μOR激动剂与拮抗剂时的关键结构特征,说明模型的预测不仅具有较高准确性,也具备明确的化学可解释性。
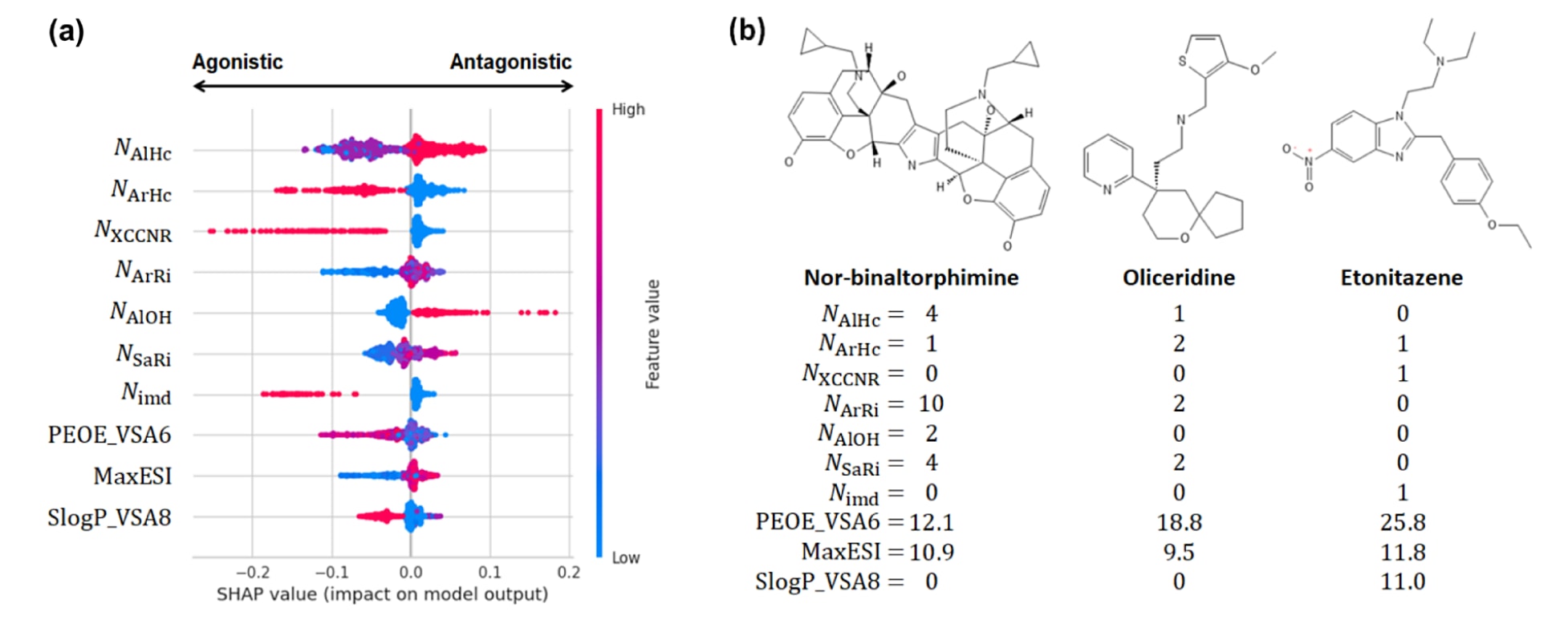
图3|ET模型关键特征分析。 (a) SHAP蜂群图(beeswarm plot)展示了最佳ET模型(见表1)中平均绝对SHAP值最高的10个特征,按重要性由高到低排列。图中每个点代表数据集中的一个样本,颜色梯度表示对应特征值的大小。前七个最重要的特征分别为:脂肪族杂环数量、芳香杂环数量、XCCNR基团数量(即三级胺结构)、芳香环数量、脂肪族羟基数量、饱和环数量以及咪唑环数量。后三个特征依次为:电荷描述符(charge descriptor)、最大电拓扑状态指数(maximum electrotopological state index)以及logP描述符(logP descriptor)。各特征的详细说明见表S2。(b) 三种代表性分子的结构及其对应特征值:拮抗剂nor-binaltorphimine、激动剂oliceridine和激动剂etonitazene。这些分子的特征分布验证了模型通过结构特征区分激动剂与拮抗剂的能力。
2.5 MPNN模型的性能评估
图神经网络(GNN)通常在大数据集上表现优于传统树模型。为验证这一假设,研究团队基于SMILES分子结构图并使用ChemProp默认参数训练了定向消息传递神经网络(MPNN)模型。结果显示,其初始性能与ET模型相当,平均平衡准确率(BA)为83.1%,F1(拮抗剂类)为75.4%,AUC为90.8%(表2)。
随后,研究者测试了多种优化策略以提升模型表现,包括:
- 加入RDKit分子特征;
- 优化分类阈值(classification threshold);
- 超参数调优(hyperparameter tuning)(表S3)。
与初始MPNN相比,
-
优化分类阈值虽提高了拮抗剂的召回率(recall),但略微降低了F1分数;
-
加入分子特征则使拮抗剂召回率提高0.5%,激动剂召回率提高0.7%,两类的精确率(precision)分别提升1.2%与0.3%,整体F1提升1.0%(表S4)。
在进一步调整阈值以最大化F1后,F1再提升0.2%,BA提升0.8%。这主要得益于拮抗剂召回率提高3.1%及激动剂预测精确率提升0.6%,尽管拮抗剂精确率下降2.9%、激动剂召回率下降0.6%(表S4)。
在集成训练(ensemble training)中,
-
10折交叉验证(10-fold CV)与5折CV的结果仅有0.2%的F1差异,
因此为节省计算时间,研究采用5折CV进行超参数优化。
该步骤显著提升了模型性能,F1与BA分别提高1.1%与1.8%。
最终优化后的MPNN(模型6,表S4)较最佳ET模型表现更优:
- F1:77.5 ± 6.7%(vs ET的75.4 ± 7.2%)
- BA:85.1 ± 5.0%(vs ET的83.3 ± 5.0%)
- AUC:91.8 ± 4.4%(略高于ET的91.5 ± 3.9%)
表2|初始与优化后MPNN模型在留出数据集上的性能评估汇总
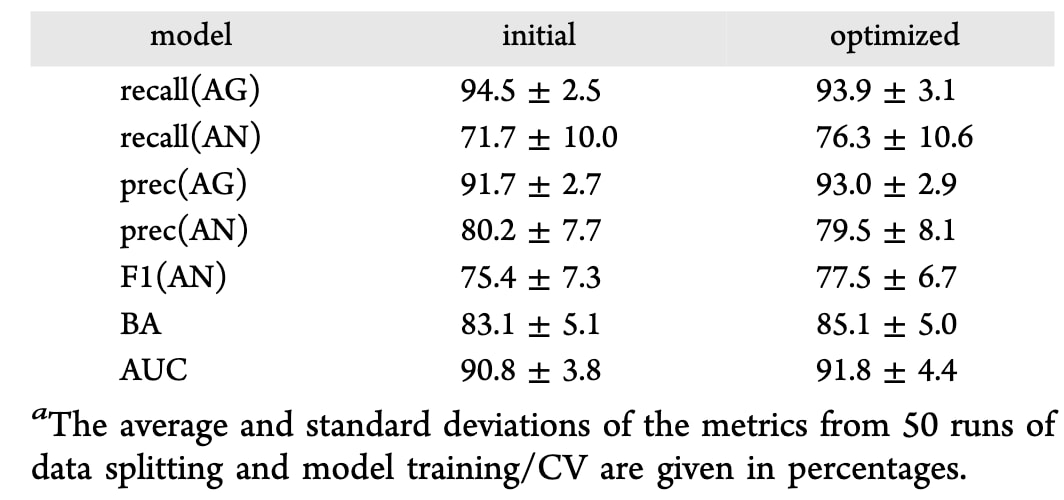
表3|最终ET与MPNN模型在包含26个化合物的外部数据集上的预测性能
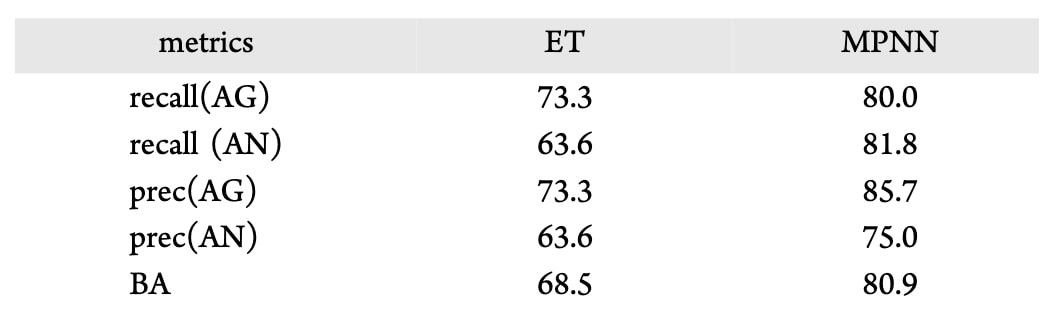
外部验证与模型预测分析
为进一步验证模型的泛化能力,研究者将ET与MPNN模型在全部983个μOR配体(755个激动剂 + 228个拮抗剂)上重新训练,并测试其在外部验证集(16个激动剂 + 11个拮抗剂)上的表现(表S5)。
结果显示,MPNN在所有指标上均优于ET模型,与留出测试(hold-out test)的结果一致。
- ET模型正确预测了15个激动剂中的11个与11个拮抗剂中的7个,召回率分别为73.3%与63.6%,BA = 68.5%;
- MPNN模型正确预测了12个激动剂与9个拮抗剂,召回率分别为80.0%与81.8%,BA = 80.9%(表3)。
值得注意的是,MPNN对少数类拮抗剂的召回率提升了18.2%,精确率也提高11.4%,说明其对类别不平衡问题具有更强的鲁棒性。
对外部预测结果的结构解释
为进一步理解模型的判别依据,研究团队采用ChemProp中实现的蒙特卡洛树搜索(Monte Carlo Tree Search, MCTS)算法,识别模型判定“拮抗剂”时的关键分子子结构。
外部验证集中包括5个苯蒽(phenanthrene)类拮抗剂:6β-naltrexol、nalodeine(N-allylnorcodeine)、(17S)-methylnaltrexone、naloxazone与orvinol 14(见图4)。
MPNN正确识别了其中4个,仅将orvinol 14误判为激动剂,推测原因是其与训练集中激动剂BU08028结构极为相似(Tanimoto相似度=0.99)。此外,外部数据集中还包含3种黄酮类化合物(flavonoids):apigenin(4′,5,7-三羟基黄酮)、4′,7-二羟基黄酮与4′-羟基黄烷酮(见图4)。黄酮为植物次生代谢产物的重要类别,具有典型多酚骨架(polyphenolic structure)。MCTS算法识别出其拮抗性预测的关键结构为色酮(chromone, 1,4-benzopyrone),即黄酮核心骨架的组成部分。MPNN正确分类了前两者(apigenin与4′,7-二羟基黄酮)为拮抗剂,但将4′-羟基黄烷酮误判为激动剂。
外部验证集中还包括4种苯蒽类激动剂:3-monoacetylmorphine、morphinone、codeinone与buprenorphine hemiadipate。
其中,MPNN全部正确分类,而ET模型错误地将buprenorphine hemiadipate预测为拮抗剂(见图5)。这些激动剂均为吗啡(morphine)衍生物,而吗啡本身已包含在训练集内。它们在C3位具有甲基或乙酰取代(如codeine、ethylmorphine、diacetylmorphine/heroin等),这一结构特征增强了模型识别的准确性。
然而,MPNN与ET模型都误判了morphiceptin(肽类激动剂)以及两种生物碱matrine与sophocarpidine(见图5)。研究者推测,这些误判可能源于训练集中缺乏与其结构相似的激动剂样本,导致模型在这类结构上泛化能力有限。
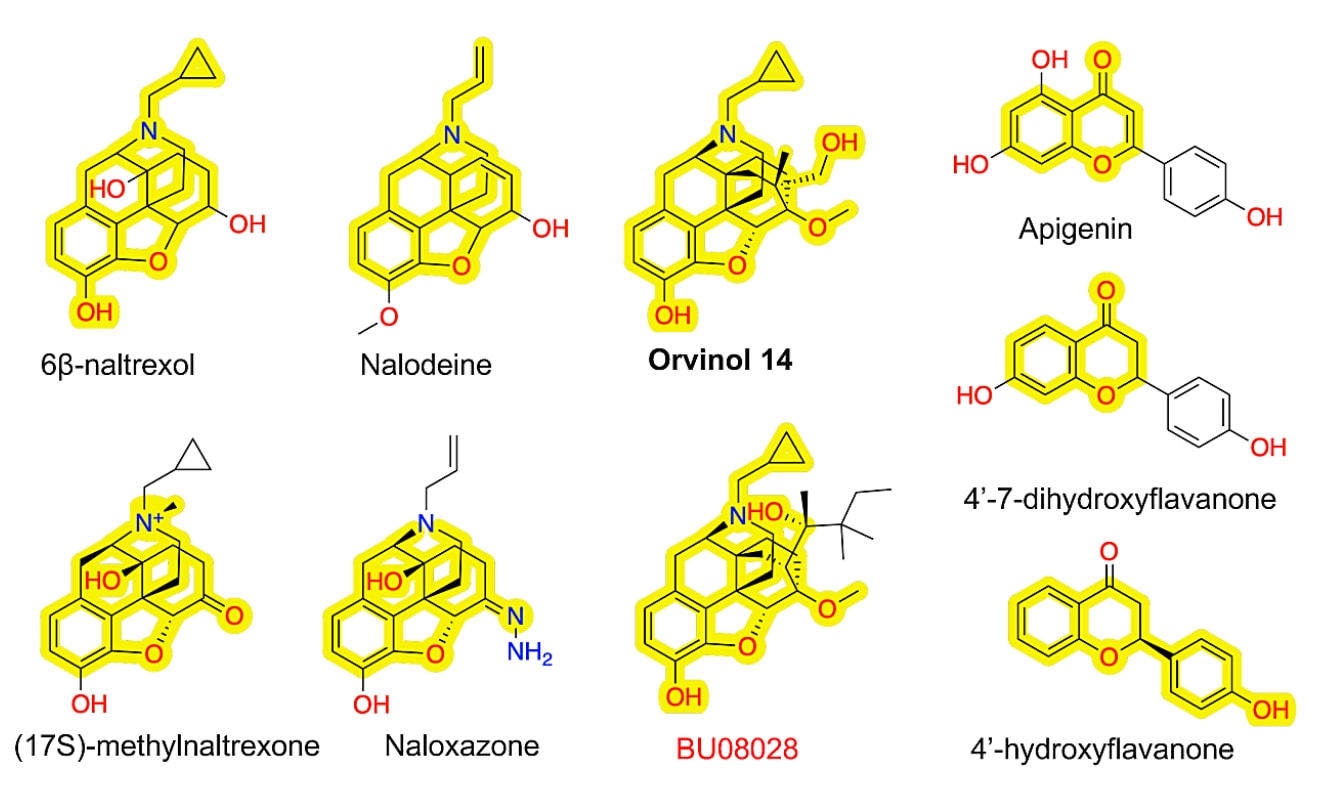
图4|外部验证集中基于菲类(a)与黄酮类(b)拮抗剂的化学结构。 图中标出了模型用于预测拮抗活性的关键子结构。加粗的分子名称表示被模型错误预测为激动剂的化合物。在菲类化合物中,orvinol 14的关键子结构也存在于训练集中激动剂**BU08028(红色标示)**中,共同的结构单元已在图中高亮显示。
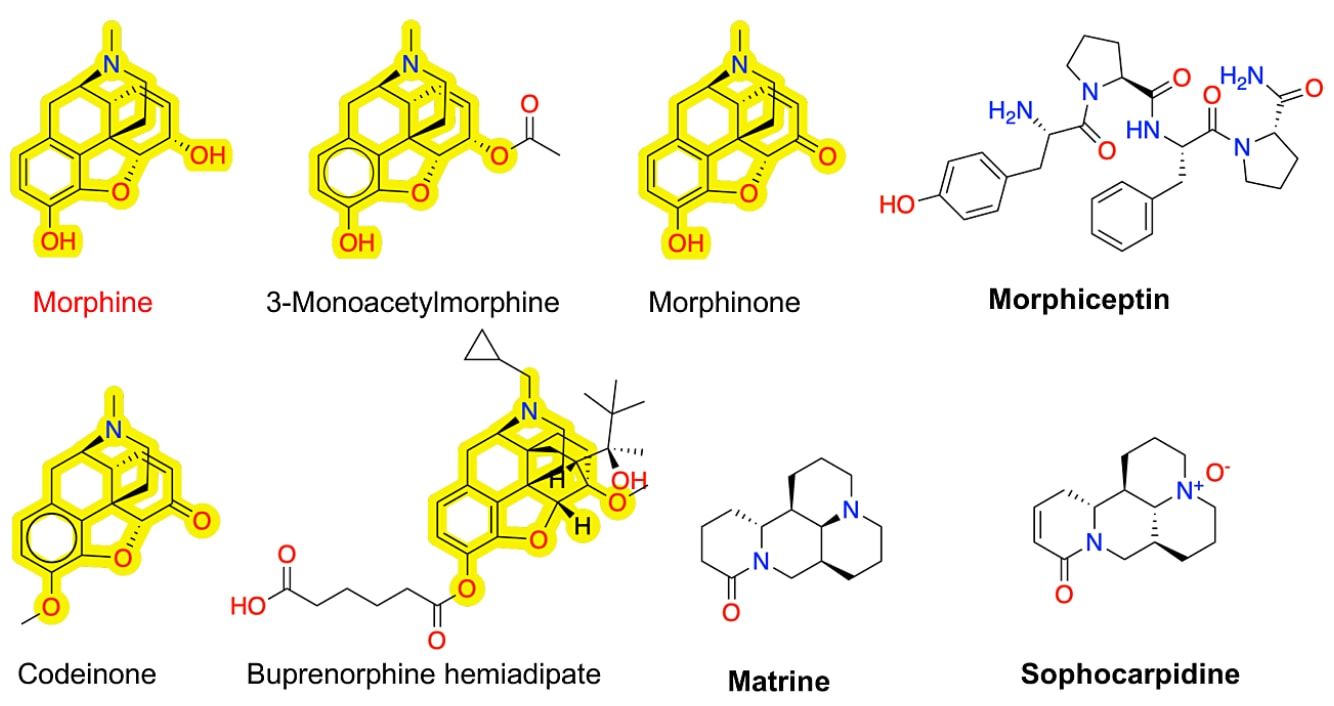
图5|外部验证数据集中基于菲结构的激动剂及三种被错误分类的激动剂的分子结构。 这些基于菲结构的激动剂均为吗啡(红色)衍生物,图中已高亮显示各化合物中共同的吗啡骨架结构。加粗标注的三种化合物为被模型错误分类的激动剂。
2.6 利用无标签数据的三重训练(Tritraining)策略验证:提升模型性能的概念验证实验
为克服数据集规模较小的问题并提升模型性能,该研究引入了一种半监督教师–学生学习(teacher–student learning)方法,称为“带分歧的三重训练(tritraining with disagreement)”。首先,研究者构建了一个包含15,816 个配体的无标签数据集,这些配体来源于 人类、小鼠及大鼠的 μOR、κOR 和 δOR,数据取自 bindingDB 数据库。该无标签数据集的规模约为带标签训练集(983 个配体)的 15 倍。
三重训练的流程如下:
- 首先将带标签数据以 9:1 的比例分为训练/交叉验证集与留出测试集。
- 接着,利用三种不同模型——ET(极端随机树)、XGBoost与**MPNN(消息传递神经网络)**对无标签分子进行分类。
- 当其中一个模型的预测结果与其他两个模型不一致时,该模型的原始训练集将被补充入该分子及由其他两模型一致预测出的伪标签;若三者预测一致,则丢弃该分子。
- 当所有无标签分子被处理完后,三个模型使用新的训练集重新训练并进行交叉验证,随后在留出数据上进行测试。
- 之后,新的模型再对先前丢弃的数据进行预测,如此反复迭代,直到模型的 AUC 不再提升、无标签数据被耗尽,或三模型间不再出现分歧为止。
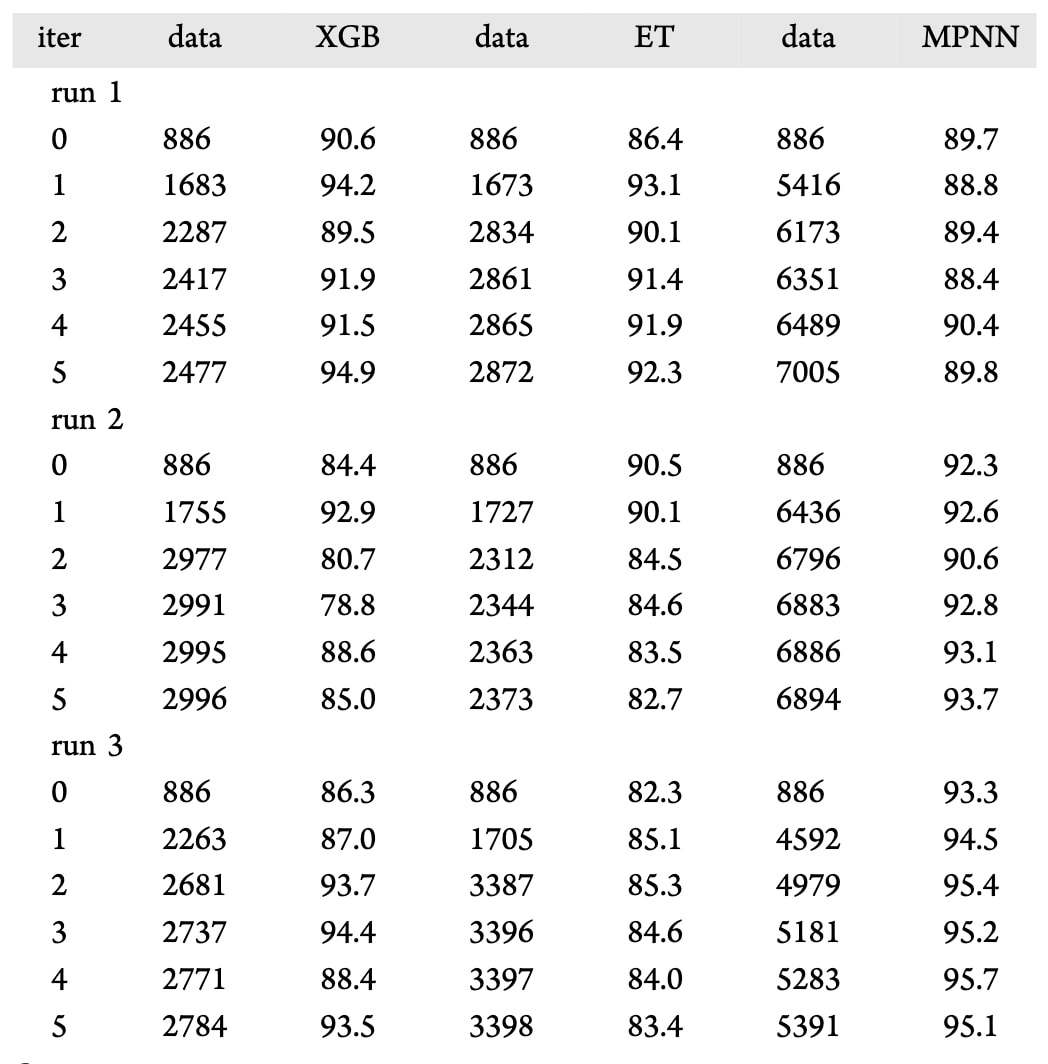
作为概念验证,研究进行了 三组三重训练实验,均采用 9:1 的训练/测试比例,且未进行超参数调优。每次迭代后,模型不仅进行了留出测试,还使用前述的 26 个外部化合物(表 S6)进行外部验证。在第一次运行中,初始的 XGB、ET 与 MPNN 模型在留出测试中的 AUC 分别为 90.6%、86.4% 和 89.7%(表4)。经过第一次迭代后,XGB 与 ET 的 AUC 分别显著提升至 94.2% 与 93.1%。在随后的迭代中,AUC 呈现出轻微的升降波动,但至第 5 次迭代时,AUC 达到 94.9%(XGB)与 92.3%(ET),均明显高于初始值。然而,仅有 XGB 模型在外部验证中表现出稳定提升——第一次迭代后的 AUC 达到 87.9%,相比初始值 69.6% 明显提高(表 S6);ET 模型的外部验证 AUC 则未优于初始结果。有趣的是,MPNN 模型的三重训练表现与树模型(XGB、ET)明显不同:其测试 AUC 仅在第 4 次迭代后略有提升。
这一差异与三模型在各轮训练中获得的新增数据量有关:至第 5 次迭代,XGB 与 ET 仅分别新增 22 与 7 条数据,而 MPNN 在第一次迭代后获得了大量新增样本,表明树模型间预测过于相似,常常一致,而 MPNN 则能提供更多分歧信息。在第二次运行中,初始 AUC 分别为 84.4%(XGB)、90.5%(ET) 与 92.3%(MPNN)。数据增长趋势与第一次运行一致,再次证明树模型相似而 MPNN 差异较大。XGB 模型在第一次迭代后测试与外部验证 AUC 均显著上升,而 ET 模型在任何迭代中均未改善(表4与表S6)。MPNN 模型的 AUC 变化较小,但除第二次迭代外,其余轮次测试 AUC 均高于初始值——第一次迭代后测试 AUC 提升至 92.6%(初始为92.3%),对应外部验证 AUC 从 83.0% 提升至 84.2%。在第三次运行中,三模型的初始 AUC 分别为 86.3%(XGB)、82.3%(ET) 与 93.3%(MPNN)。数据增长趋势与前两次类似,但 MPNN 获得的新增样本略少。随着迭代次数增加,三模型的测试 AUC 均显著上升,最高分别达到 94.4%、85.3% 与 95.7%。然而,外部验证的 AUC 并未保持同步,仅在 XGB 模型的第一次迭代中出现明显提升。这种留出测试与外部验证结果间的不一致,可能源于外部数据集样本量过小。
总体而言,这些结果表明:
带分歧的三重训练(Tritraining with disagreement)是一种在标注数据有限的情况下提升模型性能的有前景策略,尤其能帮助充分利用无标签分子信息,改善机器学习模型在 μOR 配体活性分类中的泛化表现。
表4|三次“带分歧三重训练(Tritraining with Disagreement)”运行中留出测试(Hold-Out Test)的AUC结果汇总
3 结论与讨论
该研究基于人工整理的 983 种人源 μ-阿片受体(μOR)配体数据集(其中包含 755 个激动剂与 228 个拮抗剂,文献中均报告了通过 [³⁵S]GTPγS 功能实验测得的 Emax 值),建立了用于分类小分子配体固有活性的机器学习(ML)模型。在 100 次留出测试中,极端随机树(ET)模型与消息传递神经网络(MPNN)模型均表现出较高的预测性能,AUC 分别为 91.5 ± 3.9% 与 91.8 ± 4.4%,显示出模型能够有效区分激动剂与拮抗剂,其中 MPNN 表现略优。
在类别指标方面,ET 模型预测激动剂的召回率/精确率分别为 94.1 ± 3.0% / 92.0 ± 2.6%,拮抗剂为 72.6 ± 9.9% / 79.5 ± 8.1%;MPNN 模型对应为激动剂 93.9 ± 3.1% / 93.0 ± 2.9%,拮抗剂 76.3 ± 10.6% / 79.5 ± 8.1%。拮抗剂预测性能略低,主要归因于数据集中约 3:1 的类别不平衡。在基于完整数据集训练的最终模型中,ET 与 MPNN 在 26 个外部验证分子(15 个激动剂与 11 个拮抗剂)上的平衡准确率(BA)分别为 68.5% 与 80.9%。MPNN 明显优于 ET,能够以超过 80% 的召回率准确识别两类配体。为克服数据稀缺与类别不平衡问题,研究进一步引入了**“带分歧的三重训练(tritraining with disagreement)”教师–学生学习策略,利用来自人类、小鼠与大鼠的 μOR、κOR 与 δOR 配体的无标签数据**辅助训练。
三组独立运行结果显示,该方法能在多次迭代后显著提升模型的测试与验证 AUC,验证了三重训练策略的潜力。
特别地,XGB 模型在第一次迭代后测试与外部验证 AUC 即显著提升,其训练数据量几乎翻倍,主要得益于两位“教师模型”(ET 与 MPNN)生成的伪标签数据。结果还表明,两种树模型(XGB 与 ET)在结构上相似,而 MPNN 差异较大,这导致 MPNN 在第一次迭代后获得了约 4500 条伪标注数据,远多于树模型的约 800 条。然而,MPNN 的 AUC 提升幅度有限,可能源于初始树模型性能较低;相反,XGB 模型的改进最为显著,归因于其“教师”模型质量更高(即 ET 与 MPNN 提供的标签更准确)。目前模型仅限于预测已知活性配体(actives),因为训练数据集中仅包含实验验证过的 μOR 结合分子。未来可通过引入实验确认的非活性(inactive)化合物来扩充数据集,并将任务扩展为多分类问题,使模型能区分三类药理活性:μOR 的非活性化合物、激动剂与拮抗剂。此外,当前模型仅考虑能结合(binder)的分子。
未来可设计两步预测流程:
第一步预测化合物是否能与 μOR 结合(binder / nonbinder 分类),
第二步再判断其药理作用类型(激动剂或拮抗剂)。
在第一步中,可结合分子对接模拟与虚假化合物(decoy set)以增强模型性能。值得注意的是,以经验阈值(如 Emax 14%)或文献结论来划分激动剂与拮抗剂,可能过度简化配体的药理特征,忽略了从**反向激动(inverse agonism)到完全激动(full agonism)的连续谱,例如弱部分激动剂与拮抗剂之间的微妙差别。理论上,可构建回归模型直接预测 Emax 值,但该方案受限于不同研究间报告值差异较大,这些差异可能源于受体表达水平、细胞系类型及实验条件的变化。
尽管存在上述局限,ET 与 MPNN 模型在留出测试中均取得 超过 91% 的 AUC,表明它们已能准确区分 μOR 小分子配体的固有活性类型。
此类模型在未来具有重要应用潜力:
一方面,可辅助发现新的 μOR 拮抗剂以应对阿片类药物过量问题;另一方面,也可用于评估尚未表征的化学物质,以提前识别其潜在的公共安全风险。