NBE 2025 | PepMimic: 通过结合界面模拟的肽设计
近年来,基于肽的药物因其高特异性、强亲和力以及较低毒性而成为靶向治疗的重要方向。与小分子和抗体相比,肽分子不仅具备更好的细胞通透性和潜在的口服可利用性,还能通过精确调控生物通路展现出卓越的治疗优势。然而,传统的肽设计依赖专家知识与实验筛选,不仅效率低下,还难以应对复杂的非连续结合界面。
发表于Nature Biomedical Engineering 2025的研究提出了创新方法PepMimic,通过模拟靶蛋白与已知结合物的结合界面,实现了全原子水平的肽分子序列–结构联合设计。该方法在PD-L1、CD38、BCMA、HER2和CD4等多个关键靶点上成功生成高亲和力肽,其中部分候选的解离常数(KD)达到

获取详情及资源:
0 摘要
肽在靶向治疗中具有显著优势,包括口服可利用性、细胞通透性以及高度特异性,这些特征使其区别于传统的小分子和生物制剂。在此研究中,开发了一种人工智能算法PepMimic,其核心思想是通过模拟靶点与已知结合物之间的结合界面,将已知受体或现有抗体转化为短肽结合物。PepMimic被应用于多个重要药物靶点,包括PD-L1、CD38、BCMA、HER2和CD4。表面等离子体共振成像结果显示,约8%的肽分子表现出在
此外,PepMimic还被用于缺乏现有结合物的靶蛋白。其策略是先利用现有算法设计蛋白质结合物,然后通过模拟这些人工界面进一步生成肽分子。在乳腺癌、骨髓瘤和肺癌的小鼠模型中,通过尾静脉注射对排名靠前的候选肽进行了广泛验证。实验结果表明,这些肽能有效结合细胞膜,并显示出在临床诊断成像和靶向治疗应用中的巨大潜力。
1 引言
目前已有超过80种基于肽的治疗药物获批,用于糖尿病、癌症、骨质疏松症、多发性硬化症、HIV感染以及慢性疼痛等多种疾病。肽类治疗的主要优势在于能够实现高特异性和高效力,从而精确调控生物通路。与传统小分子和抗体相比,肽药物通常表现出更佳的安全性和治疗特征,原因包括更低的毒性、更强的细胞通透性以及潜在的口服可用性。
在肽设计过程中,一个关键观察是大多数蛋白靶点往往已有结合物存在,这些结合物可能来源于天然,或通过大规模文库的高通量筛选及动物免疫实验获得。此外,机器学习算法也已被用于设计与蛋白靶点具有高亲和力的小型结合物。这些已知的结合物为功能性肽的设计提供了宝贵的相互作用模板。药物化学家长期以来通过模拟靶蛋白的天然结合物来开发肽药物,而模拟特定蛋白–蛋白相互作用的肽分子也被广泛用于研究细胞功能调控。由此,通过模拟现有结合物来设计肽分子自然成为一种极具吸引力的策略。
然而,当前方法存在两大局限。首先,这类设计通常依赖专家知识,需要深刻理解氨基酸间的相互作用模式,并往往依赖实验手段人工提取结合基序或界面锚定氨基酸。其次,药物化学家更擅长模拟连续氨基酸序列,而难以处理非连续结合界面,这限制了已知结合物界面的利用效率。现有模型大多先生成骨架,再进行反向折叠,或同时设计骨架与序列,再借助第三方软件补充侧链。这些方法本质上是建模氨基酸类型与三维骨架的条件概率,却忽略了侧链几何的重要性。氨基酸侧链的原子位置决定了氢键和π–π堆积等关键相互作用,因此全原子建模对于精确捕捉几何约束至关重要。虽然AlphaFold和RosettaFold等突破性工具实现了高分辨率结构预测,但要将其适配至生成模型,尤其是在界面模拟肽设计的场景中,仍然是巨大挑战。
近期的工作如DiffPepBuilder、PepFlow和PPFlow开始考虑侧链,但仍依赖于扭转角度作为主要表示方式。这种间接编码方式难以让模型显式学习原子级相互作用,更不具备利用现有界面来指导生成的能力,这正是当前方法的一大限制。
在此背景下,研究者提出了PepMimic,一种基于人工智能的创新方法,能够在给定蛋白靶点和已知结合物的条件下,进行全原子肽结合物的序列–结构联合设计。该模型可以自动生成模拟结合界面的肽分子,即便这些界面涉及非连续氨基酸。PepMimic通过在潜在空间中学习氨基酸类型与全原子结构的联合分布,提供了更精确高效的全原子肽设计策略,尤其适合用于界面模拟型肽设计。其关键优势在于引入基于能量的界面相似性引导,利用对比模型学习界面特征,从而在生成过程中直接整合相似性信号,实现灵活而高效的联合设计。
在评估中,研究团队首先在93个蛋白–肽复合物上进行了无界面引导的模拟测试,将PepMimic与dyMEAN、HSRN和RFDiffusion等前沿方法进行对比。随后,PepMimic被应用于五个药物靶点(PD-L1、CD38、BCMA、HER2和CD4),设计出384条肽分子,均基于天然受体、抗体或纳米抗体的界面模拟。此外,还设计了针对TROP2和CD38的肽,部分肽分子的结合亲和力达到甚至超过
这些验证结果充分证明了PepMimic能够成功设计界面模拟型肽分子,不仅为功能性肽的发现提供了新途径,也展现了其在生物学研究和治疗应用中的巨大潜力。
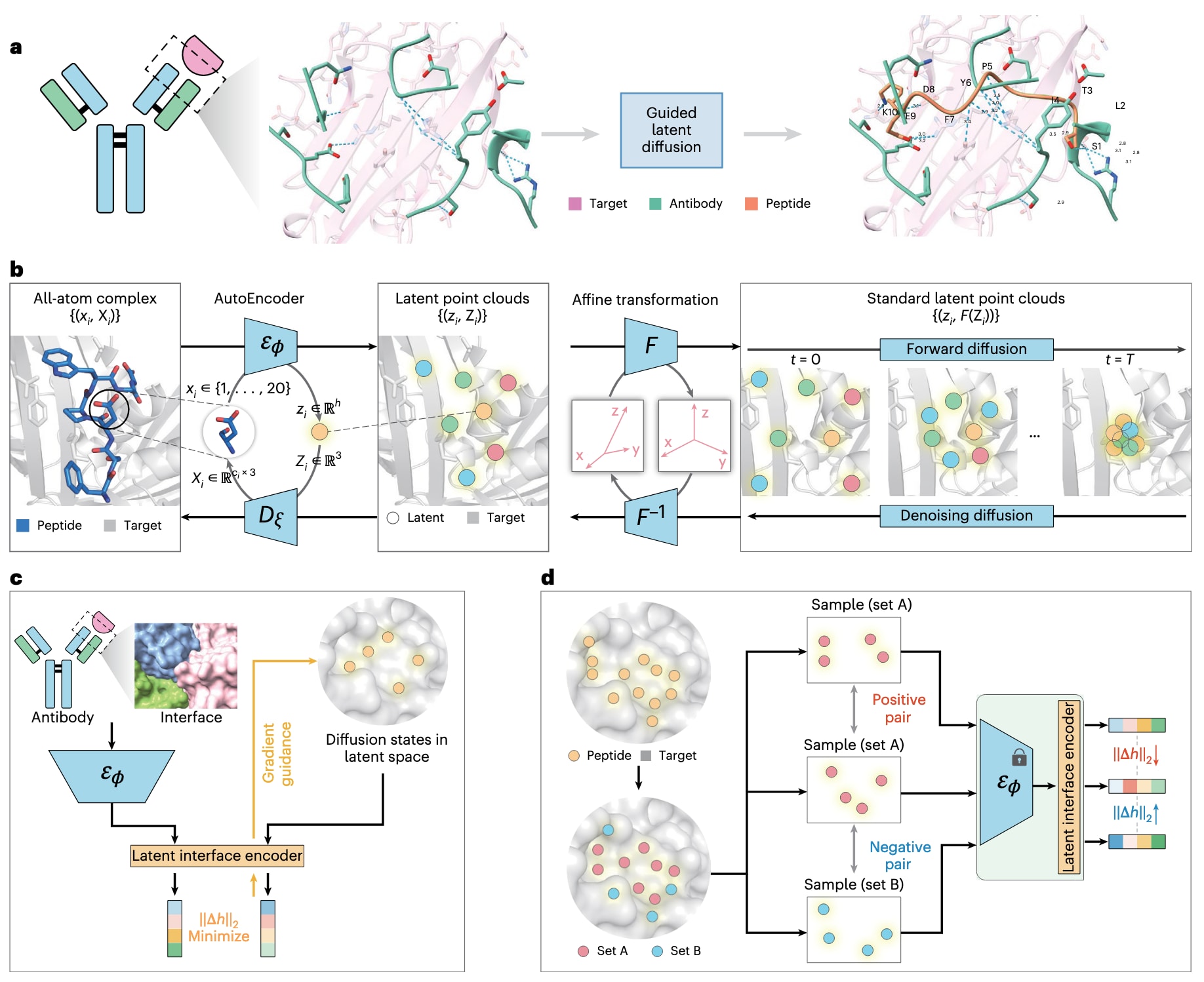
图1 | PepMimic算法示意图 在给定一个靶蛋白和特定结合物的条件下,PepMimic通过整合三个核心模块——全原子自编码器、潜在扩散模型和潜在界面编码器——来设计模拟结合界面的肽分子。a,界面提取与引导:从需要模拟的结合物(例如抗体)中提取界面,用于在潜在空间中引导扩散过程。设计得到的肽结合物通过自动识别并线性连接关键相互作用,从而模拟参考结合界面。b,生成组件:PepMimic的生成部分由全原子自编码器和潜在扩散模型组成。首先,自编码器建立起全原子几何与残基层面低维潜在表示的可逆映射。随后,在潜在空间中训练扩散模型,同时生成E(3)-不变特征与E(3)-等变特征,形成潜在点云。最终,肽分子的残基类型和全原子几何结构分别由自编码器从不变特征和等变特征中解码得到。c,界面编码与梯度引导:提取的参考界面通过潜在界面编码器转化为向量表示,从而能够利用梯度引导最小化已知界面与生成界面在潜在空间中的距离,实现高效对齐。d,界面编码器的训练数据构建:通过对蛋白–肽界面进行下采样生成训练数据。肽残基被随机划分为两个不重叠的集合A和B,其中集合A被下采样两次以形成正样本对(因其共享部分氨基酸),而集合B仅下采样一次以构造负样本。训练目标是最小化正样本对表示之间的距离,并最大化负样本对表示之间的距离,从而优化潜在界面编码器的判别能力。
2 结果
2.1 PepMimic总体框架
PepMimic由三个核心模块构成:全原子自编码器、潜在扩散模型以及用于引导模拟设计的界面编码器(Fig. 1a)。其训练目标是在特定结合位点同时联合设计肽结合物的序列与全原子结构(Fig. 1b)。
全原子编码器将与每个氨基酸相关的所有原子压缩为一个嵌入向量,从而建立起肽分子全原子几何与氨基酸层面潜在点云之间的可逆映射。其训练损失是基于潜在嵌入重建氨基酸的类型及其原子坐标。接着,训练了一个概率扩散模型,用于逐步将标准高斯噪声转化为条件于结合位点的合理潜在点云。在生成过程中,潜在点云首先由高斯噪声在潜在空间中采样,经扩散模型变换后,再通过解码器模块解码为全原子肽结构。为增强模型鲁棒性,研究还引入了一个大规模增强数据集(Methods),在对高质量蛋白–肽复合物进行微调之前,先进行预训练。
在模拟已知结合物时,PepMimic采用相同的编码器函数,将参考结合界面的全原子几何投射到潜在空间中的低维点云。其中结合界面被定义为靶蛋白与结合物之间Cβ距离低于10 Å的残基。基于这些点云,界面编码器进一步将其表示为一个数值向量,参考结合物与生成肽分别对应一个向量,两者在向量空间中的距离揭示了它们在同一蛋白靶点上的结合模式相似性(Fig. 1c)。因此,可以利用这两个向量之间的距离,通过梯度下降引导生成性扩散过程,逐步将潜在点云对齐至目标界面。
为实现有效引导,潜在界面编码器的训练目标是赋予其区分相似与不相似界面的能力(Fig. 1d),采用对比学习进行优化。这样一来,模型便具备了在不同界面间的泛化能力,无需针对新参考结合物重新训练即可完成模拟。
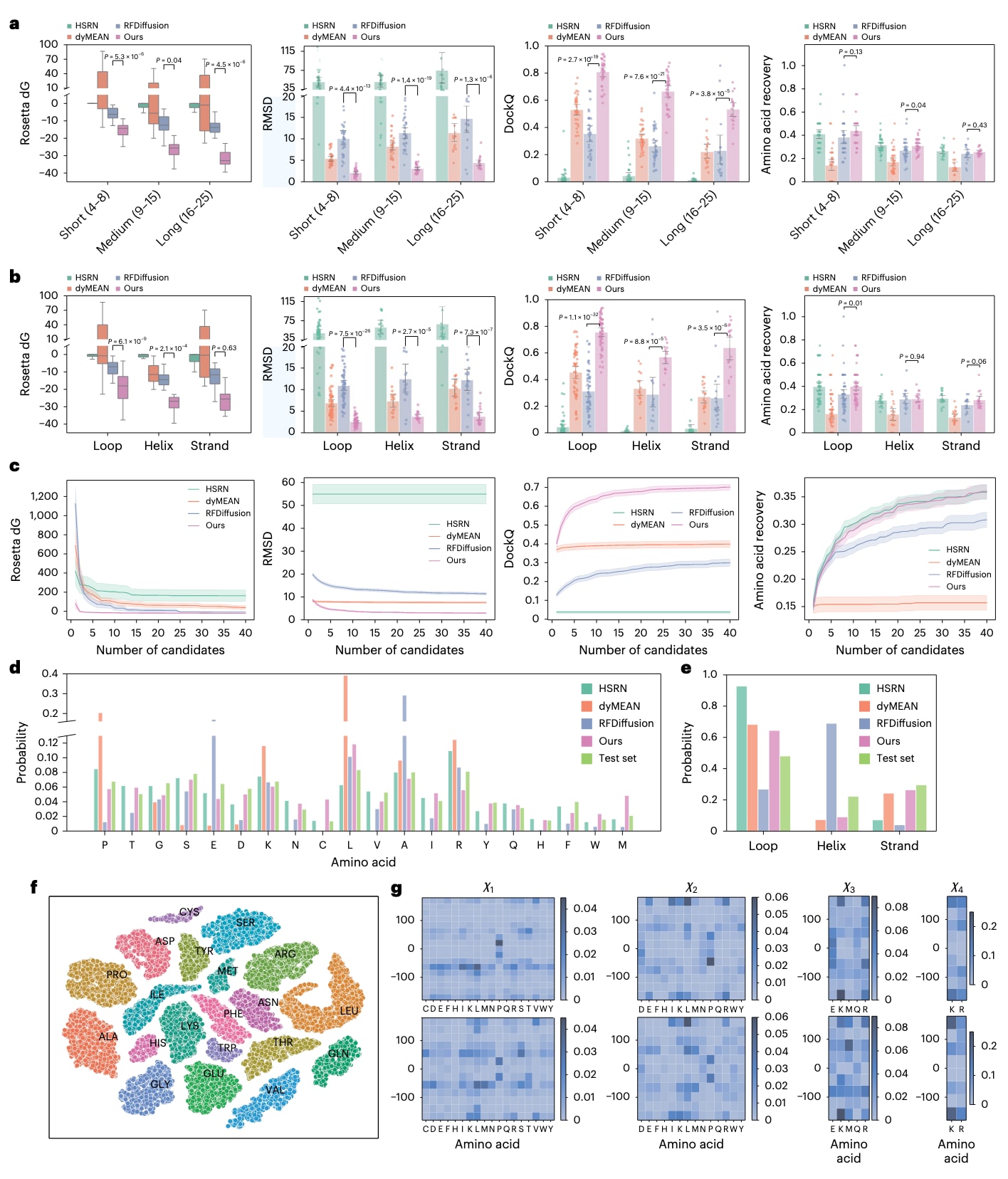
图2 | 基于文献收集的非冗余测试集上靶点特异性肽设计的评估 该图展示了不同模型在靶点特异性肽设计任务中的表现,评估指标包括Rosetta界面能量(ΔG)、Cα坐标的均方根偏差(r.m.s.d.)、全原子DockQ以及氨基酸恢复率(AAR)。a,长度分组表现:将测试集中的天然肽分为短肽(N=34)、中等长度肽(N=35)和长肽(N=17)三类。箱线图展示了数据的最小值、第一四分位数、中位数、第三四分位数和最大值;柱状图则给出平均值±标准误差(s.e.m.)。显著性通过双尾独立样本t检验确定,P<0.05视为统计学显著。b,二级结构分组表现:依据参考肽的二级结构分类,包括环(loop, N=59)、螺旋(helix, N=18)和链(strand, N=16)。同样使用箱线图与柱状图表示分布及均值差异,统计检验方式与(a)一致。c,采样数量饱和趋势:展示随着生成候选数量增加,指标表现逐渐饱和的趋势,轨迹以平均值±标准误差(s.e.m.)表示。d,氨基酸类型分布:比较不同模型生成的氨基酸类型分布与参考分布之间的差异,揭示模型在氨基酸生成偏好上的差异性。e,二级结构分布:比较生成肽与参考肽在二级结构类型上的概率分布差异。f,潜在空间中的t-SNE可视化:展示氨基酸在潜在空间中的表示聚类,类别调整兰德指数(ARI)=1.0。表示距离与BLOSUM62替代矩阵的Pearson相关系数为−0.47 (P<0.001),表明潜在空间保留了氨基酸的功能与理化相似性。g,侧链扭转角分布(χ1–χ4):上排为测试集中的参考分布,下排为PepMimic生成肽的分布。两者高度一致,说明PepMimic能够生成准确的全原子几何结构。
2.2 在靶点特异性肽设计中的表现
为了评估PepMimic在肽结合物设计方面的能力,研究团队在一个包含93个精心整理蛋白复合物的非冗余测试集上进行了验证。这些复合物均来自文献,并排除了通过mmseqs2判定序列同一性超过40%的靶蛋白,以确保对不同靶点的泛化能力。对于每个靶蛋白,所有基线模型均生成40个候选肽进行比较。
生成的三维肽分子通过以下指标进行评估(Methods):(1) Rosetta界面能量(ΔG),(2) Cα坐标的均方根偏差(r.m.s.d.),(3) 全原子界面的DockQ得分,以及(4)氨基酸恢复率(AAR)。除ΔG外,其余指标均通过与测试集中已知肽结合物对比计算。对比方法包括当前最前沿的蛋白设计模型RFDiffusion,以及两种针对抗体CDR区的序列–结构联合设计模型HSRN和dyMEAN。其中HSRN与dyMEAN均重新基于蛋白–肽复合物训练,与PepMimic保持一致;而RFDiffusion因未开源训练代码,仅使用其官方权重和管线进行设计。所有模型生成的候选肽在计算ΔG前均经Rosetta快速松弛一次,以保证公平性。
结果显示,PepMimic在所有指标上均稳定优于基线模型,且不受肽长度影响(Fig. 2a)。随着肽长度增加,氨基酸恢复率有所下降,这是由于更大的组合搜索空间带来难度提升;而Rosetta ΔG则相反,较长肽通常形成更大界面,因此界面能量更低(Supplementary Fig. 1)。基于DSSP算法识别的不同二级结构类别的分组评估进一步验证了PepMimic的优越性(Fig. 2b)。值得注意的是,在无二级结构的肽中,PepMimic的AAR为40%,显著高于RFDiffusion的33%(P ≤ 0.05),这与RFDiffusion预训练于一般蛋白、偏向具有稳定二级结构的靶点一致。
随着采样数目的增加,指标表现逐渐趋于饱和,30个样本以上的采样池即可保证至少得到一个接近模型上界性能的候选肽(Fig. 2c)。HSRN虽然在AAR上表现较好,但在生成准确结合结构方面表现不足;而dyMEAN则因缺乏多样性,导致指标在少量样本时便快速饱和(Fig. 2c)。
在氨基酸与二级结构分布方面,PepMimic生成的肽与测试集更为接近,其KL散度仅为0.06,而dyMEAN(0.89)和RFDiffusion(0.38)更倾向于生成脯氨酸、谷氨酸、赖氨酸和丙氨酸(Fig. 2d)。在二级结构分布上,PepMimic略偏向环状结构,而RFDiffusion则强烈偏向螺旋肽,这与文献报道一致(Fig. 2e)。
进一步的t-SNE可视化表明,PepMimic在氨基酸潜在表示中形成了连续且结构良好的潜在空间,不同氨基酸聚类紧密可分(ARI=1.0),并且具有相似理化性质与生物功能的氨基酸在潜在空间中邻近分布。例如,天冬酰胺、苏氨酸和谷氨酰胺(均为极性不带电侧链)紧密聚集;酪氨酸、苯丙氨酸和色氨酸(芳香族侧链)也彼此邻近。氨基酸间潜在空间的距离与BLOSUM62替代评分呈中度相关(Pearson r=−0.47, P<0.001),与基于分子指纹Tanimoto相似性的化学相似性亦呈一致趋势(r=−0.50, P<0.001)。这类相似性驱动的潜在空间有助于扩散过程生成准确或功能相近的氨基酸。
此外,PepMimic生成的侧链扭转角(χ1–χ4)分布与测试集高度一致(Fig. 2g),表明其具备生成准确全原子几何结构的能力。
总体而言,这些广泛的评估结果充分证明,PepMimic在给定特定结合位点条件下,能够稳定实现肽结合物的序列与全原子结构的联合设计,在各项计算指标上显著优于现有最先进方法。
2.3 界面模拟性能
为了评估在潜在引导机制下进行界面模拟生成的效果,研究首先提出了一个计算指标——Interface Hit,用于衡量设计界面与目标界面对齐的质量(Fig. 3a)。在一对三维界面中,假设靶蛋白的结合位点包含
为了验证这一指标的可靠性,研究在测试集中通过下采样构建了具有已知氨基酸对齐关系的界面对(Fig. 1d, Supplementary Methods 1.2.1)。结果显示,interface hit与重叠氨基酸数量高度相关(Pearson相关系数0.99, P<0.001),证明其能有效度量界面对齐程度。
接下来,为评估PepMimic模拟给定界面的能力,研究利用界面划分和下采样策略(Supplementary Methods 1.2.2),检验模型是否能区分相似与不相似界面对。结果表明,界面潜在表示之间的距离随重叠氨基酸数量的增加而下降,哪怕仅有一个重叠氨基酸对,也能显著(P≤10⁻⁵)降低距离(Fig. 3c, 左)。潜在编码器能有效区分正样本(超过两个氨基酸对重叠)与负样本(无重叠),其表示距离分布差异显著(P≤10⁻⁵),证明其可作为界面相似性的可靠可微代理(Fig. 3c, 右)。
在引导生成实验中,研究选取了PD-L1的四种结合伙伴——其天然受体PD1、由MaSIF套件设计的小蛋白结合物、纳米抗体以及抗体durvalumab(Fig. 3d)。它们与PD-L1的结合亲和力分别为:PD1(KD=8.2 μM)、小蛋白(374 nM)、纳米抗体(44 nM)、抗体(0.667 nM)。实验结果表明,引导过程显著降低了界面表示距离(P<0.01),尤其在100步扩散中的前50步后效果尤为明显。生成肽分子的interface hit分布也进一步确认了潜在引导的有效性(Fig. 3e, Supplementary Fig. 2a)。不同结合物的引导效果存在差异:PD1和小蛋白的界面更容易模拟,而纳米抗体与抗体因界面更大更复杂,涉及多环结构和二级结构接触点,模拟难度更高(Fig. 3f, Supplementary Fig. 2b)。
为了公平对比,研究还设计了一种后排序(postranking)的方法,将HSRN、dyMEAN和RFDiffusion生成的候选肽根据界面相似性排序,取前K条作为结果。结果显示,虽然经过后排序,基线模型的表现有所提升,但其interface hit率仍显著低于PepMimic(Supplementary Fig. 3e),且界面表示距离下降趋势也不及PepMimic (Supplementary Fig. 3a–d)。
此外,对比有无引导的ΔG评估结果表明,引导生成的候选肽在FoldX和Rosetta计算下表现出更优或相当的ΔG值(Fig. 3g),证明潜在引导不仅提升了界面模拟精度,同时保持了生成结果的合理性。
研究进一步探讨了参考界面特征与interface hit分布之间的相关性,旨在识别哪些类型的界面更容易在计算机模拟中被模仿。分析共涉及九项界面特征:螺旋长度、螺旋比例、环长度、环比例、链长度、链比例、最大片段长度、片段数量以及界面氨基酸之间的平均距离,二级结构均通过DSSP算法分类(Fig. 3h, Supplementary Fig. 4a–d)。
在四个不同靶蛋白(PD-L1、HER2、病毒神经氨酸酶NA和CD38)上的结果表现出一致趋势:生成肽的interface hit与界面残基平均距离呈负相关,与片段数量和链比例呈正相关(Fig. 3h)。平均残基距离越小,界面越紧凑,结合越紧密,这使得生成模型更容易将相邻的结合氨基酸连接为一条肽,从而提升模拟效果。统计结果还显示,片段数量与平均残基距离呈负相关(Supplementary Fig. 4e),这进一步降低了模拟设计的难度。然而,当片段分布稀疏、界面庞大而复杂时(如PD-L1的抗体结合界面),肽模拟设计就会变得更具挑战性。
链状结构提供了线性延展的相互作用表面,这种模式相对简单,生成模型更容易模仿,因此链比例升高往往对应更高的interface hit。然而需要特别注意的是,线性肽分子在自然条件下很少形成稳定的链状二级结构。因此,这类具有高interface hit的链模拟结果很可能是伪象,这一点将在后续的表面等离子体共振成像(SPRi)实验中得到验证。
其余特征未表现出显著相关性,或在不同靶点间呈现不一致的趋势(Supplementary Fig. 4a–d)。
总结而言,更紧凑的界面、更高的片段数量及链比例往往使界面更易于被PepMimic准确模拟,而分散而复杂的界面则显著增加了模拟难度。
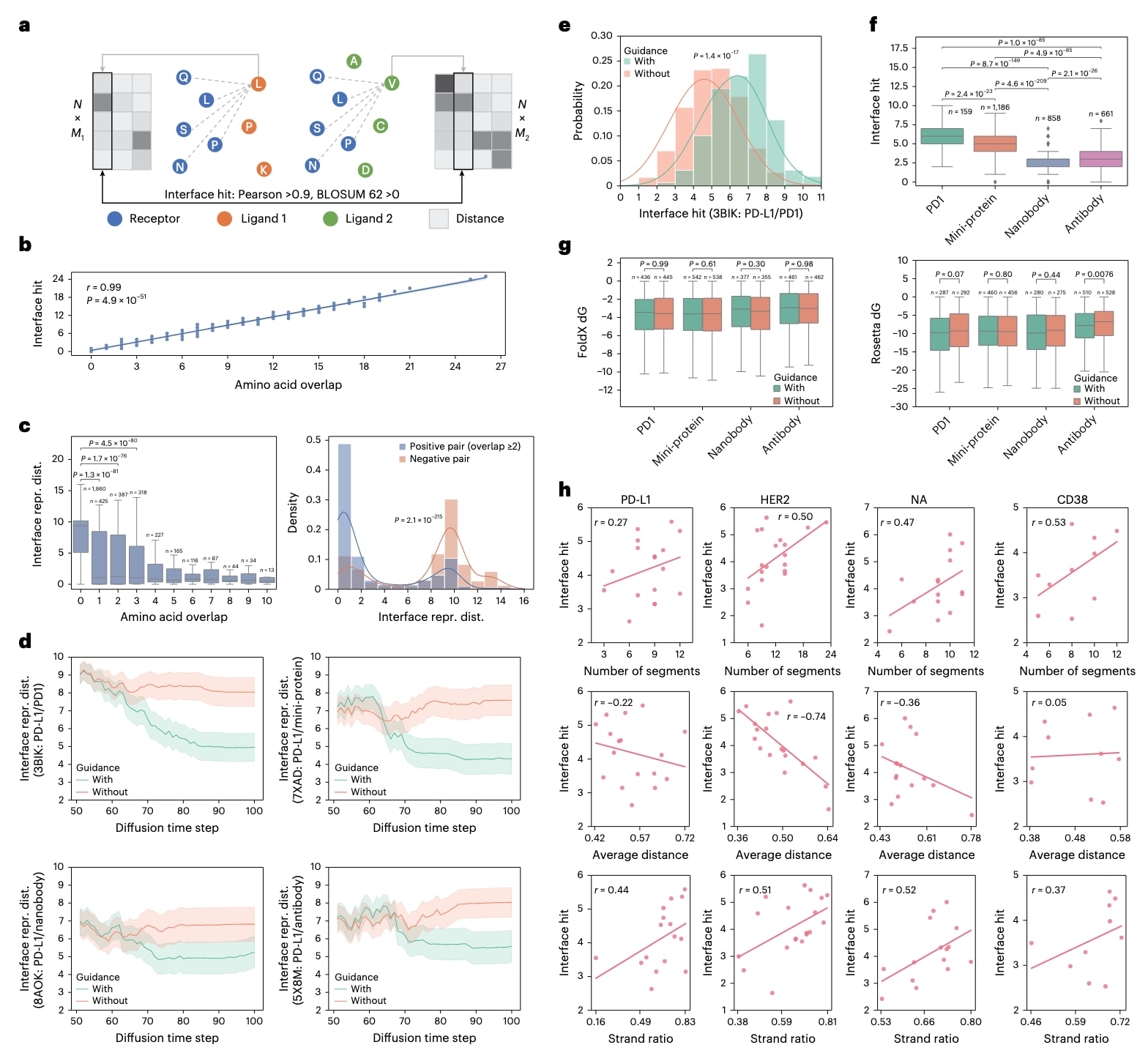
图3 | 潜在界面编码器的评估及PepMimic在界面模拟能力上的分析 a,Interface hit的定义:用于定量衡量两个界面相似性的指标。假设靶蛋白结合位点包含
2.4 模拟靶蛋白已知结合物的结果
研究在五个靶点(PD-L1、CD38、BCMA、HER2和CD4)上进行了SPRi实验,以评估生成肽的结合亲和力(Methods)。对于每个靶点,收集了包含靶点及其结合物的已解析三维复合物(Supplementary Tables 1–5)。这些结合物包括抗体、纳米抗体和天然受体,并作为PepMimic的界面模拟参考。利用Rosetta、FoldX以及AlphaFold Multimer对生成的候选肽进行排序,选取前384个进入SPRi实验,借助高通量能力进行筛选。实验中,将KD低于100 nM的候选视为成功命中。
结果显示,PepMimic在CD38、HER2和CD4的成功率超过10%,而在PD-L1和BCMA上则相对较低(Fig. 4a)。在模拟不同结合物类型时,抗体与天然受体的成功率约比纳米抗体高2%(Fig. 4b)。对于成功的候选肽,结构层面上存在一定聚类,而序列则展现出更高的多样性(Supplementary Fig. 5)。
进一步分析表明,界面命中(interface hit)数量与螺旋或链上的成功率呈中度负相关(P<0.05),而环(loop)上的命中则无显著相关性(P=0.32)(Fig. 4c)。这可能源于线性肽的构象柔性,使其难以稳定形成螺旋或链状二级结构。尤其是连续螺旋或链上的命中与成功率呈更显著的负相关(Fig. 4d, P<0.05)。这与之前的计算分析不同:虽然in silico结果显示interface hit与链比例正相关(Fig. 3h),但SPRi验证表明这些命中可能是伪象,因为缺乏周围链片段的环境支撑,生成肽难以真正形成稳定链结构。这也解释了PD-L1和BCMA成功率较低的现象,它们的参考结合物界面中螺旋与链结构比例更高(Fig. 4e, P<0.05),从而增加了模拟难度。此外,PD-L1的结合位点较为平坦,本身已知难以找到小分子结合物。
统计结果还表明,当生成肽通过环结构来模拟界面,或参考界面主要由环组成时,成功率更高(Fig. 4f)。其余界面特征未观察到显著相关性(Supplementary Figs. 6, 7a,b)。在能量指标方面,Rosetta界面能量对区分正负结合物有中等效果(Fig. 4g),而FoldX能量、pLDDT等常用指标则未展现出明显区分能力(Supplementary Fig. 7c–f)。
在实验中,研究选择了一条与PD-L1结合亲和力良好的候选肽RIWAYRKFMD (KD=43.0 nM)进行进一步验证。该肽被标记为FITC,激光共聚焦成像证实FITC标记的肽能有效结合PD-L1阳性的H1975细胞,表现出高亲和力和高特异性(Fig. 4h, Supplementary Fig. 8a)。进一步的体内实验中,将该肽与DOTA偶联以螯合放射性核素^68Ga,进行小动物PET-CT成像。静脉注射30分钟后,在MDA-MB-231肿瘤中的摄取量是A549肿瘤的8.85倍(6.02 ± 0.4% ID cm⁻³ 对比 0.68 ± 0.1% ID cm⁻³)(Fig. 4i)。肿瘤–血液比值分别为A549 (0.75 ± 0.09)与MDA-MB-231 (6.67 ± 0.26)。随着时间推移,大部分探针在60分钟内经肾脏清除。免疫荧光染色进一步证实该肽能特异性识别PD-L1阳性肿瘤,而对PD-L1阴性的A549几乎无结合(Fig. 4i)。这说明该肽在体内外均表现出卓越的靶向性,显示了其在癌症影像中的应用潜力。
类似的体内外验证还在CD38和HER2的肽模拟物中完成(Supplementary Figs. 8b,c, 9)。此外,研究构建了一个容量为
在对不同靶点成功设计的进一步探索中,研究发现了多样化的界面模拟模式,包括:(1) 单条肽跨越β-折叠片段(Fig. 5a);(2) 模拟连续β-折叠之间的环(Fig. 5b);(3) 单条肽连接线性分布的环片段(Fig. 5c);(4) 跨越两条平行位移的β-折叠(Fig. 5d);(5) 模拟抗体CDR区(Fig. 5e);以及(6) 连接两个独立界面(Fig. 5f)。
综上,PepMimic不仅能在多个靶点上生成高亲和力肽结合物,还展现出高度的多样性和特异性,其体内外验证结果进一步凸显了其在靶向治疗与分子影像中的应用潜力。
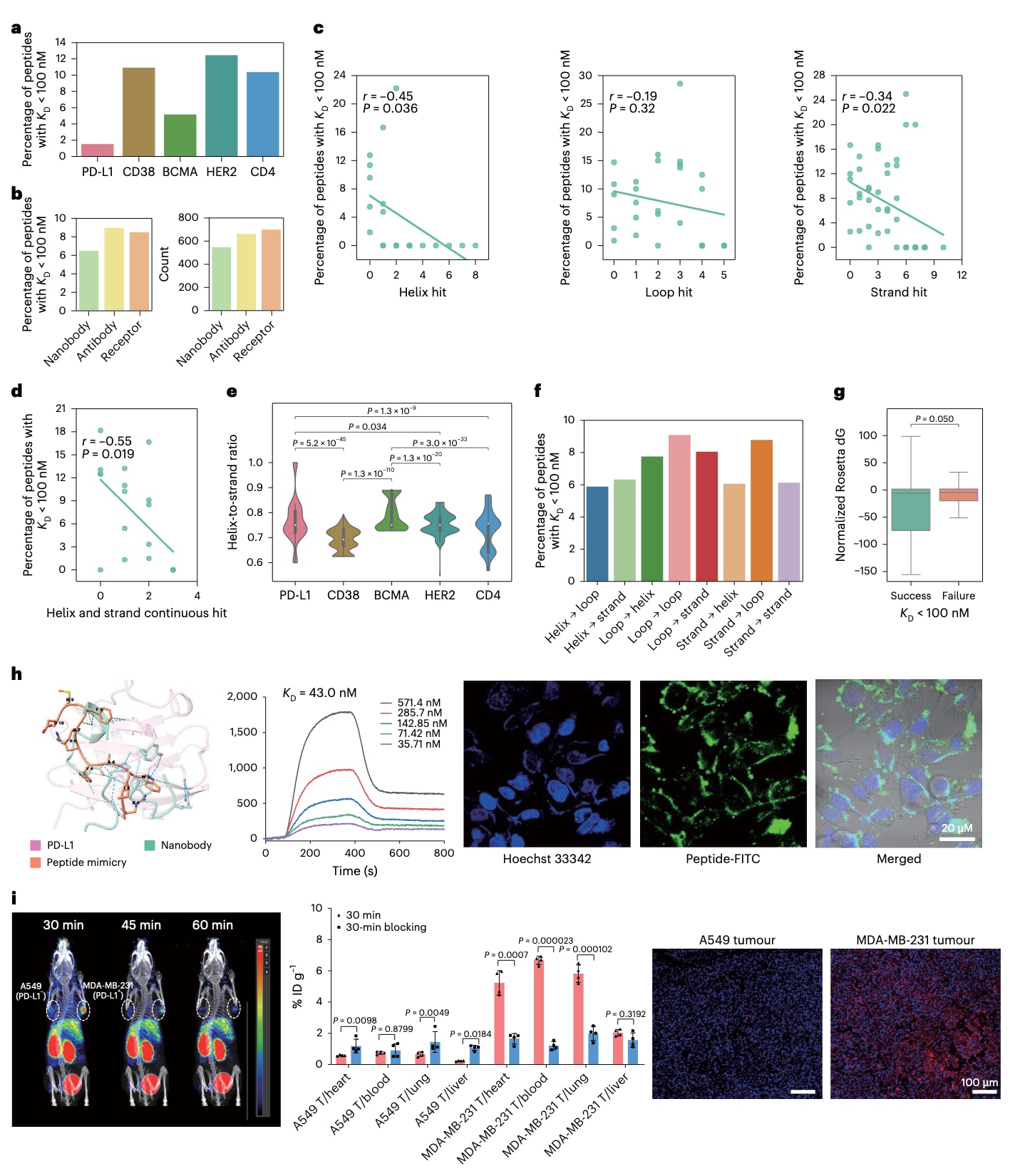
图4 | 肽模拟的SPRi实验、细胞实验与体内验证结果及分析 a,靶点成功率:在不同靶蛋白上的SPRi实验成功率统计(KD < 100 nM),展示各靶点的整体表现。b,结合物类型差异:按参考结合物类别(抗体、纳米抗体、天然受体)对成功率与实验测试候选数量进行比较。结果显示抗体与天然受体模拟的成功率略高于纳米抗体。c,成功率与界面命中的相关性:利用“interface hit”算法评估参考结合物上的二级结构(螺旋、链、环),统计其命中数量与成功率的Pearson相关性。结果显示螺旋与链的命中数量与成功率呈负相关,而环无显著相关性。d,连续命中对的相关性:进一步分析参考结合物中连续螺旋与链命中数量与成功率的相关性,显示更强的负相关性,说明线性肽难以稳定形成连续二级结构。e,界面结构组成:按靶点统计参考结合物界面中螺旋与链的比例。PD-L1与BCMA的界面中螺旋和链比例更高,解释了其成功率较低的原因。f,二级结构映射类型:统计不同二级结构映射关系下的成功率。例如“Helix → loop”表示生成肽以螺旋结构模拟参考界面的环。结果表明使用环进行模拟时成功率更高。g,能量区分度:对比成功候选(N=156)与失败候选(N=1,762)的Rosetta界面能量(已按参考结合物平均能量归一化)。成功组能量显著低于失败组,表明Rosetta能量对区分正负结果具有一定效力。h,体外特异性验证:SPRi与共聚焦成像结果表明,筛选出的肽(RIWAYRKFMD)在PD-L1阳性H1975细胞中表现出特异性结合。比例尺20 μm,共聚焦实验重复3次。i,体内成像与组织验证:注射^68Ga–DOTA–RIWAYRKFMD后,在双侧A549与MDA-MB-231肿瘤荷瘤小鼠中进行小动物PET成像。结果显示MDA-MB-231肿瘤中摄取显著高于A549,肿瘤–正常组织比值差异显著。免疫荧光进一步确认该肽特异性识别PD-L1阳性肿瘤。数据以平均值±标准差(N=3)表示,统计学显著性通过Welch’s t检验评估(P<0.05)。整体结果表明,PepMimic生成的肽在体外与体内均展现出高亲和力与高特异性,验证了其在靶向治疗和分子影像中的潜力。
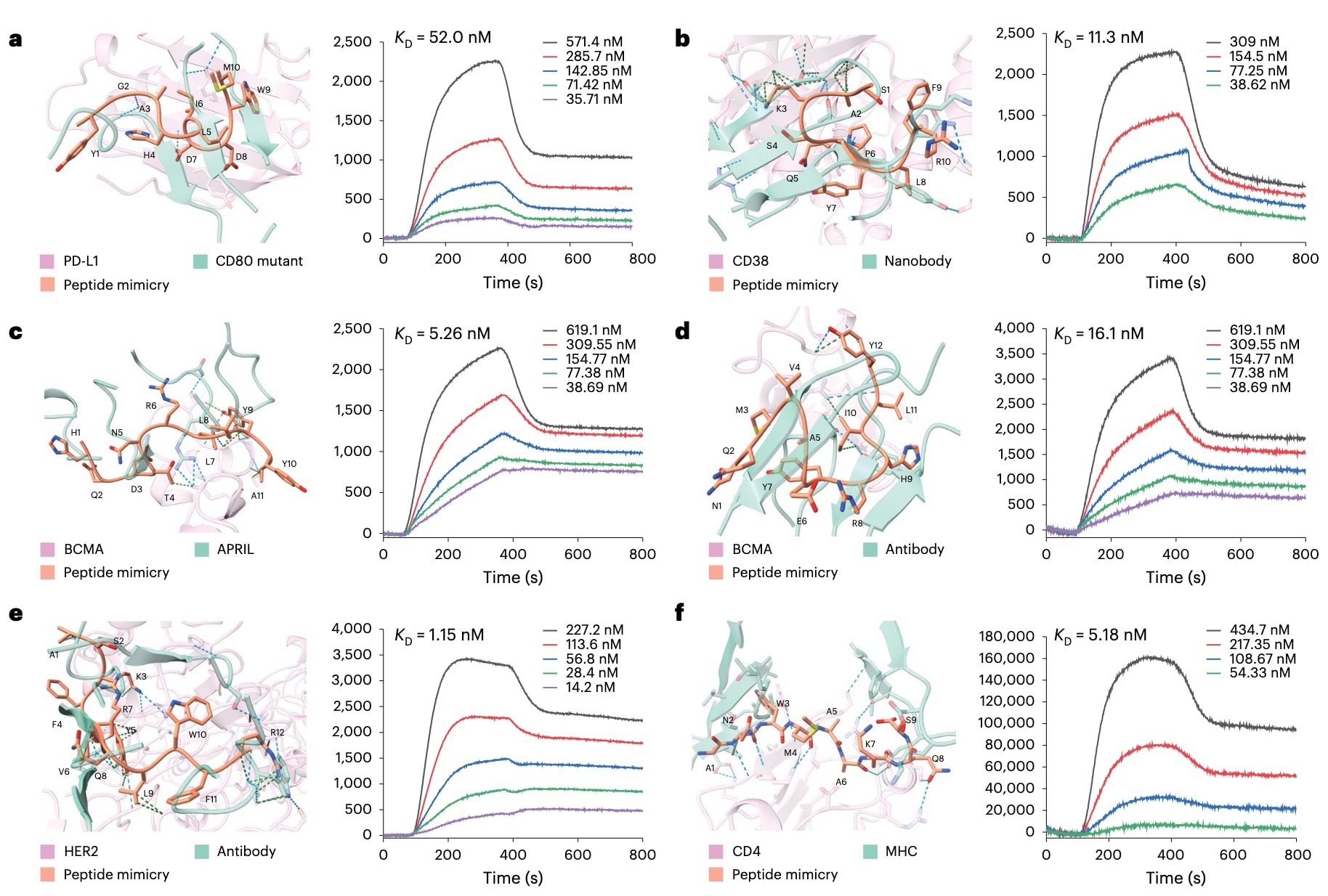
图5 | 不同靶蛋白上设计肽模拟物的结构与SPRi结果 a, PD-L1结合物:源自CD80突变体结合PD-L1 (PDB: 7TPS),设计肽的结合亲和力为KD = 52.0 nM。b, CD38结合物:源自纳米抗体结合CD38 (PDB: 5F1K),设计肽的KD = 11.3 nM。c, BCMA结合物:源自APRIL结合BCMA (PDB: 1XU2),设计肽的KD = 5.26 nM。d, BCMA结合物:源自抗体结合BCMA (PDB: 4ZFO),设计肽的KD = 16.1 nM。e, HER2结合物:源自抗体结合HER2 (PDB: 3N85),设计肽的KD = 1.15 nM。f, CD4结合物:源自MHC结合CD4 (PDB: 3T0E),设计肽的KD = 5.18 nM。
这些结果表明,PepMimic在不同靶点上均能生成高亲和力肽模拟物,其中部分候选肽的KD达到纳摩尔级甚至接近亚纳摩尔水平,充分展示了其在多靶点高效模拟结合界面方面的能力。
2.5 模拟AI生成结合物的结果
在前述研究中发现,即便在未使用引导的情况下,PepMimic在序列恢复率和结构r.m.s.d.方面依然优于RFDiffusion(Fig. 2a–c)。因此进一步探索了是否可以通过模拟RFDiffusion生成的肽与mini-binder来改进RFDiffusion本身的性能(Fig. 6a)。
研究选择了两个靶点——CD38与TROP2。依据已有文献的结构–功能分析手动选择了表位(Supplementary Table 6)。对于每个靶点,利用RFDiffusion生成了12,000个候选,并用Rosetta与FoldX进行排序,从中筛选前1,500个进入后续分析。结果表明,RFDiffusion生成的mini-binder在理化性质上与天然肽存在显著差异,包括等电点、pH 7.0下电荷状态及疏水性(Supplementary Fig. 11a–c),这些特征可能进一步增加其聚集倾向(Supplementary Fig. 11d)。此外,RFDiffusion更倾向生成螺旋结构,而抑制了链与环的比例,与天然肽差异明显(Supplementary Fig. 11e–g)。
基于这些mini-binder的PepMimic肽模拟结果显示:CD38的成功率约为16%,TROP2约为8%(KD<100 nM)(Fig. 6c)。进一步分析CD38的成功率后发现,使用AI生成的mini-binder作为参考时往往能获得更高的成功率(Fig. 6d)。推测原因在于AI生成的mini-binder通常在靶蛋白上形成更小、更紧凑的界面,相比抗体或纳米抗体这类天然结合物,对肽模拟施加的结构约束更少(Supplementary Fig. 11h)。
深入分析表明,生成的肽模拟在理化性质和二级结构上比AI生成的mini-binder更接近天然肽(Fig. 6e–g)。例如,在TROP2的实验中,一个高亲和力肽结合物的结构被解析,其在细胞实验中表现出极佳的特异性(Fig. 6h)。共聚焦成像验证了该肽在高水平表达TROP2的BxPC3细胞膜上的高效、特异结合(Fig. 6h, Supplementary Fig. 12a)。该肽还被用于肿瘤小鼠模型的体内验证,结果显示,静脉注射30分钟后,该探针在BxPC3肿瘤中的摄取量是A549肿瘤的16.18倍(6.15 ± 0.9% ID cm⁻³ 对比 0.38 ± 0.07% ID cm⁻³)(Fig. 6i)。双瘤小鼠模型中,BxPC3肿瘤侧出现明显放射性信号,而A549侧几乎无摄取。免疫荧光结果与PET–CT成像一致,证实了BxPC3中高表达TROP2,而A549中表达极低(Fig. 6i)。这些结果说明,^68Ga–DOTA–PDMWTEAQLRFL作为一种对TROP2具有高亲和力与高特异性的PET–CT探针,展现出在肿瘤成像和个体化治疗策略指导中的应用潜力。另一条结合TROP2的候选肽也在体内外实验中表现优异(Supplementary Fig. 12b–e)。
为对比,研究还利用容量为
综上,PepMimic不仅能成功模拟AI生成的mini-binder,还能产生更接近天然肽特征的候选物,并在体外与体内实验中展现出极高的亲和力和特异性,进一步证明了其在复杂靶点上的应用潜力。
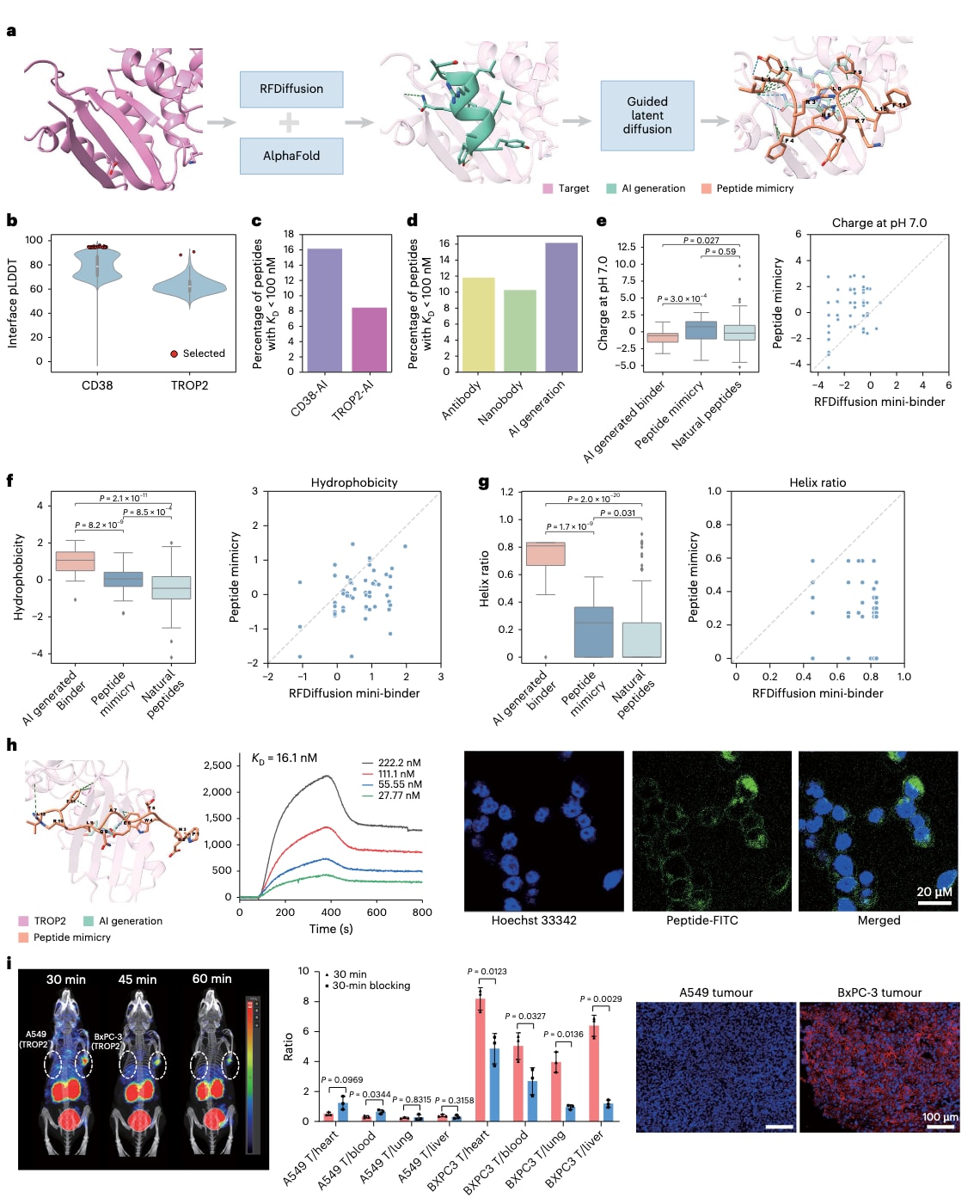
图6 | 基于AI生成结合结构的肽模拟结果 a,总体流程:在缺乏参考结合物的情况下,先使用RFDiffusion生成mini-binder,再通过AlphaFold Multimer进行筛选与结构预测。最终选取界面pLDDT评分较高的结合结构作为肽模拟设计的基础。b,界面pLDDT分布:展示了用于模拟设计的结合结构在CD38 (N=2939)与TROP2 (N=2787)上的界面pLDDT得分分布。界面pLDDT定义为距离结合伙伴6.0 Å以内的残基平均pLDDT。c,成功率:展示了该流程在不同靶点上的成功率(KD < 100 nM)。d,参考结合物类型分类结果:比较了不同参考类型的成功率,其中“AI generation”指利用上述流程得到的AI生成结合物。结果显示AI生成的mini-binder通常带来更高的成功率。e–g,理化性质与结构特征对比:对比了AI生成结合物(N=32)、肽模拟物(N=55)与天然肽(N=93)在等电点、GRAVY疏水性以及螺旋比例等特征上的差异。箱线图展示了分布范围(最小值、四分位数、中位数、最大值)。显著性通过双尾独立样本t检验确定(P<0.05)。散点图进一步描绘了通过模拟RFDiffusion生成mini-binder后,这些特性在肽模拟物上的变化。展示的肽模拟物均为SPRi验证成功(KD < 100 nM)的候选。h,TROP2特异性肽验证:SPRi结合曲线显示了候选肽与TROP2的结合动力学。共聚焦成像证实所选肽能高效、特异性结合TROP2阳性的BxPC3细胞。比例尺20 μm,共聚焦实验重复3次。i,体内成像与组织验证:在双侧A549与BxPC3肿瘤荷瘤小鼠中注射^68Ga–DOTA–PDMWTEAQLRFL后,30、45与60分钟的micro-PET成像显示,BxPC3肿瘤的探针摄取显著高于A549(6.15 ± 0.9% ID cm⁻³ 对比 0.38 ± 0.07% ID cm⁻³),肿瘤–正常组织比值亦显著提升。免疫荧光染色结果与PET成像一致,证实BxPC3肿瘤中高水平TROP2表达,而A549极低。数据以平均值±标准差(N=3)表示。整体而言,该策略不仅能利用AI生成的mini-binder作为参考生成高亲和力肽,还能得到在理化性质和结构上更接近天然肽的候选物,并在体外与体内实验中展现出卓越的特异性和应用潜力。
3 讨论
该研究开发了一种创新的几何生成模型,能够在原子级别上同时设计肽序列及其全原子结构。在给定复杂靶蛋白结构与已有结合物的情况下,PepMimic能够生成结合界面的肽模拟物,并再现关键分子相互作用。与AlphaFold3、RosettaFold All-Atom等专注于由序列预测结构的模型不同,PepMimic聚焦于序列与全原子结构的联合设计,因而特别适合用于复杂生物环境下的界面模拟。
在文献收集的高质量蛋白–肽复合物测试中,PepMimic超越了当前最先进的蛋白设计模型。SPRi实验结果显示,所设计肽在五个靶点上的平均成功率达到8%(KD<100 nM),远高于传统随机文库筛选。而在缺乏复合物结构的靶点上,研究提出了一条新流程:先利用已有的蛋白结合物设计模型生成结合界面,再由PepMimic设计肽模拟物。该流程在两个靶点上的平均成功率为14%,显示其在困难场景下的潜力。推动这一流程的动机在于mini蛋白的低可开发性,它们常因非人源氨基酸序列而引发免疫反应,而肽模拟物往往免疫原性更低,可以作为替代方案。
当前框架主要针对线性肽设计,但在方法和数据层面上天然可扩展至环肽设计。例如,在数据构建时,可筛选出末端距离较短的线性肽(如Cα–Cα距离<6 Å),作为类似环肽的训练样本;同时,还可利用AlphaFold预测的高置信度单体(pLDDT>80)来丰富训练数据。在算法层面,可以引入**无分类器引导(classifier-free guidance)**等条件生成技术,在采样过程中引导模型形成头–尾闭合。这些扩展将有助于序列–结构的联合设计,推动大环肽结合物的发展,展现出重要的研究前景。
PepMimic有望在生物学与治疗学中广泛应用,特别是在系统性地将现有抗体或纳米抗体药物转化为具有更高口服可利用性与效力的肽类似物方面。同时,该计算框架也可扩展至抗体或纳米抗体的设计,使其模拟天然或AI生成的结合物。该研究主要聚焦于肿瘤成像肽的开发,未来还将进一步探索抗癌治疗肽的潜力。
尽管PepMimic在湿实验验证中已取得积极成果,但仍存在若干亟需解决的局限:
首先,需要开发更有效的排序标准来区分真实结合物与模型生成的假阳性。在回顾性分析中发现,现有的统计能量评分、结构松弛稳定性以及基于深度学习的结构预测模型均未能显著区分结合物与非结合物。
其次,体内稳定性仍是扩展至治疗应用的一大挑战。改研究设计的肽主要优化于成像应用,易受体内降解(如蛋白水解)与快速肾脏清除的影响。因此未来需要设计或筛选更稳定的候选肽。
第三,目前方法仅依赖单一参考结合物。然而在实际情况中,常常存在多个结合物作用于同一靶点,它们可能提供互补的结构与功能优势。未来可扩展模型以同时利用多种参考,从中提取共享的相互作用模式,并整合不同特征,从而实现更鲁棒、更高效的肽设计。
综上,PepMimic不仅展示了强大的界面模拟与肽生成能力,也为未来多肽药物的开发提供了新的可能性。