Commun. Chem. 2025 | 从定制化虚拟分子数据库到真实有机光敏剂的迁移学习:催化活性预测研究
今天介绍的是一项发表于《Communications Chemistry》的研究,主题为**“从定制化虚拟分子数据库到真实有机光敏剂的迁移学习:催化活性预测”。这项工作针对长期困扰催化科学的核心问题——实验数据稀缺——提出了一种创新解决方案。研究构建了基于分子片段组合的虚拟分子数据库**,并利用这些人工生成的分子,通过图卷积神经网络(GCN)进行预训练,将学习到的结构特征迁移至真实体系中,实现了对有机光敏剂(OPSs)光催化活性的高精度预测。值得注意的是,即便这些虚拟分子在真实化学意义上并不典型,模型依然展现出优于传统数据库与Transformer模型的性能,体现出数据高效、跨域泛化能力强的特征。该研究不仅展示了虚拟数据库驱动的迁移学习在分子设计中的巨大潜力,也为数据稀缺条件下的催化建模与智能化分子发现提供了全新的思路。

获取详情及资源:
0 摘要
实验数据的稀缺长期限制了机器学习在催化研究中的深入应用。该研究展示了一种基于图卷积网络(Graph Convolutional Network, GCN)的模型,通过在一种非常规的分子拓扑指数上进行预训练,可有效用于预测催化活性——这一通常需要高度专业知识才能完成的任务。
在预训练阶段,研究者利用了定制化的虚拟分子数据库,这些数据库可通过系统化分子生成方法,或使用团队自主开发的分子生成器轻松构建。值得注意的是,所使用的虚拟分子中有94%–99%未被PubChem数据库收录,但基于这些分子训练得到的GCN模型,仍然显著提升了对真实有机光敏剂催化活性的预测性能。
结果表明,这种迁移学习策略能够高效地利用来自自建虚拟分子的可得信息,为机器学习在催化科学领域的应用提供了一条切实可行的途径。
1 引言
利用机器学习(Machine Learning, ML)预测催化性质已成为化学与材料科学中的重要挑战之一,它为基于数据驱动的催化剂设计与筛选开辟了新的可能。然而,将机器学习真正应用于实际的催化研究时,训练数据的匮乏成为制约其发展的主要障碍。为了解决这一问题,迁移学习(Transfer Learning, TL)被提出——该方法通过将模型在一个任务中获得的知识迁移到另一个任务,从而在数据量有限的情况下仍能显著提升模型性能。尽管迁移学习已在分子科学领域得到广泛应用,但针对分子催化体系的专门迁移学习策略仍亟需进一步的拓展与优化。
在过去十年中,已经报道了大量有机光催化反应。随后,也有研究发展了能够预测底物反应性或光催化活性的机器学习方法。其中,光敏剂(photosensitizers)的催化性能对光反应的成功至关重要,因此对其进行机器学习预测尤为关键。由此可见,开发性能更优的迁移学习方法以预测光催化活性成为迫切需求。
研究团队此前已提出一种基于领域自适应(domain adaptation)的迁移学习策略,用于预测有机光敏剂(OPSs)的催化活性,该方法采用梯度提升模型(gradient boosting)实现。然而,这一策略并非基于深度学习(Deep Learning, DL)框架,其源任务与目标任务均集中在基于光敏剂性质的产率预测上。与此同时,基于深度学习的迁移学习方法也逐渐成为催化反应预测的重要工具,但其在光催化活性预测中的应用仍然不足。
通常而言,深度学习的迁移学习方法多采用微调(fine-tuning)的形式:首先在一个大型数据集上进行预训练,然后将预训练模型的参数迁移至新的任务中并进行微调,以实现高效适应。该方法的独特之处在于,它可以利用与目标任务并不直接相关的信息,从而弥补数据稀缺的局限。举例来说,Sunoj 使用了基于 Transformer 的模型,在 ChEMBL 数据库中的 SMILES 分子字符串上进行预训练;而 King-Smith 则采用了基于图神经网络(Graph Neural Network, GNN)的模型,在 CCDC 晶体结构数据库上完成预训练。这些研究表明,深度学习能够通过文本或结构信息显著提升对反应收率及对映体过量(enantiomeric excess)的预测性能。
除 ChEMBL 与 CCDC 外,还有多种分子数据库被广泛应用于模型训练与验证,例如 QM9、ZINC、Open Reaction Database (ORD) 以及 USPTO 等,这些数据库多来源于大量的量子化学计算、实验结果及商业化数据整合。然而,即便如此,它们所覆盖的化学空间仅占整个化学宇宙(约
该研究旨在探索这些尚未被利用的分子在催化迁移学习中的潜力。研究者构建了一个定制化虚拟分子数据库,并考察其在预测真实有机光敏剂(OPSs)催化活性时的信息可迁移性(见图1)。与以往迁移学习工作相比,该研究的核心贡献在于:通过系统化的生成方法及团队自研的分子生成器,构建了由类OPS片段组成的虚拟分子数据库。
然而,在使用这类定制数据库时,预训练标签的获取成为主要挑战。在构建大规模数据库时,若通过量子化学计算或实验获得可作为预测目标的化学性质,往往会面临极高的计算与实验成本。为此,研究者采用了**分子拓扑指数(molecular topological indices)**作为预训练标签——尽管这些指标与光催化活性并无直接关联,但与量化化学性质或实验数据相比,它们的获取成本更低、效率更高。
传统上,化合物性质常基于已知化合物的相关性质进行推断。而在该研究中,研究者进一步尝试借助深度学习引入来自多样且未被识别化合物的“非直观信息”,以增强模型对目标性质的预测能力。这一方法为机器学习在催化领域的应用提供了新的思路与更广阔的化学空间基础。

图1|所提出迁移学习(Transfer Learning, TL)策略的流程图。 (a) TL策略的图示概览;(b) TL过程的详细工作流程。该研究中,研究者利用基于图卷积网络(Graph Convolutional Network, GCN)的深度学习(Deep Learning, DL)模型,并在虚拟分子的拓扑指数(topological index)上进行预训练,从而构建用于预测有机光敏剂(Organic Photosensitizers, OPSs)催化活性的机器学习(Machine Learning, ML)模型。
2 结果与讨论
2.1 虚拟分子数据库的生成
首先,研究者通过分子片段的组合构建了虚拟分子数据库。具体而言,准备了30种供体(donor)片段、47种受体(acceptor)片段以及12种桥接(bridge)片段(见附图S1–S3),部分参考了Liu与Wang的研究。
供体片段主要由芳基或烷基氨基、具有多种取代基的咔唑基团、以及含有电子给体取代基的芳环构成,此外还包括芴基、硅芴基及二苯膦基等结构。受体片段则包括含氮杂环与含电子吸拉基的芳环,以及三环芳香体系如菲(phenanthrene)和苯并[1,2-b;4,5-b’]二呋喃。桥接片段则由相对简单的π共轭单元组成,包括苯环、炔烃、乙烯、呋喃、噻吩、吡咯等,同时也包含醚与硫醚片段。
为了避免数据库因有机化学家的主观偏好而过度偏向特定结构,研究还特意纳入了一些供体、桥接与受体边界模糊的片段,以增强分子的多样性。基于这些片段,研究者构建了Database A——通过在预设位置系统地组合片段生成;而Databases B–D则由团队自研的**分子生成器(molecular generator)**自动生成。在Database A中,研究还考虑了同一受体片段在不同位置的成键情况(见图S3)。
最终,基于30个供体片段(D)、65个受体片段(A)与12个桥接片段(B),生成了25,350个分子,其结构涵盖D–A、D–B–A、D–A–D以及D–B–A–B–D等多种组合形式(见图2a)。
用于构建Databases B–D的分子生成器基于一种表格型强化学习(tabular Reinforcement Learning, RL)系统(图2a)。强化学习是一种机器学习方法,智能体(agent)通过与环境的交互不断学习最优决策:采取动作后获得奖励或惩罚反馈,并逐步调整策略以最大化累计奖励。RL可引导分子生成过程朝特定方向优化,而在该研究中,关键的**Q函数(Q-function)**采用表格化形式实现,用于策略决策。
在奖励函数计算中,采用**Tanimoto系数(Tanimoto Coefficient, TC)评估分子相似度,该系数基于生成分子的Morgan指纹(fingerprint)计算。每个新生成分子的TC会与所有已生成分子的TC进行比较并取平均。研究将平均TC的倒数(avgTC⁻¹)**作为奖励值,以鼓励生成与已有分子差异更大的结构。
为了保证Databases B–D中的分子不偏离Database A的合理化学空间,设定了多项约束条件:例如分子量小于100或大于1000、以及含有六个以上片段的分子均被赋予负奖励(详见补充说明1)。此外,生成结束后,具有负奖励或重复canonical SMILES的分子会被剔除。
在策略设置上,采用了ε-greedy策略以平衡探索与利用,并设置了三种不同的ε值:
- Database B:ε = 1,即完全依赖随机探索,不利用任何先前行动所得知识;
- Database C:ε = 0.1,即90%时间利用已有经验进行生成,仅10%随机探索;
- Database D:ε 从1逐渐递减至0.1,使模型在训练过程中逐步增加对已学知识的依赖。
通过以上三种策略,每种方法均生成了约3万种虚拟分子。不同奖励函数与策略设定下的分子生成行为差异在补充信息(图S4与图S5)中进行了总结。
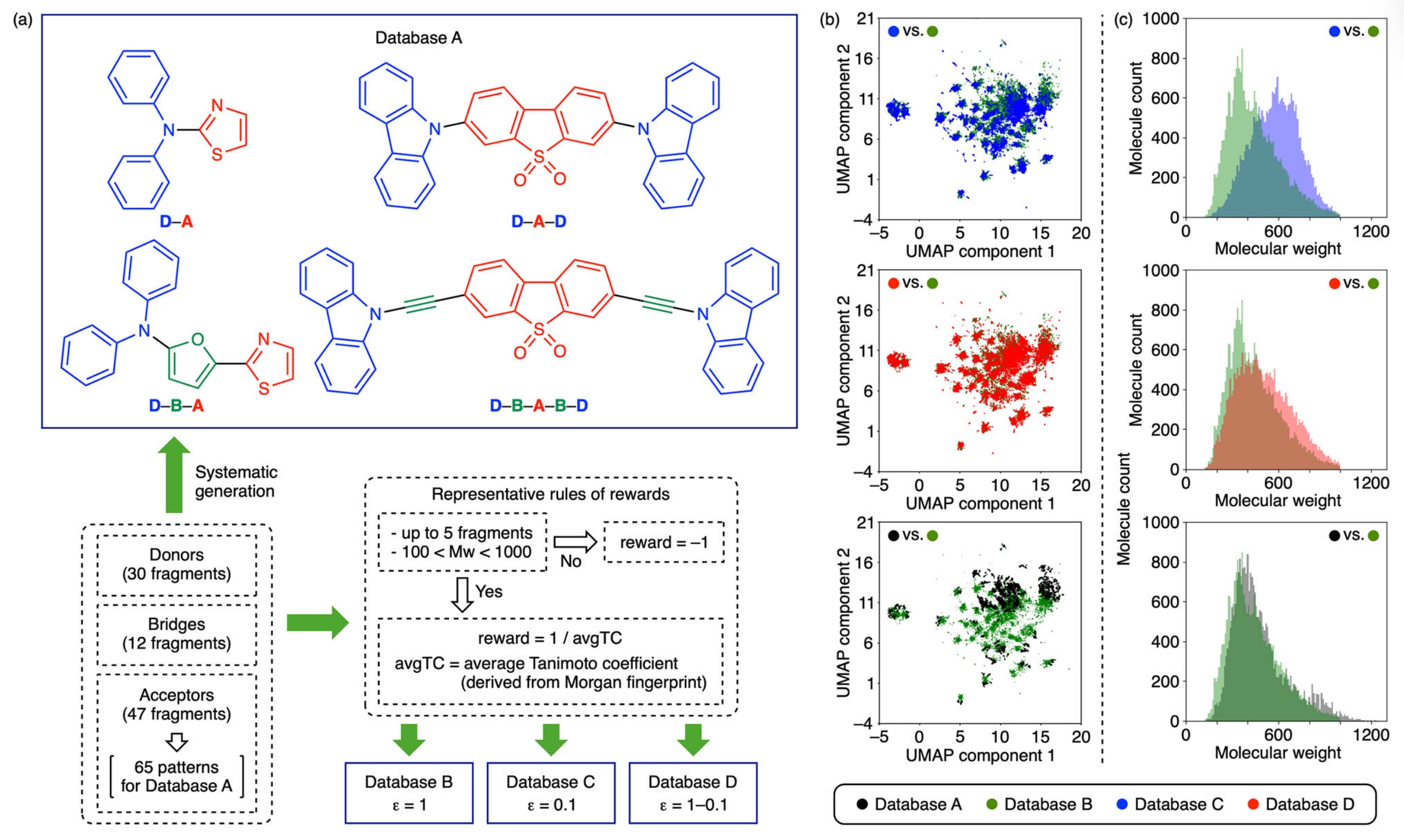
图2|数据库的生成与比较。 (a) 数据库构建规则:Database A中的分子通过在预设位置系统性地组合供体(D)、桥接(B)与受体(A)片段生成;而Databases B、C 与 D则由研究团队自主开发的分子生成器(molecular generator)构建。 (b) 各数据库的化学空间分布,基于Morgan指纹经UMAP降维所得结果。(c) 各数据库的分子量分布,用于展示不同生成策略下分子复杂度与组成的差异。
2.2 预训练标签的选择与数据库性质的比较
在该研究中,选择既有效又具成本效益的预训练标签是实现迁移学习策略的关键。因此,研究者重点关注了可从 RDKit 与 Mordred 描述符集中提取的分子拓扑指数(molecular topological indices),以下简称为RDKit与Mordred特征集。
从这些特征集中,研究者筛选出16个用于预训练的候选标签,包括:
Kappa2、PEOE_VSA6、BertzCT、Kappa3、EState_VSA3、fr_NH0、VSA_EState3、GGI10、ATSC4i、BCUTp-1l、Kier3、AATS8p、Kier2、ABCGG、AATSC3d 和 ATSC3d。
基于 SHAP(SHapley Additive exPlanations)分析 的结果显示,这些特征在研究团队内部的交叉偶联反应数据集中对产物收率预测具有显著贡献(见附图S6–S8),从而验证了它们作为预训练标签的代表性与有效性。有关预训练标签筛选的具体过程详见“方法(Methods)”部分。
在删除无法计算任一预训练标签的分子后,Database A成为最小的数据集,共包含25,286个分子。为保持数据规模一致,研究者对Databases B–D进行了随机抽样,使其样本数量与Database A相同。
随后,对各数据库的化学空间分布与分子量分布进行了比较(见图2b、c)。化学空间采用基于Morgan指纹描述符的统一流形近似与投影(UMAP)进行可视化分析。结果表明,Database C因在强化学习过程中更注重利用(exploitation),其化学空间分布比Database B更为集中,但生成的高分子量分子比例更高。这说明在该系统中,化学空间广度与高分子量分子的生成频率之间存在一定的权衡关系。
与此同时,Database D的化学空间分布与Database B相似,但其分子量分布与B、C两者均有所差异(见附图S14)。相比之下,Database A在分子量分布上与Database B较为接近,但其化学空间明显更窄。
总体而言,研究所开发的强化学习分子生成系统能够在保持广阔化学空间覆盖度的同时,生成结构复杂度多样的分子,其生成性能显著优于基于系统组合方式构建的传统Database A,即使二者使用了相同的分子片段。
2.3 利用迁移学习预测C–O键形成反应的光催化活性
在该研究中,**光反应的产率(yield)作为机器学习模型的预测目标(除预训练阶段外)。由于每个光反应的产率主要由所使用的有机光敏剂(Organic Photosensitizer, OPS)**决定,因此在后续讨论中,**催化活性(catalytic activity)**可视为产率的代表性指标。
为比较不同数据库在光催化活性预测中的有效性,研究首先构建了基于图卷积网络(Graph Convolutional Network, GCN)的深度学习(Deep Learning, DL)预训练模型。GCN是一类能够通过图卷积操作捕捉分子内部原子(节点)与化学键(边)之间关系的图神经网络。分子特征由开源工具包DeepChem中的 MolGraphConvFeaturizer 提取。
在监督预训练阶段,所采用的DL模型包括4个GCN层、1个最大池化层和3个全连接层,用于预测前述由RDKit与Mordred导出的分子性质。模型输入为各虚拟数据库中分子的SMILES字符串所生成的图特征。预训练的模型结构及过程详见附图S9与“方法”部分。
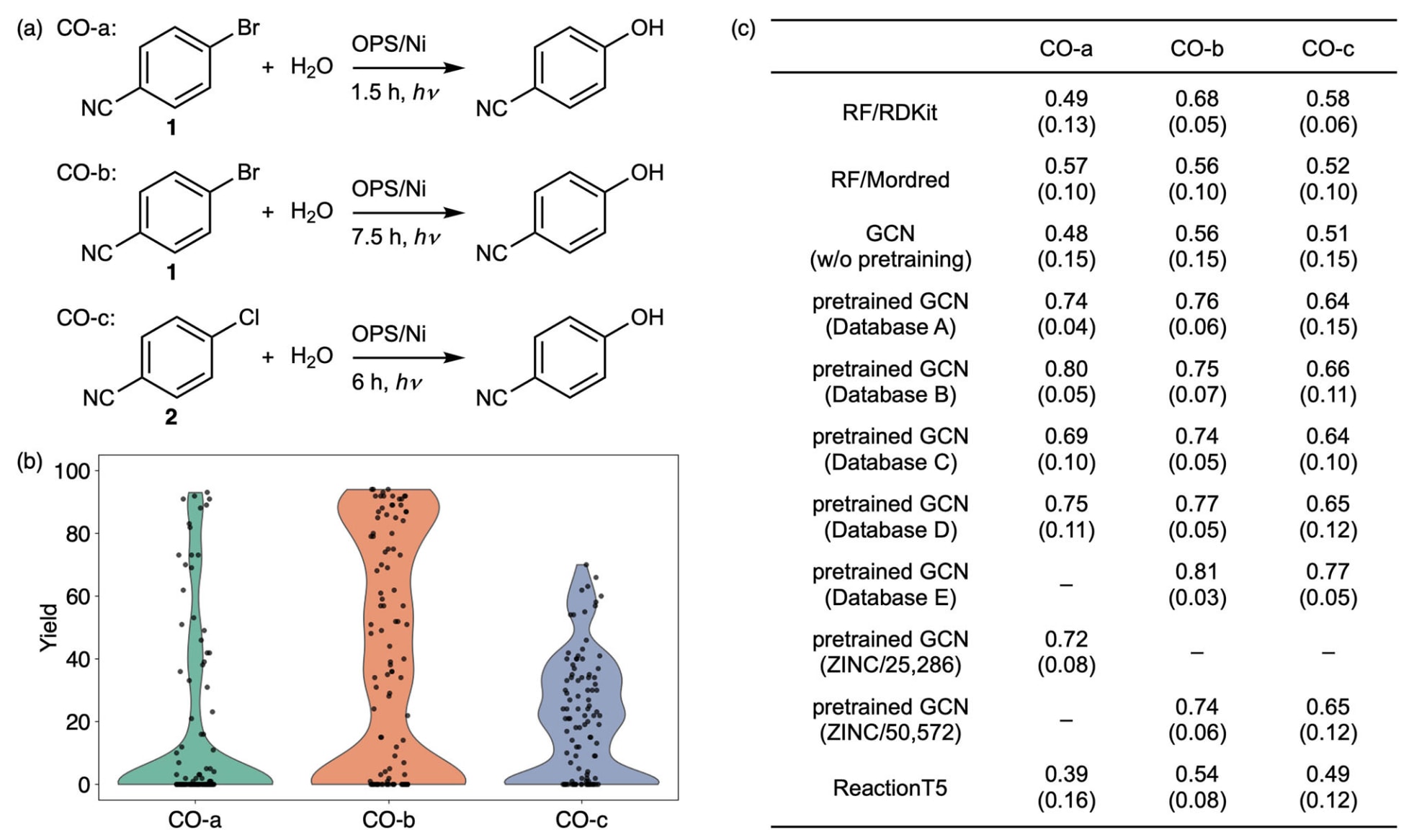
接下来,研究者对这些预训练GCN模型进行了微调(fine-tuning),以预测OPS的催化活性(即反应产率)(见图3)。所用数据集包含100种真实OPS分子,涵盖已报道的光敏剂及其带有不同取代基的衍生物(详见补充说明2)。这些光敏剂参与的C–O键形成反应由无机镍盐与OPS共同催化,相关实验数据来自团队先前的研究。由于苯酚类化合物在有机合成中的重要性,该反应体系被认为是机器学习研究的理想目标。
研究将OPS数据集随机分为两部分(50%训练集,50%测试集),并在10次不同划分下计算平均
(1)CO-a 反应体系
第一个微调任务为CO-a,以**4-溴苯甲腈(1)**为底物,反应时间为1.5小时,在可见光照射下进行。
基准模型包括:
- 随机森林(RF)结合RDKit描述符(
) - RF结合Mordred描述符(
) - 无预训练的GCN模型(
)
结果显示,以BertzCT为预训练标签的模型表现最优。在所有情况下,预训练均能显著提高预测精度,其中以Database B为基础的模型效果最佳:
- Database A:
- Database B:
- Database C:
- Database D:
同时,Database B的模型在不同训练集划分下表现出更低的方差(见图3c与表S10),说明其具有更稳定的预测性能。
(2)CO-b 与 CO-c 反应体系
随后进行了两个扩展案例研究。
- CO-b:底物仍为1,但反应时间延长至7.5小时;
- CO-c:底物改为4-氯苯甲腈(2)。
数据分布显示(见图3b):
- CO-a中,54个数据点产率为0%,仅8个≥80%;
- CO-b中,27个为0%,25个≥80%;
- CO-c中,23个为0%,仅5个≥60%,整体产率偏低。
基准模型的平均
- CO-b:RF/RDKit=0.68,RF/Mordred=0.56,GCN无预训练=0.56;
- CO-c:RF/RDKit=0.58,RF/Mordred=0.52,GCN无预训练=0.51。
在微调后,CO-b的最佳模型来自Database D(
- A: 0.76, B: 0.75, C: 0.74。
研究进一步尝试将Database A与C连接成新的Database E(共50,572个分子),其预测性能显著提升(CO-b:
对于CO-c,尽管A–D的模型表现较弱,但Database E依然取得最佳结果(A=0.64, B=0.66, C=0.64, D=0.65, E=0.77)。与CO-a相同,BertzCT依旧是最优预训练标签(见表S11与S12)。
(3)化学空间与模型比较
虽然Database A与C的Morgan指纹化学空间较Database B更窄,但经连接后的Database E覆盖范围更广(见附图S15a),并包含更宽分子量分布(见S15b)。其中重复分子仅占0.6%(共323个),剔除后预测性能无显著变化。
作为对照,研究还使用ZINC15数据库中相同数量的分子进行预训练(25,286用于CO-a,50,572用于CO-b与CO-c)。尽管ZINC含有类药性商业分子,但其预训练模型性能不及Database B与E(CO-a=0.72, CO-b=0.74, CO-c=0.65)。
此外,还测试了基于Transformer结构的ReactionT5模型(预训练于ZINC与ORD数据库)。经微调后,其平均
- CO-a:0.39
- CO-b:0.54
- CO-c:0.49。
这些结果表明,尽管Transformer模型在通用迁移学习中表现强劲,但在小数据集、专用光催化任务中,所提出的GCN迁移学习体系展现出更高的适用性与预测稳定性。

图4|迁移学习(TL)策略在C–S键、C–N键形成反应及[2 + 2]环加成反应中的应用。 (a) 目标反应列表,展示三类典型光催化转化反应:C–S键形成反应、C–N键形成反应以及**[2 + 2]环加成反应**。(b) 各任务的预测性能汇总表。对于GCN模型:
- 在**C–S键形成反应(CS)与[2 + 2]环加成反应(CA)**中,采用 ABCGG 作为预训练标签;
- 在**C–N键形成反应(CN)**中,采用 Kappa3 作为预训练标签。
预训练所使用的数据库分别为:
- Database E(用于CS与CA任务);
- Database F(用于CN任务)。
表中 平均
2.4 迁移学习策略在其他光催化反应中的应用
为了进一步验证所提出迁移学习(TL)策略的普适性,研究者将其应用于其他类型的光催化反应体系,包括C–S键形成反应(CS)、C–N键形成反应(CN)以及[2 + 2]环加成反应(CA)(见图4a)。
在此前的研究中,这些反应的产率数据已被系统收集。然而,与C–O键形成反应相比,基准模型(包括随机森林RF模型与未经过预训练的GCN模型)的预测效果明显较差:
- C–S反应的
仅为 0.11–0.21; - C–N反应为 −0.02–0.19;
- [2 + 2]环加成反应为 −0.04–0.15(见图4b)。
在监督预训练阶段,研究采用了不同的数据库与标签设定:
- 对于 C–S与CA反应,使用 Database E 的分子数据,并选取 ABCGG 作为预训练标签;
- 对于 C–N反应,采用 Database B与D合并形成的Database F,并以 Kappa3 作为预训练标签。
经过微调(fine-tuning)后,模型的预测性能在所有任务中均有提升:
- C–S反应:
- C–N反应:
- CA反应:
与未预训练的GCN模型相比,迁移学习显著改善了模型表现。然而,整体预测精度仍未达到理想水平(见图4b)。
值得注意的是,这些基于虚拟数据库预训练的GCN模型在预测性能上,明显优于在ZINC数据集或ReactionT5模型上预训练的同类模型(见表S22)。
这些结果表明,尽管所提出的迁移学习方法能在C–O键形成反应中取得令人满意的表现,但当任务本身在基于SMILES特征的信息提取方面存在内在困难时,该方法的预测能力仍受到限制。换言之,在反应机理更复杂、数据表达信息较弱的体系中,模型构建的精确性成为主要挑战。
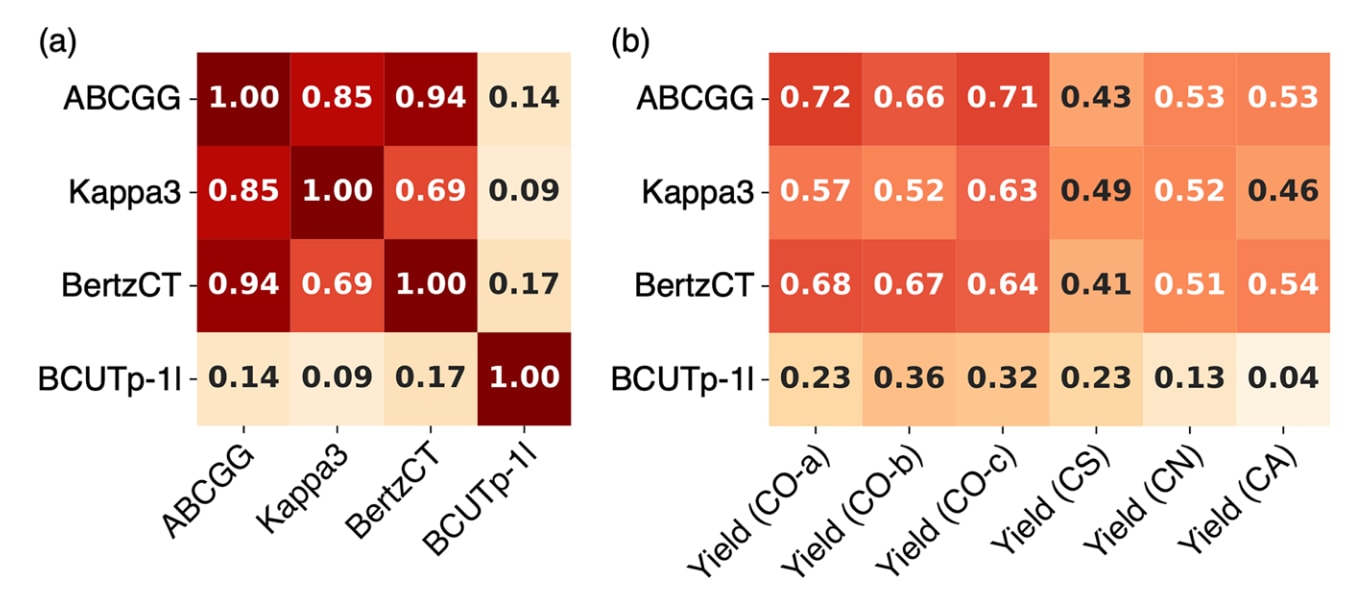
图5|预训练标签与反应产率的相关性分析。 (a) 各预训练标签之间的相关系数;(b) 预训练标签与反应产率之间的相关系数。所有分析均基于**100种真实有机光敏剂(OPSs)**的实验数据进行,展示了不同拓扑指标(如ABCGG、BertzCT、Kappa3等)之间及其与光催化反应产率的关联程度。
2.5 对有效与无效预训练标签的分析
在该研究中,为提升GCN模型的预测性能,研究者选用了ABCGG、BertzCT与Kappa3作为预训练标签。
- ABCGG代表Graovac–Ghorbani原子–键连通性指数(atom–bond connectivity index);
- BertzCT表示Bertz复杂度指数(Bertz complexity index);
- Kappa3为Hall–Kier三阶κ指数(third-order kappa index)。
这些指标均反映了分子的拓扑特征,包括其复杂性(complexity)、**形状(shape)及支化程度(branching)**等结构信息。
尽管三者基于不同的数学计算公式而得,但在100种真实OPS分子中表现出高度相关性(见图5a;Databases A–D的相关性结果见附图S16–S19)。此外,这些拓扑指数与C–O键形成反应的产率之间也存在较强的相关性(见图5b;相关系数为0.52–0.72),而与C–S、C–N及[2+2]环加成反应产率的相关性则处于中等水平(0.41–0.54)。
相比之下,若采用BCUTp-1l作为预训练标签,则迁移学习后的模型性能并未优于未使用迁移学习的基准模型(见表S10–S12及S19–S21)。BCUTp-1l代表Burden矩阵的极化率加权第一最低特征值,其与ABCGG、BertzCT及Kappa3的相关系数较低,仅为0.09–0.17(见图5a)。同时,该指标与产率的相关性也较弱(见图5b;0.04–0.36),这与其迁移学习性能不佳的结果相一致。
根据“方法”部分的说明,预训练标签最初是依据其在相关光反应机器学习模型中的**高SHAP值(特征重要性)**选出的。然而,从上述结果可以看出,选择与预测目标(即产率)相关性较高的分子性质,亦是一种有效的预训练标签筛选策略。
综上所述,能够灵活选择与任务密切相关的分子特征作为监督预训练标签,是该迁移学习框架的一大优势。
但需要指出的是,尽管SHAP分析与相关性分析可用于估计潜在的候选指标,要精确识别单一最优的预训练标签仍是一项具有挑战性的任务。
2.6 虚拟分子数据库的补充信息
图6展示了从Database A与Database B中随机选取的10个代表性分子(Databases C与D的示例见附图S21与S22)。
由于研究团队开发的分子生成器在生成过程中并未对片段进行供体(donor)、桥接(bridge)或受体(acceptor)标注,因此Database B中的分子并不全都具有典型的供–受体组合结构。
相对而言,Database A中的分子结构更加系统化,但其中也包含了一些不适用于OPS的化学键,例如P–S键与N–O键。
换言之,尽管这些数据库中的分子均由“类OPS片段”构建,但其中仍存在大量非自然键型或非典型片段组合,与常见的供–受体型光敏剂(如4CzIPN与Acr⁺–Mes)存在差异。
截至2025年5月16日,研究数据集中100种OPS分子中,有84种可在PubChem数据库中检索到;而虚拟数据库中的分子则大多未被收录——
- Database A:未注册率 97.5%
- Database B:94.1%
- Database C:98.9%
- Database D:96.6%
此外,尽管ABCGG、BertzCT与Kappa3等拓扑指数与目标性质——光催化活性——之间呈中等至高相关性,但从有机化学意义上来看,这些指数并不直接对应具体的化学反应特征。
值得强调的是,即便这些虚拟分子在结构上并不完全符合真实有机体系,基于其拓扑指数进行预训练的GCN模型仍能在预测性能上超越基于真实分子数据库的GCN与Transformer模型。
在计算效率方面,研究指出:以普通个人计算机(单核Intel Xeon w32423 2.11 GHz,64 GB RAM)生成Database B的全部分子并计算其BertzCT值仅需约40分钟(2529秒)。
这一高效的数据构建过程表明,所提出的策略可在实验数据稀缺的催化研究领域中,提供一种**数据高效(data-efficient)**的迁移学习途径,从而显著提升模型的预测能力与实用价值。

图6|从Database A与Database B中随机选取的分子示例。 左侧为Database A中的分子,右侧为Database B中的分子。在图中,最初被标记为不同功能片段的结构单元以颜色区分:蓝色表示**供体(donor)片段,绿色表示桥接(bridge)片段,红色表示受体(acceptor)**片段。
2.7 潜在局限与未来展望
在该研究提出的策略中,虚拟数据库是通过分子片段组合方式构建的,这一方法在多种分子相关任务中被证明是有效的。然而,该策略也存在潜在局限性——当研究体系中无法明确定义用于组合的结构单元(building blocks),或**缺乏可计算的标签(如拓扑指数)**时,该方法的适用性可能受限。
此外,研究结果表明,尽管该迁移学习(Transfer Learning, TL)方法能显著提高预测精度,但在某些本身难以构建高精度机器学习模型的任务(如C–S、C–N键形成反应及[2+2]环加成)中,预测性能仍未达到理想水平。在这些情况下,可能需要引入更具物理意义的特征信息,如来自量子化学计算的分子物性参数,以作为描述符或预训练标签,从而提升模型表现。
此外,研究发现最有效的虚拟数据库取决于具体的目标反应类型。因此,未来的重要方向之一,是探索如何设计更具普适性的定制虚拟分子数据库,使其不仅在光催化活性预测中表现优异,也能适用于更广泛的化学反应体系与分子现象。
与此同时,将当前的预训练方法与监督多任务学习 或 自监督学习 进行对比研究,可能为进一步优化基于虚拟分子的迁移学习策略提供新的见解。
此外,探讨该TL策略能否应用于 机器学习驱动的反应优化框架(如贝叶斯优化) 中,用于提升替代模型(surrogate model)的性能,也具有重要的现实意义。
3 结论
该研究首次验证了基于自主构建的定制虚拟分子数据库进行迁移学习的概念性可行性,证明无需额外实验或依赖现有数据库,即可实现对光催化活性(photocatalytic activity) 的有效预测。
通过分子生成器在不同策略下生成的数据库,无论单独使用或组合使用,都能提供一种高数据效率(data-efficient)的迁移学习途径。值得注意的是,尽管这些分子中存在非典型化学结构,但其包含的类OPS片段仍使模型获得了优于现有数据库分子的预测性能。
此外,即便ABCGG、BertzCT与Kappa3等拓扑指数与光催化活性在化学意义上并无直接关联,它们仍作为高效预训练标签显著提升了模型表现。
总体而言,该研究展示了一种以虚拟数据库为核心的迁移学习新范式,为分子科学领域的数据辅助建模提供了新的方向,也为加速光催化剂与功能分子设计开辟了潜在的新途径。