JACS 2023 | 口服可利用肽类靶向细胞内蛋白的开发:从初始化合物到临床级KRAS抑制剂
细胞内蛋白–蛋白相互作用(PPI)长期以来被视为“不可成药”的顽固靶点,而环肽因兼具小分子药物的口服潜力与大分子药物的特异性识别能力,逐渐成为破解这一难题的重要方向。今天介绍的一项研究发表于《Journal of the American Chemical Society》,报道了首个从类药性环肽出发,经系统化学优化获得口服临床候选物LUNA18的完整研究路径。
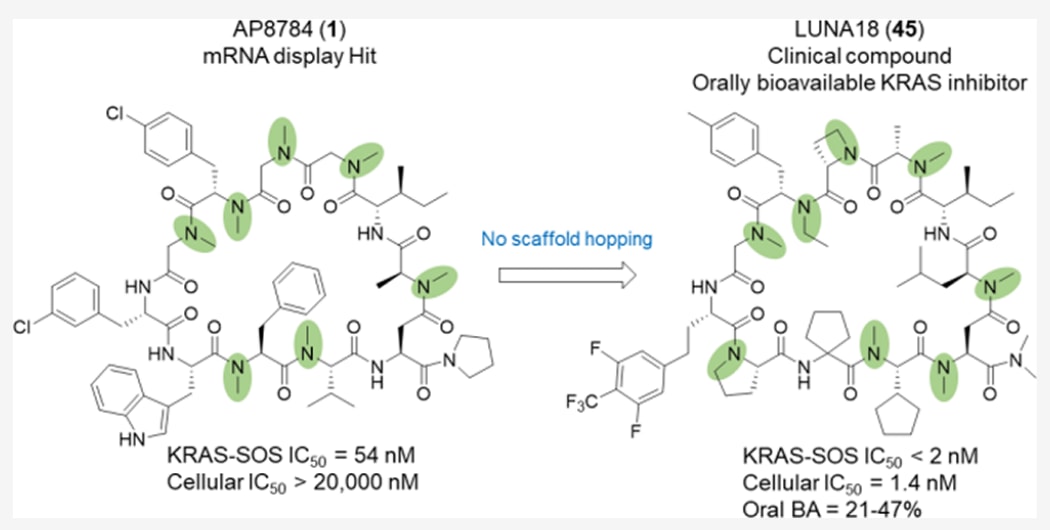
研究团队以mRNA展示文库筛选的KRAS–SOS抑制肽AP8784为起点,利用N-烷基化模式优化、骨架刚化及侧链诱导契合等策略,显著提升了分子的膜通透性与活性。最终产物LUNA18为一款口服可利用的11肽环肽KRAS抑制剂,在动物模型中展现出优异的抗癌活性与21–47%的口服生物利用度,且已进入日美联合Ⅰ期临床试验。
该研究不仅建立了中等分子肽药物的可口服化设计范式,也为靶向细胞内蛋白提供了新的结构生物学思路,标志着环肽药物化学迈入可临床化的新阶段。

获取详情及资源:
0 摘要
环肽作为一种具有治疗潜力的分子形式,因其可经口吸收与进入细胞内难以成药靶点的能力而备受关注。该研究以通过mRNA展示文库筛选得到的类药性初始化合物为起点,阐述了其化学优化过程,最终获得了LUNA18——一种可口服的临床候选化合物,即针对细胞内顽固靶点RAS的11肽环状抑制剂。
研究的关键发现包括:
-
确定了两条肽侧链,它们各自均可使RAS的结合亲和力提高超过10倍;
-
通过侧链修饰可调控包括Clog P在内的理化性质(PCP),从而增强膜通透性;
-
限制环肽构象被证实是调节PCP并提高生物活性的有效手段;
-
在Caco-2通透性实验中,当通透率达到约0.4 × 10⁻⁶ cm/s或更高时,环肽表现出显著的细胞内活性;
-
在保持环肽主链构象的同时,研究者还发现了一个实例:RAS蛋白通过诱导契合(induced-fit),其结构因肽侧链的结合而发生显著变化。
这一研究展示了**无需骨架跃迁(scaffold hopping)即可对生物活性肽进行化学优化的可行途径,过程与小分子药物发现中遵循Lipinski“五规则”**的策略类似。此方法为从类药性起始化合物出发,发展为可成药肽类分子提供了一种灵活而有效的新策略。
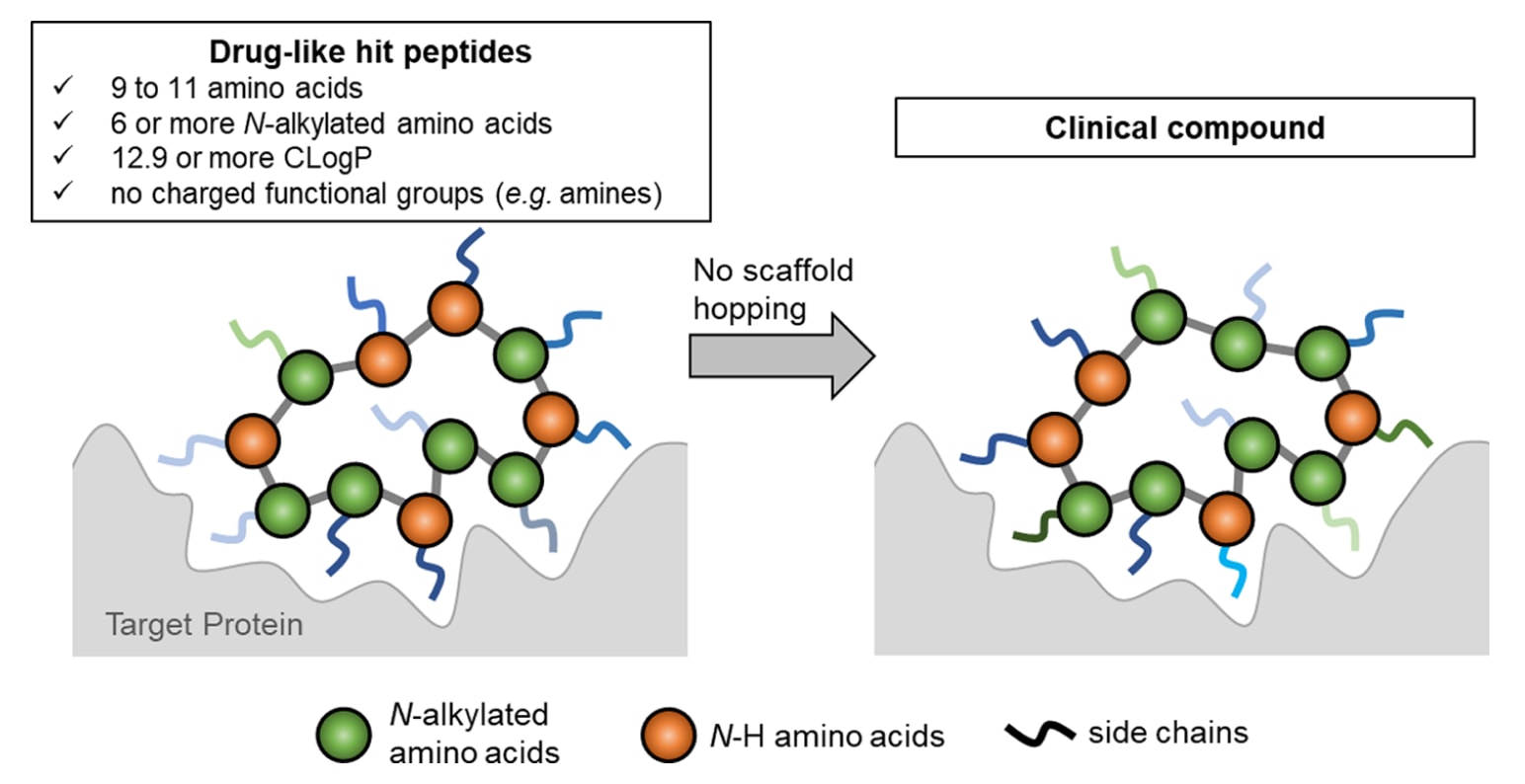
图1|以类药性肽初始化合物为起点的药物发现流程。 拥有类药性肽骨架(drug-like peptidic scaffold)能够避免对骨架结构进行剧烈改造,同时实现足够的生物活性与良好的类药性质。
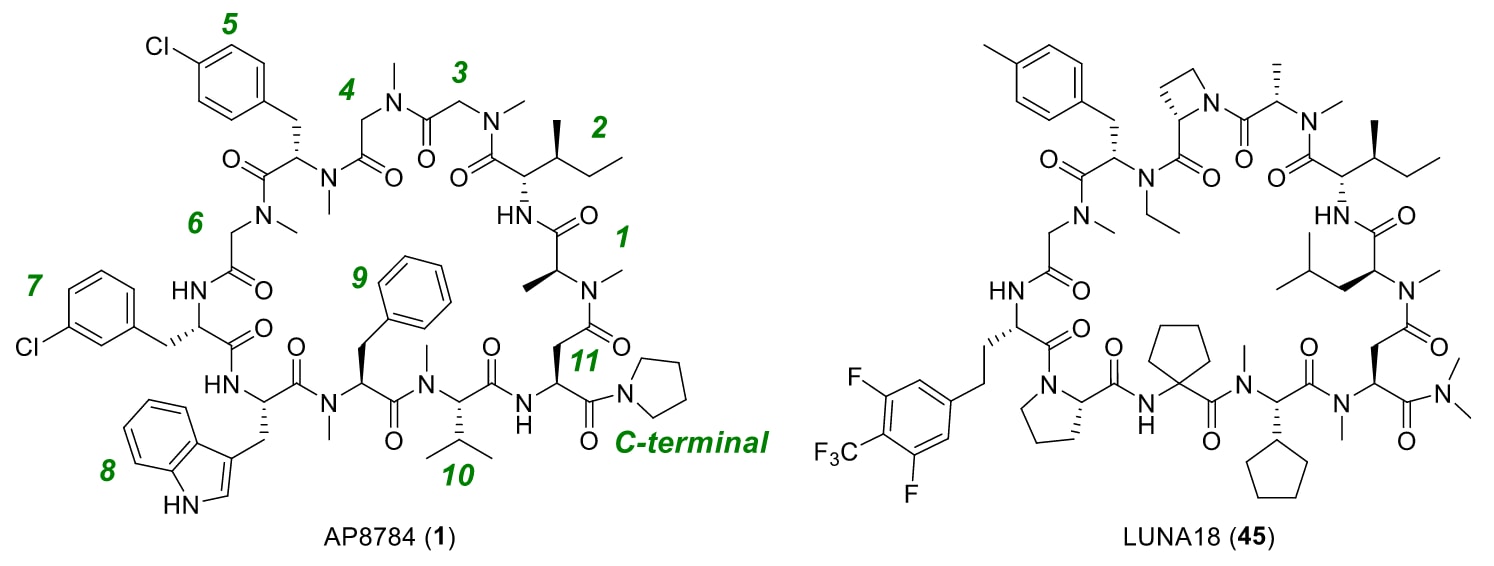
图2|AP8784(1)与LUNA18(45)的结构。 图中绿色数字表示氨基酸的位置编号。
1 引言
能够抑制细胞内蛋白−蛋白相互作用(PPIs)等顽固靶点的分子,有望成为一种全新的强效药物类型,从而大幅拓展可治疗的靶点范围。其中,一种潜在的治疗形式是中等分子量的环状肽。已有多项研究表明,分子量高达1200 g/mol的化合物仍然可以实现细胞内靶点抑制并具备口服可利用性。
一个经典例子是环孢素A(Cyclosporin A),它是一种口服生物利用度约为30%的人用免疫抑制剂。该药物通过与环孢素结合蛋白(Cyclophilin)形成复合物,进而与钙调神经磷酸酶(Calcineurin)发生细胞内PPI作用,从而实现药理效应。
mRNA展示文库是一种极为有用的技术手段,可用于获得具有活性的环肽初始化合物。以此为起点的口服中等分子药物发现中,具有代表性的研究来自默克公司开发的前蛋白转化酶PCSK9抑制剂MK-0616(目前处于Ⅱb期临床试验阶段)。PCSK9是一个胞外蛋白,通常可被抗体药物靶向。尽管默克的研究成功展示了该肽类药物在体内经口给药的有效性,但其口服生物利用度仍仅为2.9%,且需要与肠道渗透促进剂联合使用才能弥补其较差的被动膜通透性。
这一研究证明,通过对mRNA展示筛选得到的肽结构进行优化,确实可以获得口服可用的临床化合物。然而,其低被动通透性仍暴露出中等分子肽类药物在靶向细胞内蛋白及提高口服可利用性方面的挑战。
为了建立一种可通用的中等分子药物平台技术,研究者们近期验证了一种新的药物发现策略:从具有类药性质的肽初始化合物出发(如图1所示)。这种**类药性肽骨架(drug-like peptidic scaffold)**能够避免剧烈的骨架变更,同时保证足够的生物活性、细胞通透性与口服生物利用度。
为实现这一概念,研究团队对环肽的类药性范围进行了定义,并基于此构建了具有以下三项特征的mRNA环肽展示文库:
- 肽链长度约为11肽;
- 高N-烷基化;
- 高疏水性(lipophilicity)。
将这一类药性环肽文库应用于致癌蛋白KRAS的筛选后,研究者发现了KRAS-SOS相互作用抑制剂AP8784(1)作为初始化合物(见图2)。对该化合物的进一步优化,最终得到临床KRAS抑制剂LUNA18(45)。LUNA18在小鼠异种移植肿瘤模型中表现出显著且剂量依赖的抗癌活性。在小鼠、大鼠、猴及犬等动物实验中,其口服生物利用度达21−47%,且无需任何特殊制剂。
该研究将重点介绍该化合物的先导优化过程,包括其KRAS抑制活性、**理化性质(PCP)以及药代动力学(PK)**方面的关键成果。
图式1|环肽合成流程概述。
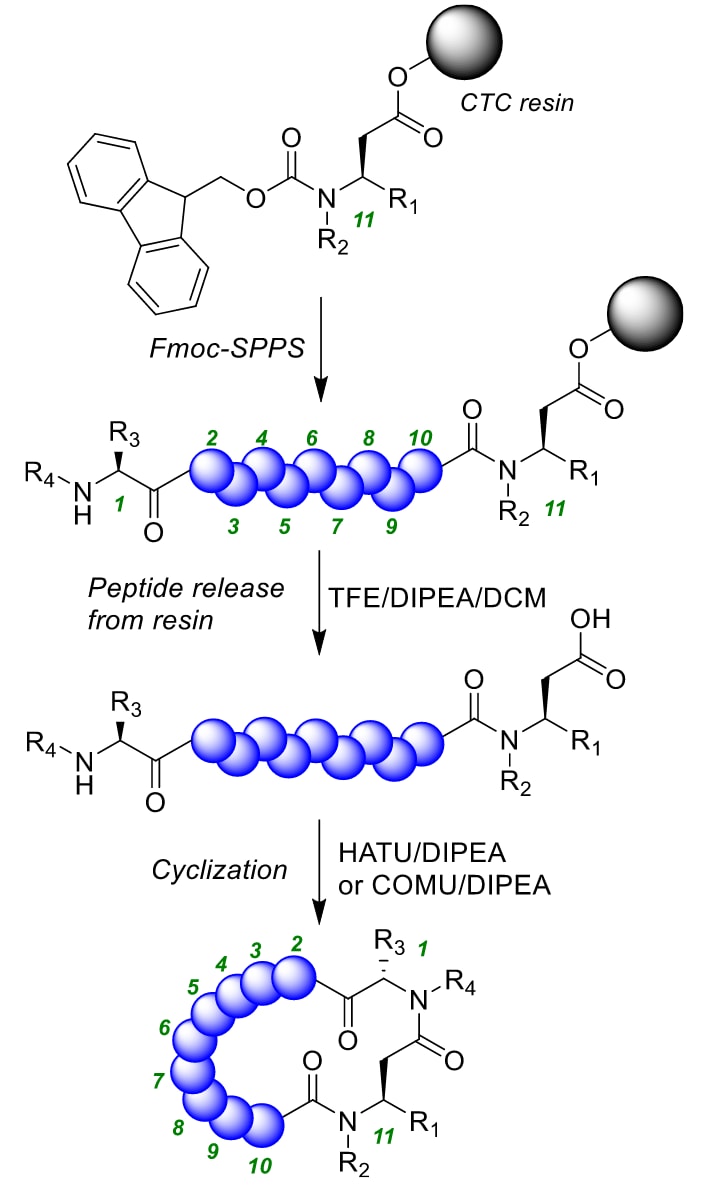
2 结果与讨论
AP8784(1)的性质
化合物1源自类药性环肽文库,在AlphaScreen检测中表现出显著的PPI抑制活性(KRAS^G12D–SOS1,IC₅₀=54 nM)。然而,在细胞水平上(AsPC-1胰腺癌细胞株,KRAS^G12D背景)未观察到活性(IC₅₀>20,000 nM),推测原因在于膜通透性较差。
在结构优化初期获得的化合物1与KRAS^G12D的X射线共晶结构(PDB编号:7YUZ)显示,该化合物结合于Switch II与α3螺旋之间的疏水沟槽。化合物1几乎满足研究者为环肽设定的类药性标准,具体包括:
-
含有11个氨基酸残基(理想范围为9–11个);
-
其中7个为N-烷基化氨基酸(理想值为6个或以上);
-
Clog P=12.7(理想值≥12.9);
-
无带电官能团,如羧酸或胺基。
研究者推测,通过修饰侧链结构可进一步提升Clog P值,从而改善膜通透性。
肽的制备
该研究所报道的45种肽均采用固相合成法制备,并针对高N-烷基化环肽进行了优化(合成路线见图式1)。
肽链的延伸采用HOAt/DIC或Oxyma/DIC体系;延伸完成后使用TFE/DCM混合液将肽从树脂上裂解下来。随后通过HATU/DIPEA或COMU/DIPEA在溶液条件下进行环化反应,得到目标环肽。所得粗产物经**制备型反相高效液相色谱(RP-HPLC)**纯化后,用于后续的性质评估。
侧链探索性修饰
为确定侧链优化策略,研究者首先进行了探索性修饰实验(见表1)。
将位于第8位、面向溶剂的Trp侧链分别替换为苄基(化合物2)与甲基(化合物3),结果两者的PPI抑制活性(IC₅₀)与化合物1相当,说明不与RAS直接相互作用的侧链位置可以自由修饰。因此,研究者计划通过第8位残基修饰来调节化合物的理化性质(PCP)。
此外,研究者还确定第11位同样可用于PCP调节,因为缺乏C末端酰胺的化合物10仍保持PPI IC₅₀=150 nM的活性。
在化合物2的第2、5、9与10位进行丙氨酸扫描(Ala scanning)后(化合物4、7、8、9),活性均下降超过10倍。这表明这些侧链与RAS之间存在关键相互作用,这一结论与共晶结构所显示的各侧链与KRAS接触模式完全一致。
将第3、4位的MeGly替换为MeAla后(化合物5与6),活性下降超过70倍,推测原因是MeGly构象的改变。该位置的构效关系(SAR)详见表4与图6。
化合物11与12虽具有Clog P=13.2(符合类药性标准),但在Caco-2渗透实验中表现出较低的表观通透系数P_app,提示初始化合物1的N-烷基化模式通透性相对较低(详见后文)。在AsPC-1细胞中检测化合物12的活性时,未观察到细胞效应(IC₅₀>20,000 nM)。
对于化合物13,其氯代苯基基团被认为嵌入了一个狭窄的结合口袋,因为即使是微小的结构变化(如卤素与氢的置换),也导致活性下降超过2倍(化合物14–17)。
然而,在第7位引入新的侧链(化合物18、19、20),这些侧链预期会与化合物1的Phe(3-Cl)所占据的口袋发生“顶撞”,结果分别获得IC₅₀=28 nM、120 nM、640 nM。其中化合物18活性显著提升,其原因在于KRAS蛋白结构因肽侧链的诱导契合(induced-fit)形成了一个新的空腔(SII-hole)。
化合物18与KRAS^G12D的共晶结构(PDB编号:8JJS)显示,其Hph(4-CF₃)基团正位于Switch II区域形成的新腔体内,因此Switch II向外扩张(见图3A–C)。除Switch II外,KRAS的其余结构未见显著变化。第7位与第10位的侧链共同占据了SII-hole,而该空腔明显大于化合物1中Phe(3-Cl)所在的结合口袋(见图3D)。
表 1 | 侧链修饰
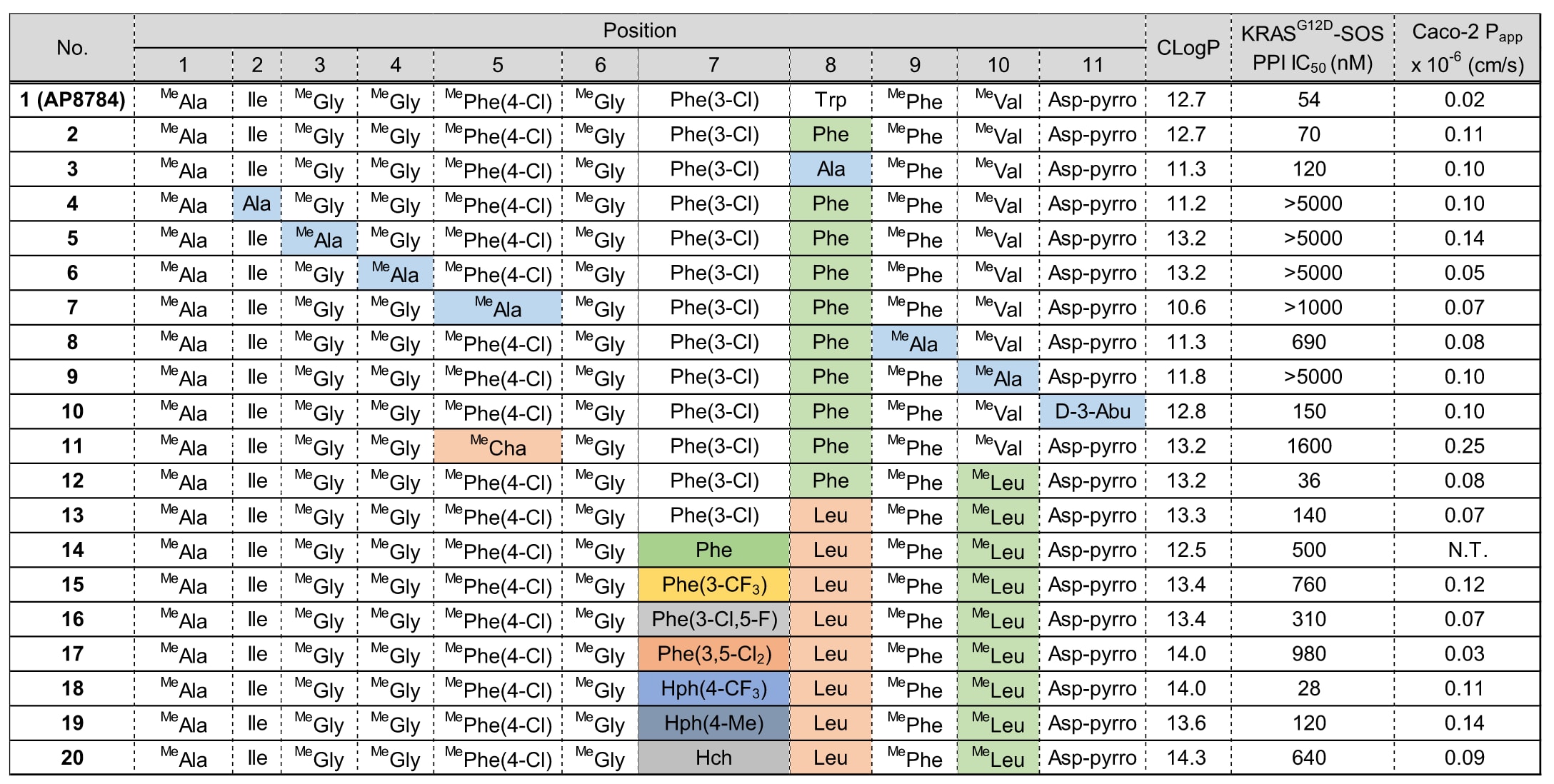
该节所展示的侧链修饰研究为后续的衍生化方向提供了重要指导,结论如下:(1)通过修饰第8位与第11位不与RAS相互作用的氨基酸,可以有效调节理化性质(PCP);(2)位于SII-hole中的第7位与第10位可作为增强生物活性的关键位置(见图4)。
主链调控以获得膜通透性(第8与第9位)
由于侧链修饰后获得的最高Caco-2表观通透系数(Papp)仅为0.25 × 10⁻⁶ cm/s,研究者进一步尝试通过主链调控来改善膜通透性。此前已有研究表明,N-烷基化氨基酸的排列模式对膜通透性有显著影响,其中约63%的序列显示较高通透性,而其余37%则表现较差。
为寻找可能的主链修饰位点,研究者分析了化合物18与KRAS的X射线共晶结构的φ/ψ二面角分布图(Ramachandran图)(见图5A)。结果显示,含N-H氨基酸的φ/ψ分布与N-甲基化氨基酸的分布明显不同,这表明**主链改性(如将N-H氨基酸甲基化或去甲基化)**可能引起构象变化,从而降低与RAS的亲和力。
在可供N-烷基化的第2、7、8与11位中,研究者推测第8位最有潜力在保持RAS亲和力的同时被修饰,因为该位点周围存在足够空间,且其N-H未与KRAS形成任何相互作用(见图5B)。此外,位于第9位的MePhe残基(φ=+51°,ψ=+54°)处于α,α-二烷基氨基酸的优选构象范围,因此也被认为是可行的修饰点。
通过对化合物18的第8与第9位主链进行修饰,研究者成功获得了具有良好膜通透性的化合物24,其Caco-2 Papp达到了0.83 × 10⁻⁶ cm/s,超过了标准阈值0.4 × 10⁻⁶ cm/s(见表2)。
化合物21为18在第8位的N-甲基化衍生物,其生物活性下降约6倍;在第8位引入Pro残基后得到化合物22,其活性提升至IC₅₀=25 nM。尽管化合物21与22的效力均处于可接受范围,但通透性并未提升。
随后,研究者在第9位引入非N-烷基化的α,α-二烷基氨基酸以实现主链酰胺去甲基化。化合物23(第9位为环亮氨酸cLeu,第1位为MeLeu用于调节Clog P)在Caco-2实验中显示Papp=0.39 × 10⁻⁶ cm/s,同时保持与化合物18相近的生物活性。
进一步地,将化合物23的第5位4-氯苯基取代为环己基,得到化合物24,其Caco-2通透性提升至0.83 × 10⁻⁶ cm/s,表现出优异的膜渗透性能。
对于化合物21–24的代谢稳定性,通过人肝微粒体实验(hLM assay)进行评估。结果显示,这四种化合物的内在清除率(CLint,mic)均≤100 μL/min/mg蛋白,符合设计预期,表现出良好的代谢稳定性。
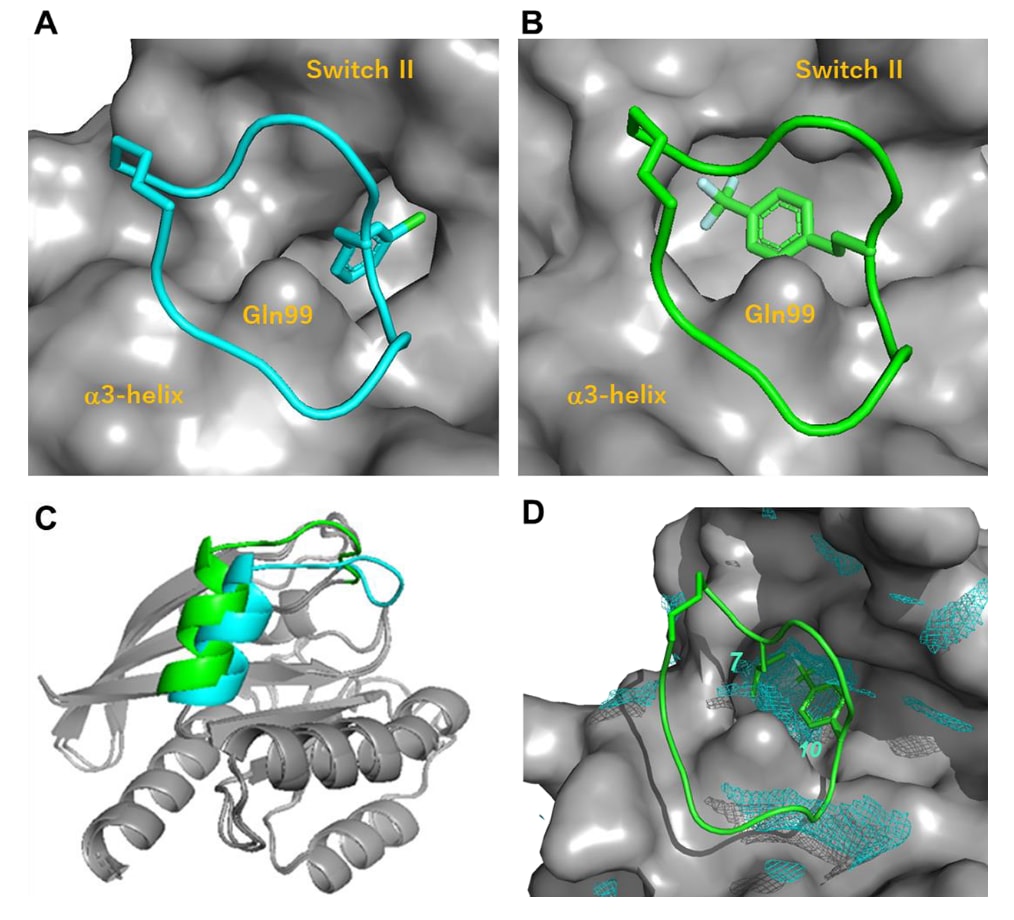
图3|(A,B) 比较KRAS^G12D与化合物1(A;PDB编号7YUZ)及化合物18(B;PDB编号8JJS)的复合结构。可以看到第7位侧链分别占据了不同的结合口袋。(C) 叠合显示KRAS分别结合化合物1(青色)与18(浅绿色)时的整体构象差异。(D) 第7位与第10位的侧链共同位于SII-hole区域。
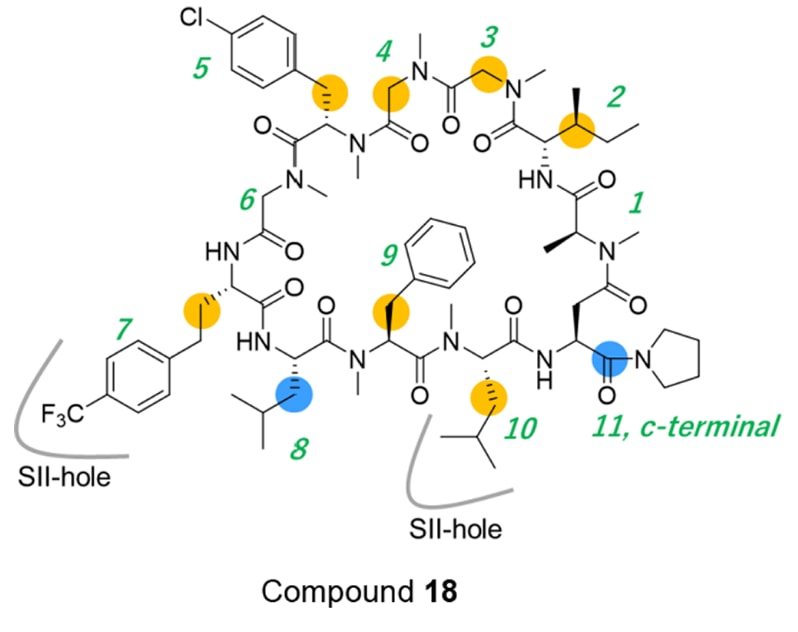
图4|侧链修饰结果示意图。 图中橙色标示的位置代表对生物活性影响显著的修饰位点,而蓝色标示的位置则为**可用于调节理化性质(PCP)**的潜在修饰点。
SII-hole区域侧链的优化(第7与第10位)
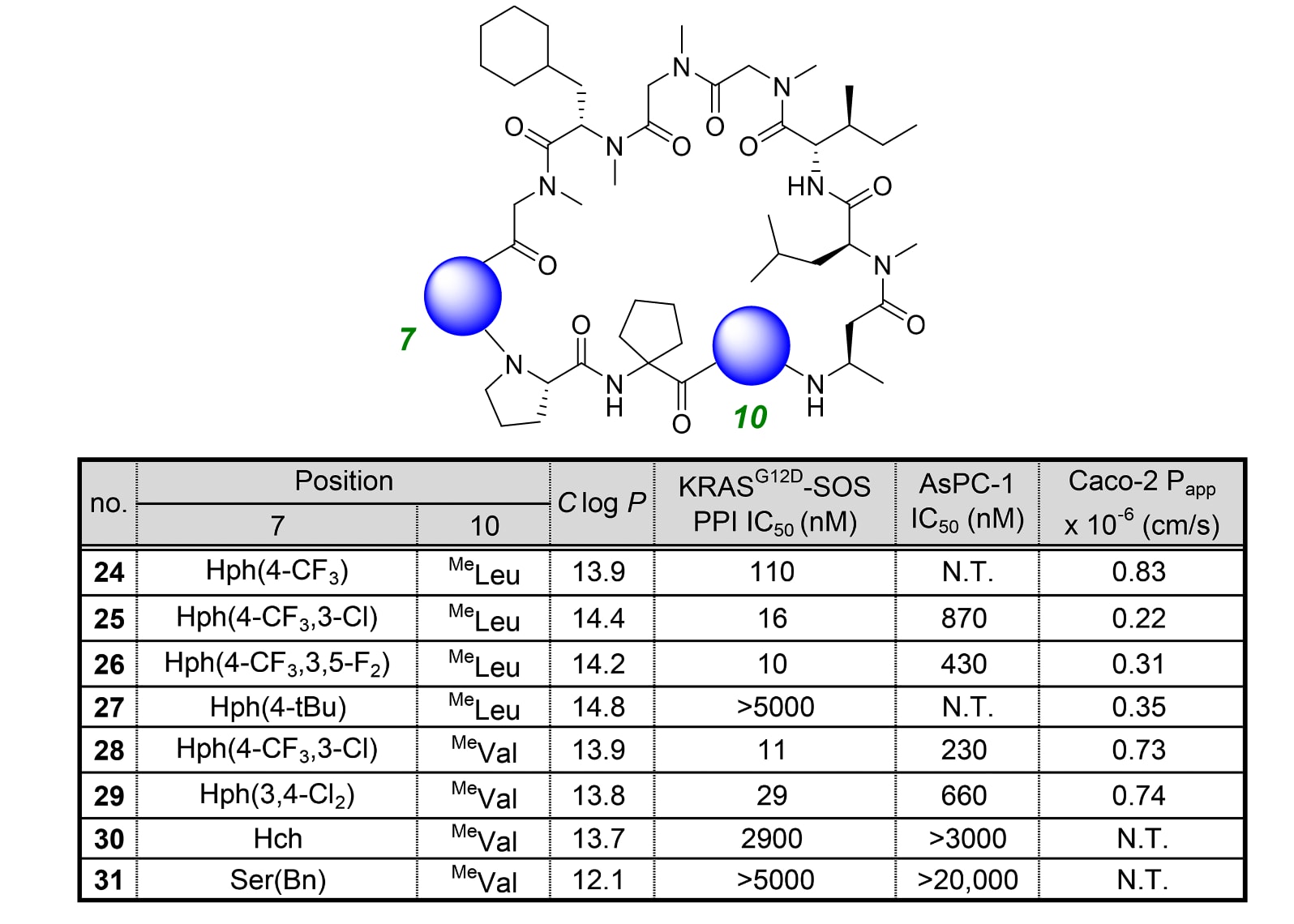
通过对SII-hole区域侧链的精细修饰,研究者实现了超过10倍的活性提升,其IC₅₀降至10 nM,同时仍保持Caco-2通透性高于0.4 × 10⁻⁶ cm/s(见表3)。
为充分利用SII-hole周围3′与5′位置的疏水空间,研究者在这些位点引入了不同的疏水取代基,合成了化合物25、26、28与29。结果显示,3′-氯取代衍生物25与28的PPI抑制活性分别较化合物24提升7倍与10倍;而3′,5′-二氟取代衍生物26表现出11倍更强的PPI抑制效果,IC₅₀达到10 nM。
相比之下,化合物27、30与31在第7位引入了比24更大的取代基,导致活性显著下降,说明SII-hole的空间无法进一步扩张。
化合物26与28兼具强PPI抑制活性与中等膜通透性,在细胞实验中表现出IC₅₀分别为430 nM与230 nM的抑制效果。这表明,尽管其分子量远高于典型小分子药物(约500 g/mol),但这些化合物仍能有效穿透细胞膜并发挥作用。
综上,研究者选定化合物28作为后续优化研究的新起始点。
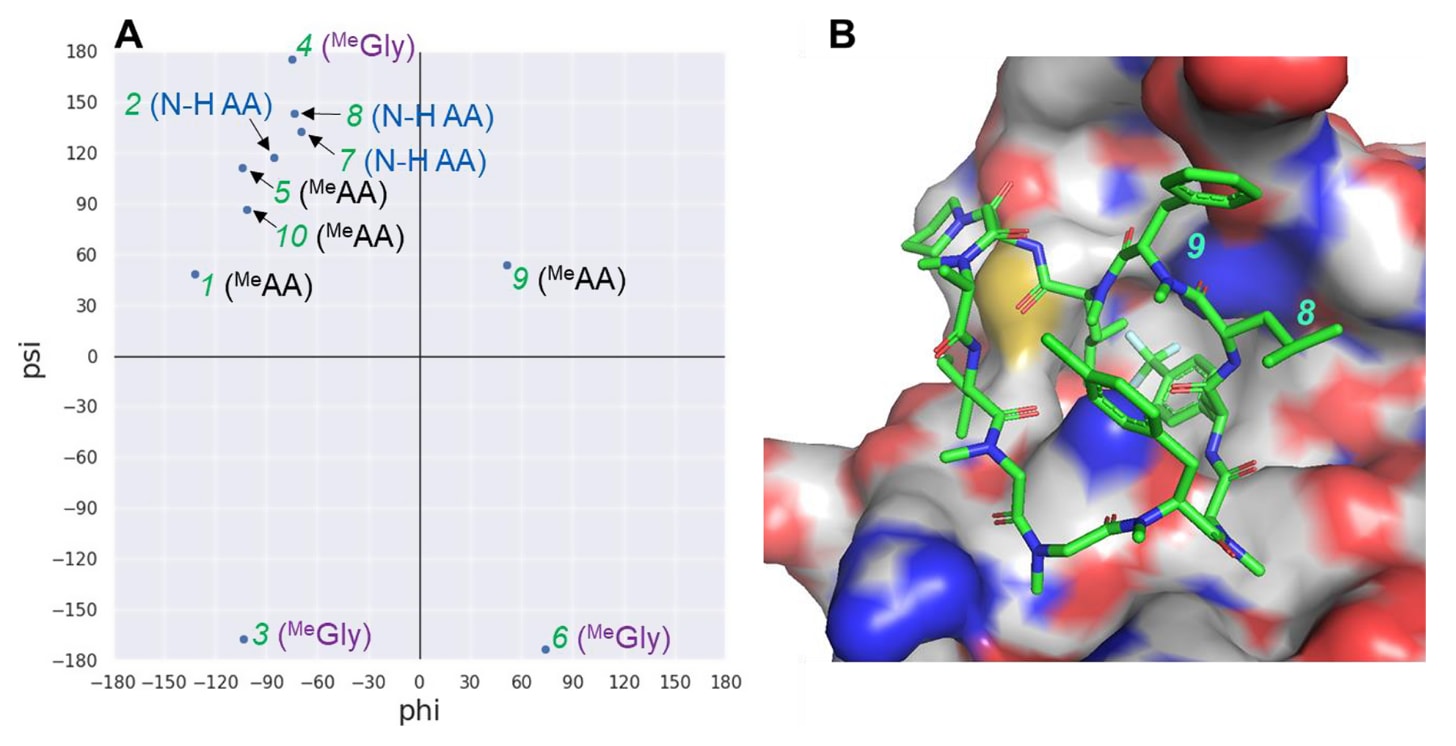
图5|(A) 化合物18与KRAS结合时的φ/ψ角分布图。其中N-H AA表示含N-H的氨基酸,MeAA表示N-甲基化氨基酸。由于第11位的ψ角仅反映C末端侧链的取向而非主链构象,因此未绘制其φ/ψ角(φ=+127°,ψ=−131°)。可见,N-H氨基酸的ψ角范围为+117°至+143°,而N-甲基化氨基酸的ψ角范围为+48°至+112°(不含无α侧链的MeGly)。(B) 化合物18的X射线共晶结构显示,第8位周围存在明显的空旷空间(open space),为后续修饰提供了潜在位点。
表2|具有膜通透性的化合物24的鉴定。

第3与第4位(MeGly–MeGly基序)的修饰
为了验证限制φ/ψ二面角的氨基酸能否稳定活性构象,研究者重点关注了位于α3螺旋沟槽上的第3与第4位柔性MeGly–MeGly基序。MeGly相比普通α-氨基酸,能够覆盖更广的φ/ψ角分布范围(见图6A、B)。
研究团队将MeGly分别替换为Aze(2)、Pro、Pic(2)与MeAla,所得衍生物表现出显著的活性差异(见表4)。当单点以MeAla取代MeGly时(化合物32与36),活性明显下降。在三种环状氨基酸中,**Aze(2)**表现出最佳IC₅₀值,且随着环的尺寸增大(化合物33–35与37–39),活性逐渐降低。值得注意的是,化合物40在第3位含MeAla、第4位含Aze(2),其活性与化合物28相当。
化合物28及32–39之间活性差异的根源通过分子动力学(MD)模拟的φ/ψ角分布得到解释(见图6)。
在化合物18中,第3位MeGly的φ/ψ角(φ=−103°,ψ=−167°)与Ac–MeGly–NMe₂及Ac–Aze(2)–NMe₂所偏好的φ/ψ角一致(见图6A、C),这一结果与化合物28与33的强PPI抑制活性相符。
相反,Ac–MeAla–NMe₂、Ac–Pro–NMe₂及Ac–Pic(2)–NMe₂的φ/ψ角与18中第3位MeGly的构象明显不同(见图6B、D、E),表明在第3位引入MeAla(化合物32)、Pro(34)或Pic(2)(35)会扰动肽链构象,导致活性下降。
第4位的构效关系(SAR)亦可由此解释:18中第4位MeGly的φ/ψ角与Ac–Aze(2)–NMe₂和Ac–Pro–NMe₂所偏好的角度一致,对应化合物37与38的较高活性(见图6C、D)。
此外,第3位β-碳的空间位阻也是影响活性的关键因素。与化合物28相比,化合物32(第3位MeAla、第4位MeGly)活性显著降低,而当第4位改为Aze(2)后(化合物40),活性恢复。
模型结构分析表明:在化合物18中用MeAla替代第3位MeGly后,其α碳引入的甲基与第3位N-甲基及第4位α-碳之间产生空间排斥(见补充图S8a)。
MD模拟比较进一步说明,在第4位引入Aze(2)可使第3位MeAla的φ/ψ角接近18中第3位MeGly的取向(见图6B、F),从而避免β-碳位阻并恢复合理的构象与活性。
最终获得的化合物45与KRAS
将MeGly–MeGly基序转化为MeAla–Aze(2)基序后,在低介电常数溶剂氯仿中形成了稳定的肽骨架构象,该溶剂可模拟脂质膜内部环境(见图7)。
通过¹H NMR与交换谱(ESXY)实验发现:化合物28存在三种主要构象,其互变速率为k=1.2 s⁻¹;而化合物40仅存在两种主要构象,互变速率显著减慢至k=0.028 s⁻¹(见补充图S7)。
类似于环孢素A及其可透膜衍生物在氯仿中维持的受限构象,这一结果表明MeAla–Aze(2)的引入增强了骨架刚性,从而提升了化合物40的Caco-2膜通透性,优于化合物28。
基于类药性肽的药物发现概念验证
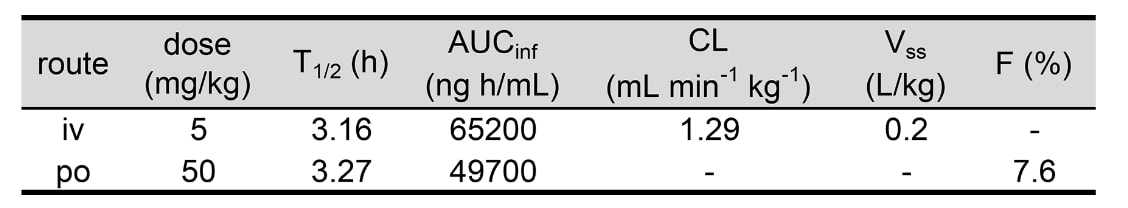
化合物40在细胞水平表现出IC₅₀=110 nM的效力,并在小鼠口服实验中展现出良好的药代动力学特性(PK):
在50 mg/kg口服剂量下,其AUC_inf=49,700 ng·h/mL,口服生物利用度(F)=7.6%,血浆清除率(CL)=1.29 mL/min/kg(见表5)。
这些结果充分验证了研究者提出的从类药性肽出发进行药物发现的策略的可行性与有效性。
表3|第7位与第10位的结构修饰。
化合物45(LUNA18)的确定
在化合物40的基础上,研究者通过基于累积构效关系(SAR)的结构精修,成功获得了临床候选化合物——化合物45(LUNA18)。参考的SAR规律主要包括以下三点:
-
第5位侧链相较脂肪族取代基,更倾向于芳香族结构(见表2中化合物23与24的对比);
-
第11位可作为调节理化性质(PCP)的可行修饰位点;
-
SII-hole中的第7位与第10位能够用于增强生物活性。
精细结构调控的结果见表6。
当将第5位的环己基甲基替换为4-甲基苄基(化合物41)时,活性提升近8倍。
随后在第11位进行N-烷基化修饰(化合物42),形成了新的N-烷基化模式,表现出显著的生物活性。
由于化合物42–45在AlphaScreen体系中已达到抑制平台,因此使用**结合常数(KD)**替代PPI IC₅₀进行测定。
在SII-hole的第10位引入环戊基侧链(化合物43)后,结合亲和力提升18倍。
通过第5位N-Me/N-Et的置换进一步优化Clog P值,化合物44的Caco-2通透性得到改善。
最终,将此前在表2中被鉴定出的Hph(4-CF₃,3,5-F₂)基团引入化合物44的第7位,获得了最终产物化合物45(LUNA18)。
该化合物在AsPC-1细胞中表现出IC₅₀=1.4 nM的极高效力,且Caco-2 Papp=2.3 × 10⁻⁶ cm/s,兼具优异的细胞活性与膜通透性。
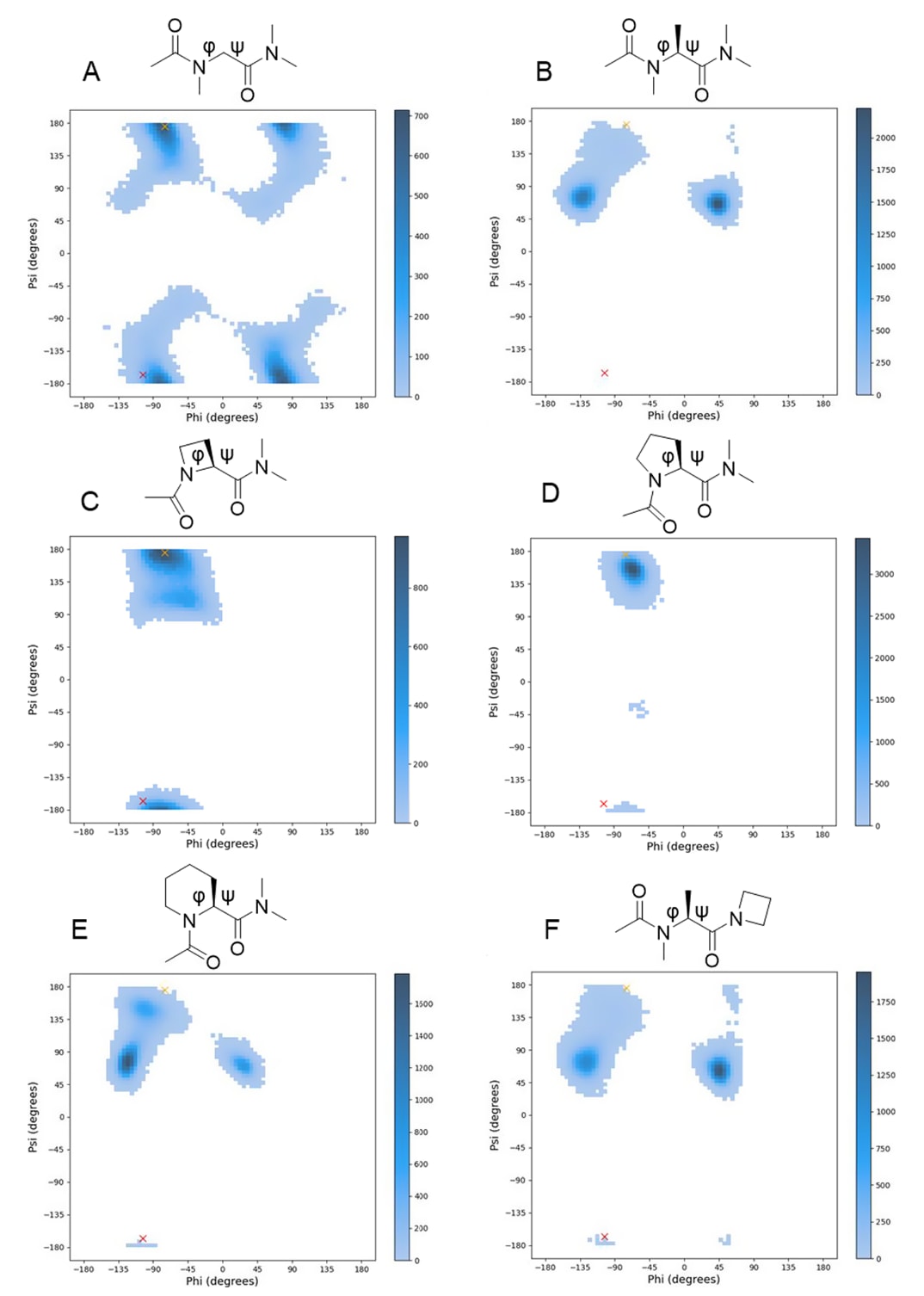
图6|分子动力学(MD)计算得到的φ/ψ角分布。
展示了以下六种模型化合物的φ/ψ角分布结果:Ac–MeGly–NMe₂ (A)、Ac–MeAla–NMe₂ (B)、Ac–Aze(2)–NMe₂ (C)、Ac–Pro–NMe₂ (D)、Ac–Pic(2)–NMe₂ (E) 与 Ac–MeAla–Azetidine (F)。
所有体系均使用Amber18软件进行溶质回火复制交换(REST)模拟。在体系平衡后,从10,000个快照构象中计算φ/ψ二面角,并以5°为区间绘制每个构象分布的直方图。每个区间内仅含1个构象的情况被视为“零构象”。
图中红色叉号表示化合物18中第3位MeGly的φ/ψ角(φ=−103°,ψ=−167°),
黄色叉号表示化合物18中第4位MeGly的φ/ψ角(φ=−75°,ψ=+175°)。
化合物45作为临床KRAS抑制剂
化合物45对多种携带KRAS突变的癌细胞系表现出极强的细胞效应,IC₅₀范围为0.17–2.9 nM。受试细胞系包括:LS180(结肠癌,KRAS^G12D)、GSU(胃癌,KRAS^G12D)、NCI-H441(非小细胞肺癌NSCLC,KRAS^G12V)、NCI-H2122(NSCLC,KRAS^G12C)、MiaPaCa-2(胰腺癌,KRAS^G12C),以及前文提到的AsPC-1。
在NCI-H441与MiaPaCa-2异种移植小鼠模型中进行了体内疗效评估。每日口服给药(10 mg/kg)连续14天后,观察到显著的肿瘤退缩,且无明显体重下降。
为了验证其作为口服药物的潜力,研究者在小鼠、大鼠、猴与犬中进行了药代动力学(PK)研究。化合物45在口服给药后表现出21–47%的生物利用度,显示出优良的跨物种口服吸收性能。
这些结果表明,化合物45具有治疗KRAS突变相关癌症的口服药物潜力。目前,该化合物(LUNA18)已在日本与美国开展I期临床试验。
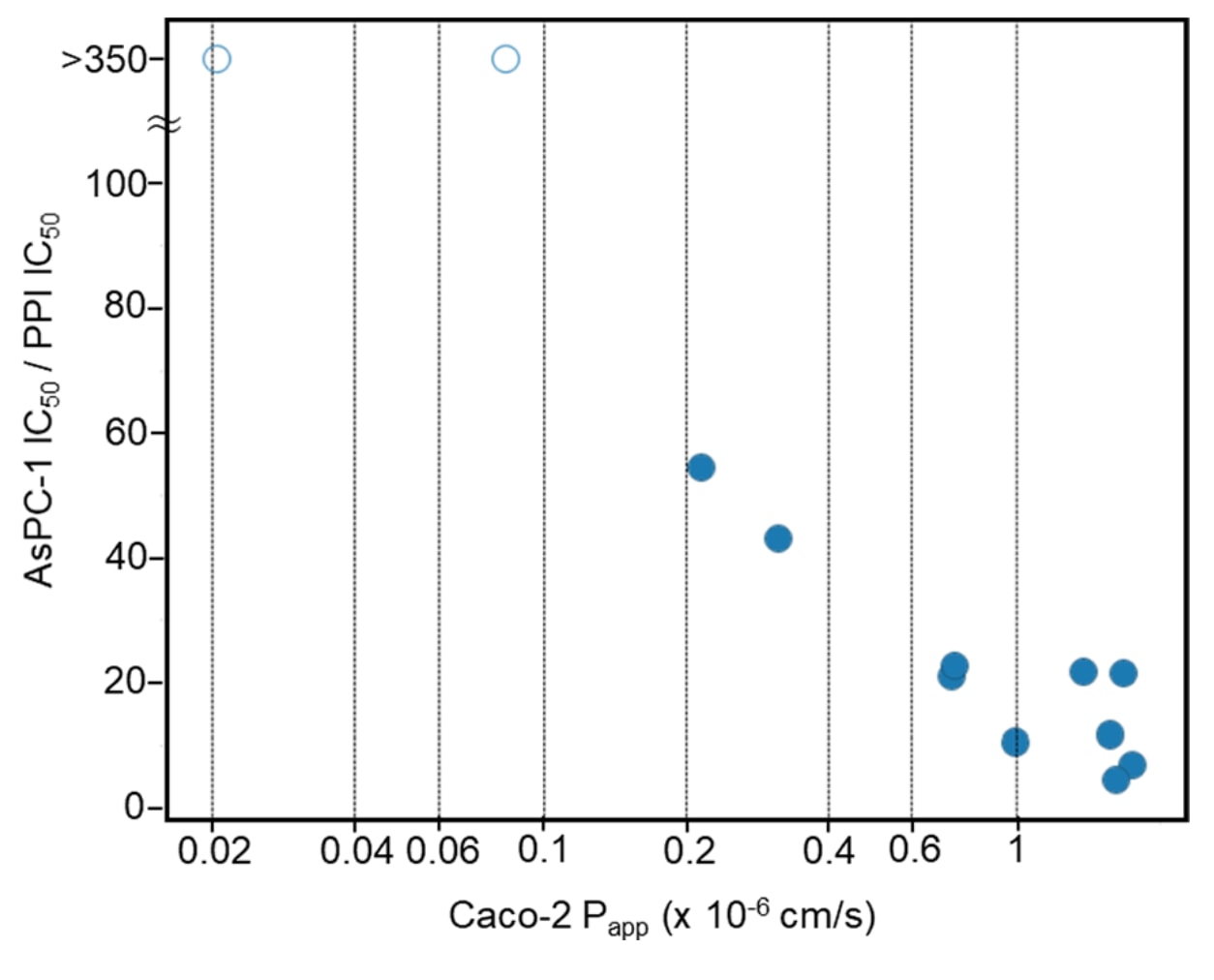
细胞效应与Caco-2膜通透性的相关性
研究还揭示了细胞效力与Caco-2膜通透性之间的显著相关性(见图8)。
细胞内转运能力可通过细胞活性与无细胞活性之比(AsPC-1 IC₅₀ / PPI IC₅₀)来衡量,该比值越小,代表化合物越容易进入细胞内部。该比值与Caco-2的P_app值呈明显关联。
具体而言:
- Caco-2 P_app<0.1 × 10⁻⁶ cm/s的化合物完全缺乏细胞活性,说明无法实现有效细胞转运;
- Caco-2 P_app≈0.2 × 10⁻⁶ cm/s的化合物开始显示有限的细胞转运,其(AsPC-1 IC₅₀)/(PPI IC₅₀)比值通常在40以上,表明进入细胞的效率仍不足;
- 当Caco-2 P_app≥0.6 × 10⁻⁶ cm/s时,化合物通常能实现充分的细胞内转运,其(AsPC-1 IC₅₀)/(PPI IC₅₀)比值约为10,体现出理想的药效表现。
这一观察结果进一步验证了研究者提出的类药性Caco-2通透性阈值≥0.4 × 10⁻⁶ cm/s的标准,该标准基于该环肽体系及已上市药物的实验数据而确立。
表4|
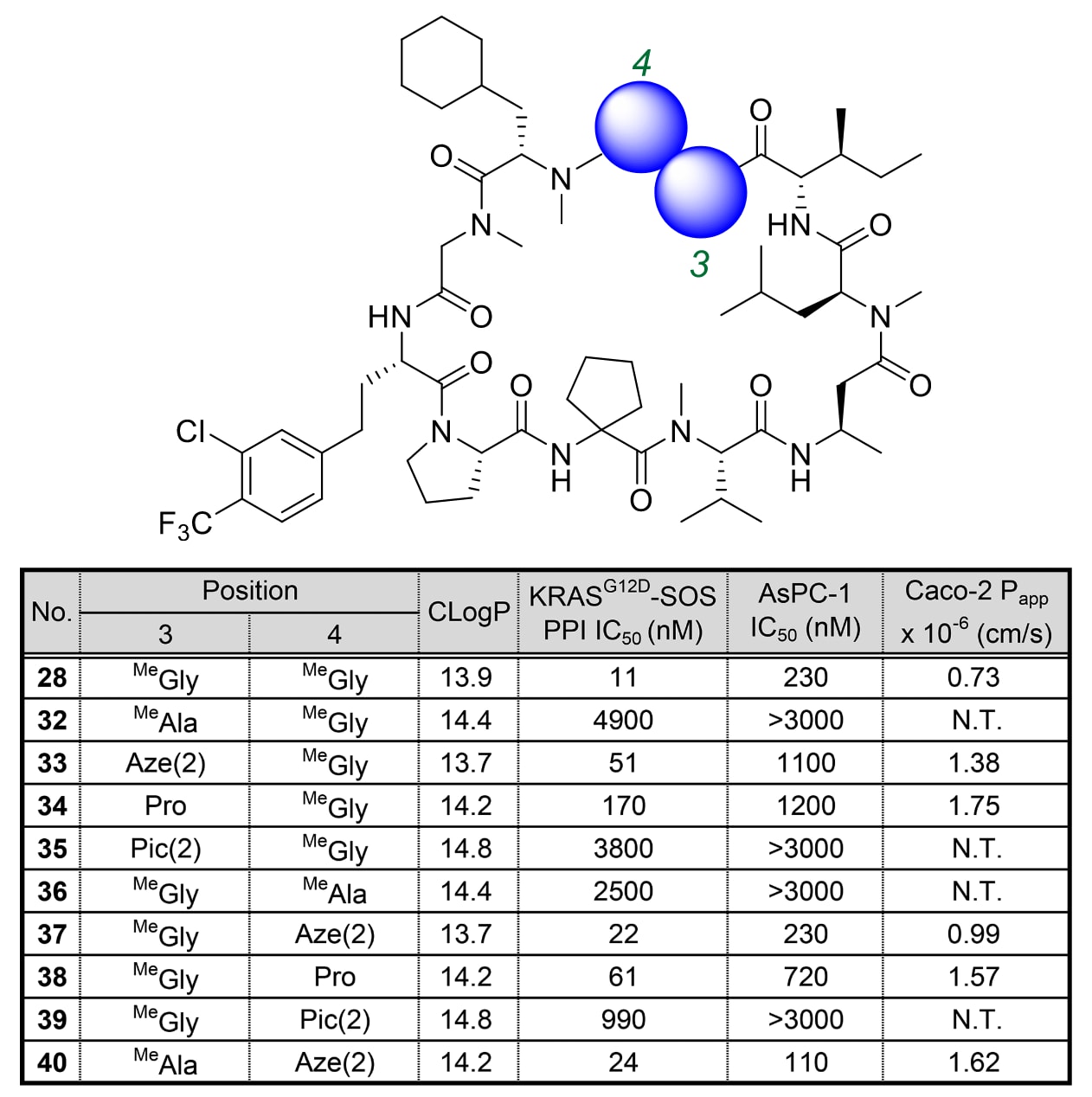
图7|化合物28(上)与化合物40(下)在氯仿-d中的¹H NMR谱图。
圆圈内数字表示主要构象体的N-甲基信号数量,虚线箭头所示峰为次要构象体的N-甲基信号。
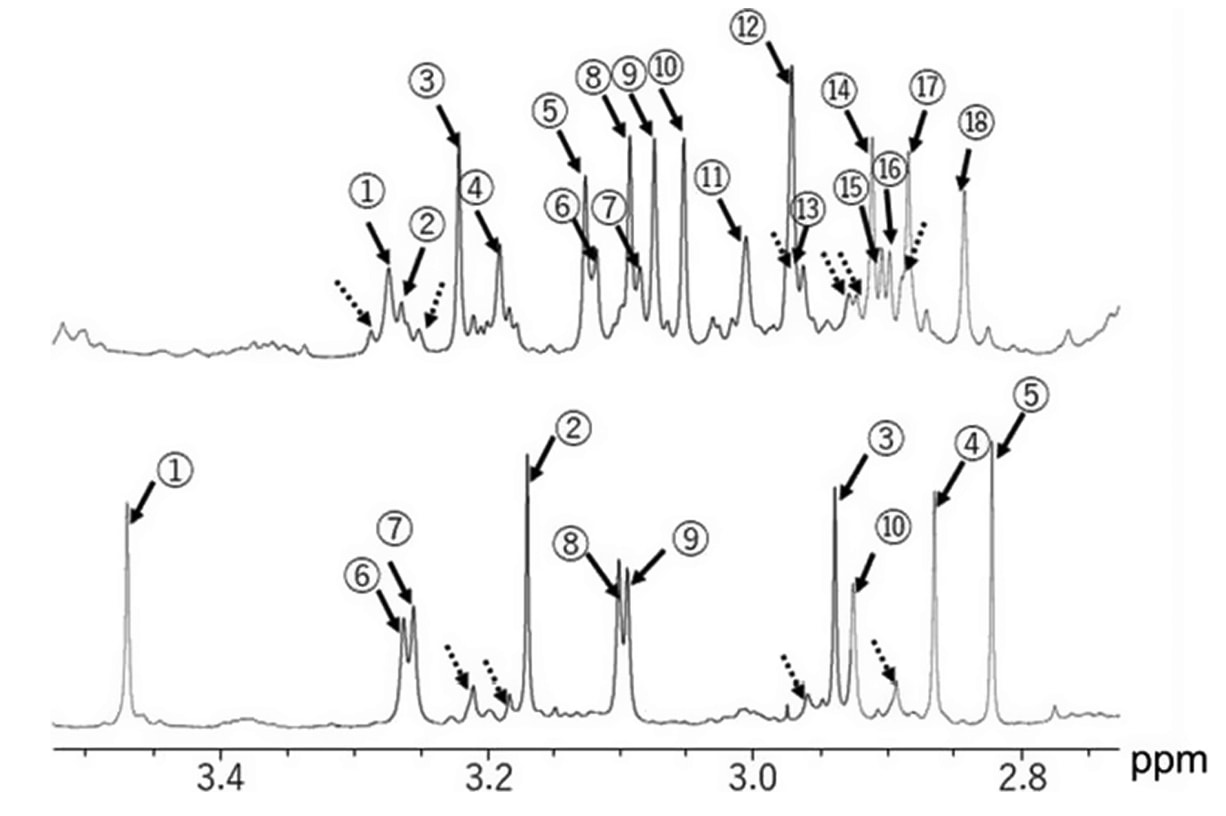
表5|化合物40在小鼠中的体内药代动力学(PK)结果。
3 结论
该研究首次展示了从类药性肽初始化合物出发,通过结构优化获得口服可利用性KRAS抑制剂的成功案例。研究成果为针对顽固靶点的环肽药物发现提供了全新的化学修饰策略。
通过部分调整N-烷基化模式与骨架刚化(backbone rigidification),研究者显著改善了肽分子的膜通透性。基于φ/ψ角图的半定量构象分析在解释构效关系(SAR)时发挥了关键作用。化合物的主要Clog P分布集中在13–15之间,这一较窄范围已足以实现良好的通透性能。
在整个优化过程中,仅使用了疏水性官能团进行侧链修饰,并无需引入极性基团(如胺基或羧酸)即可获得高生物活性。此外,研究发现一次侧链修饰即可诱导KRAS蛋白发生显著构象变化,这一现象源于诱导契合效应(induced-fit)。在所有优化步骤中,仅第7位侧链修饰导致了这种结构变化,并通过X射线晶体结构分析得到确认。
值得注意的是,当靶蛋白发生显著结构变化时,先前建立的SAR往往不再适用于后续优化。因此,对于那些表现出“异常活性”而无法用既有SAR解释的新衍生物,需警惕其可能与靶蛋白的构象重塑相关。
在这种情况下,通过X射线结构验证可有效避免结构优化过程中的不确定性。
未来的研究方向将聚焦于深入理解诱导契合在肽–蛋白识别中的作用机制,这一探索有望进一步推动具有更高亲和力与更丰富生物功能的环肽分子的设计与开发。
表6|第5、7、10与11位的精细结构调控。
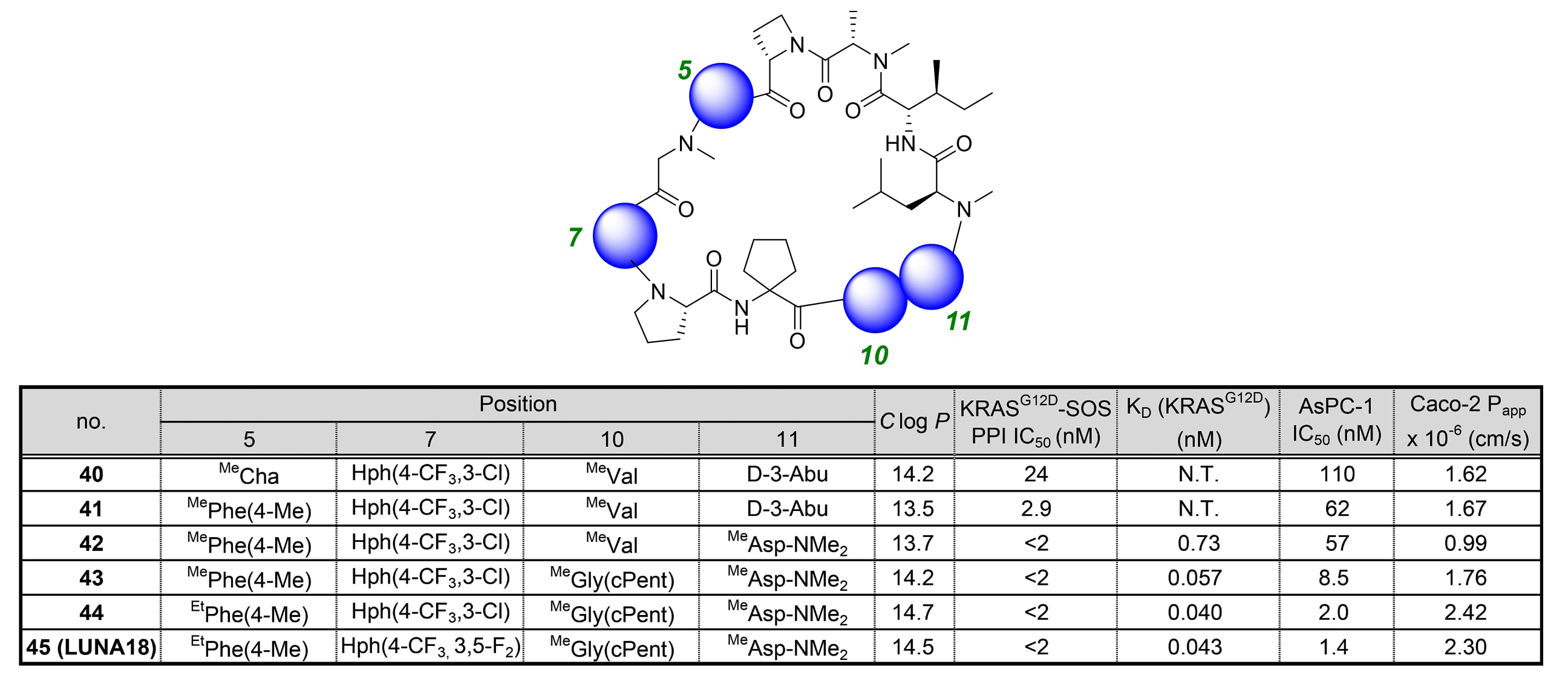
图8|AsPC-1 IC₅₀/PPI IC₅₀(纵轴)与Caco-2膜通透性(横轴)的相关性。 随着Caco-2通透性的增加,细胞活性与无细胞活性之间的差距逐渐减小。图中空心圆表示化合物1与12,它们在细胞中未表现出活性(AsPC-1 IC₅₀>20,000 nM)。