Commun. Chem. 2025 | AEV-PLIG:使用增强数据缩小机器学习评分函数与自由能微扰之间的差距
今天介绍的是发表在 Communications Chemistry 的一项研究,聚焦于如何缩小ML评分函数与自由能微扰(FEP)之间的性能差距。研究团队提出AEV-PLIG,这是一种结合原子环境向量与蛋白–配体相互作用图的全新模型,能够以更精细的结构表征方式提升结合亲和力预测能力。更具特色的是,该研究系统评估了增强数据的价值,通过引入基于模板建模与分子对接生成的额外三维结构,使模型在FEP基准中对同系配体系列的相关性与排序表现大幅提升。结合OOD Test与0 Ligand Bias等更严格的基准后,这项工作展现了对ML模型泛化能力的深度审视。总体来看,AEV-PLIG正逐步逼近FEP+的预测精度,同时在速度与计算成本上拥有压倒性优势,为结构驱动的分子设计带来更高效的技术路径。

获取详情及资源:
0 摘要
Machine learning在实现快速且精确的结合亲和力预测方面展现出巨大潜力。然而,现有模型缺乏稳健的评估体系,并在先导化优化过程中常见的关键任务上表现不佳,例如对同系配体系列进行亲和力排序。这些局限显著削弱了模型在真实药物发现场景中的适用性。
为解决上述问题,研究者首先提出一种全新的注意力机制图神经网络模型AEV-PLIG(atomic environment vector–protein ligand interaction graph),通过原子环境向量结合蛋白–配体相互作用图,强化模型对局部与整体结构特征的表征能力。其次,构建了一个更加贴近真实应用场景的全新分布外测试集OOD Test,用于严格检验模型的泛化能力。
模型在OOD Test、CASF-2016以及一个用于自由能微扰(FEP)计算的基准测试集上均进行了系统评估。结果显示,AEV-PLIG性能具有竞争力,并在与物理学驱动方法的对比中实现更为现实、严谨的性能刻画。此外,通过利用模板建模或分子对接生成的增强数据,可显著提升在FEP基准上的亲和力预测相关性与排序能力,PCC和Kendall’s τ由0.41与0.26分别提升至0.59与0.42。
这些策略共同缩小了机器学习模型与FEP计算之间的性能差距。相比之下,FEP+在该基准上的加权平均PCC和Kendall’s τ分别为0.68和0.49,但AEV-PLIG约快40万倍,展现出极具应用潜力的效率优势。
1 引言
预测蛋白与配体结合时的自由能变化是计算驱动的小分子药物发现领域中的核心任务。在先导化合物发现阶段,需要从大量候选物中筛选出对靶点具有高亲和力的配体;而在hit-to-lead与先导优化阶段,结合亲和力又需要与安全性和生物效能等其他关键性质共同优化。由于化学空间极其庞大,也因此潜在药物数量巨大,计算机辅助药物设计能够在体外实验前通过大规模体内筛选显著加速药物发现过程,从而避免消耗大量实验资源与成本。对虚拟筛选方法而言,高速且准确的结合亲和力预测是显然的核心需求。
传统的计算方法多依赖知识驱动或物理驱动策略,前者使用统计势能,后者依赖分子力场。这些方法在成本与精度之间存在取舍。例如分子对接中的评分函数通常依靠启发式规则与物理近似,因此精度可能受限。相比之下,基于显式溶剂全原子分子动力学的炼金术自由能模拟能够更严格地估计绝对结合自由能或一系列相似配体之间的相对结合自由能。其中自由能微扰(FEP)方法尤为常用,常依托FEP+流程进行,对部分体系已表现出接近化学精度极限的能力。然而,这类方法仍存在诸多限制,包括对力场选择敏感、准备流程复杂、对构效变化的适用范围有限,以及高昂的计算成本,这些因素共同限制了其在大规模虚拟筛选中的应用。
在此背景下,machine learning成为一种极具潜力的替代方案。随着结合亲和力测定数据与高分辨率结构数据不断增加,ML模型能够从标注了实验亲和力的蛋白–配体复合物中学习结构–亲和力映射关系,从而以极低的成本预测新体系的亲和力。近年来,基于特征指纹、物化描述符以及CNN与GNN等深度学习模型快速发展,并常以PDBbind作为训练数据源,再通过CASF基准进行评估。这些模型在传统基准上的表现已经超过许多经典方法。然而,大量研究表明,这些模型往往无法真正学习蛋白–配体相互作用的核心物理规律,在分布外数据上的表现显著下降,可能依赖于配体特征记忆甚至拟合噪声。深度模型的不可解释性进一步加剧了问题,使其在真实药物发现流程中的应用受到限制,而FEP仍是可靠度更高的金标准。
解决这一问题的关键在于构建能够真正考察泛化能力的分布外基准。然而,尽管算法架构有所进步,缺乏高质量数据仍是核心瓶颈。结构驱动方法既需要准确的亲和力测量,也需要高分辨率三维结构。数据增强是扩充数据量和多样性的潜在策略,但在化学和生物体系中生成有意义的合成数据具有挑战性,因为其受到复杂的物理化学约束。
该研究提出了一系列提升ML评分函数适用性的策略,同时实现更加贴近真实应用的性能评估。首先,引入一种结合原子环境向量与蛋白–配体相互作用图的全新特征化方法,称为AEV-PLIG,并构建注意力图神经网络来学习不同邻域环境的重要性,从而捕获复杂的原子间相互作用。其次,构建新的分布外基准OOD test,用于严惩模型对配体或蛋白的记忆行为,并配合多个来源于真实药物项目的测试集,这些测试集中包含典型的同系配体系列,能够更真实地反映实际药物化学场景,并允许ML方法与基于分子动力学的FEP方法直接比较。
为了应对训练数据不足的问题,研究进一步探索增强数据策略,将实验结构与模板建模或分子对接生成的复合物共同用于训练。综合评估显示,AEV-PLIG在多个基准上均表现出与主流方法相当或更优的结果。增强数据显著提升了模型在FEP基准上对同系系列的相关性与排序性能。另外,AEV-PLIG较FEP快数个数量级,准备工作量低,并可直接给出绝对结合亲和力而非相对值。
这项工作凸显了新型特征化方法在捕获蛋白–配体相互作用方面的潜力,强调更稳健基准的重要性,并展示了增强数据在训练快速、准确的ML评分函数中的价值。
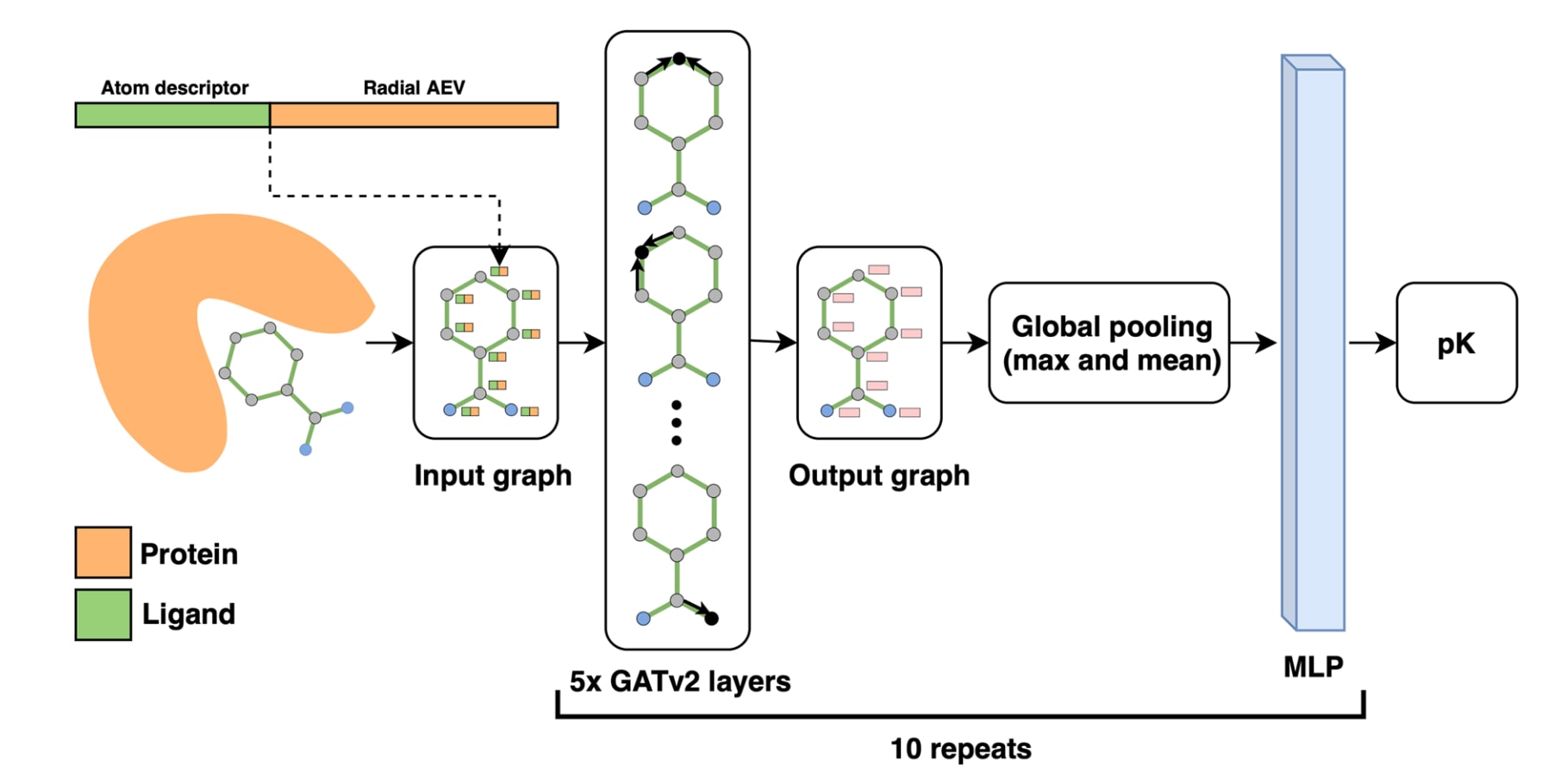
图1 | AEV-PLIG架构示意图。 输入的配体图以原子描述符与径向AEV构成节点特征向量,并依次经过五层GATv2传播,每层包含三个注意力头。随后对图进行全局池化,将max pooling与mean pooling的向量拼接后输入四层MLP。最终预测值由十个独立训练、结构相同但随机种子不同的模型输出均值计算得到。
2 结果
2.1 AEV-PLIG在更具代表性的基准上取得竞争性表现
先前模型AEScore基于AEV实现,与其他先进ML方法表现相当,但仍存在提升空间,因此促成了AEV-PLIG的开发。该模型是一种新的基于注意力的图机器学习评分函数。图模型因其能自然表征分子三维结构与拓扑,已成为蛋白–配体结合亲和力预测的重要方向。在分子复合物层面,Moesser等人提出蛋白–配体相互作用图,将两者间的接触编码为图节点特征。然而该方法仅统计一定截距范围内的蛋白原子数量,并未显式编码分子间距离信息。该研究基于PLIG概念进行了扩展,以配体原子描述符与以配体原子为中心的径向AEV作为节点特征。AEV由原子中心对称函数构成,通过基于高斯函数的径向或角度项描述局部化学环境。以往AEV在结合亲和力预测中通常仅依赖元素类型,而未显式考虑原子连通性,因此推断采用ECIF的原子类型可提供更细致的化学环境表征。此外,虽然AEV通常含径向与角度两部分,该研究使用的AEV-PLIG仅采用包含分子间成对相互作用的径向部分。模型架构进一步利用增强版的GATv2层,使图神经网络具有更强的表达能力。蛋白–配体复合物对应的图通过五层GATv2传播,再经全局池化,最后通过四层MLP输出预测的结合亲和力pK。
在CASF这一广泛使用的“评分基准”上评估AEV-PLIG后发现,其表现优异,PCC为0.86,Kτ为0.67,超过或接近多种经验与ML方法。然而,尽管CASF表现良好,已有研究指出许多ML模型会在训练集中记忆样本,从偏差甚至噪声中学习,而非掌握真实生物物理规律。CASF源于PDBbind的数据采样方式也可能导致污染,使训练集与测试集存在相似样本,从而放大记忆性。此外,两者在晶体分辨率及配体原子数量上的差异亦可能影响预测。这些因素综合造成对模型能力的乐观估计,无法真实反映其应对未见蛋白–配体体系的能力。同时,大部分ML模型并未显式编码蛋白–配体相互作用,使其易对配体或蛋白特征过拟合。
为解决这些问题,研究界提出多种构建代表性测试集的方法,包括按时间划分训练与测试,或采用基于骨架或靶标序列的划分,但这些方法仍未完全规避结构或拓扑上的相似性。该研究提出新的OOD测试集,通过最小化测试集与PDBbind训练集在配体相似度、蛋白序列相似度与蛋白口袋相似度上的重叠,从而构建更严格的分布外基准。最终得到295个复合物用于测试。对该测试集的预测显示,AEV-PLIG的相关性与排序显著低于CASF,这与以往研究一致,反映出OOD场景的挑战性。该基准也便于在统一条件下公平比较不同模型的泛化能力。
进一步在统一训练条件下比较多种模型后发现,所有模型在OOD Test上的性能均显著下降。例如OnionNet与Pafnucy的PCC分别从0.83降至0.57,0.76降至0.55,表明单一基准并不能真实反映模型能力。同时,在一个由相同配体与不同蛋白构成的“零配体偏倚”测试集中,所有模型表现均下降,显示其明显依赖配体特征。AEV-PLIG在此基准上取得最高的PCC=0.37,Kτ与OnionNet-2相近。与AEScore相比,AEV-PLIG在所有基准上均表现更优,这与其采用更合理的径向AEV、ECIF原子类型及更强的图注意力架构相关。
尽管OOD能力评估重要,ML模型的核心应用仍在早期药物发现,需要对相似结构的小化学变化进行排名,因此必须在贴近此情景的基准上检验模型性能。该研究使用Ross等人整理的FEP基准,这是目前最大规模的公共蛋白–配体系列数据集,涵盖超过50个靶点与1200多个配体。其结构特点包括大量同系系列、复杂构效关系与显著的活性悬崖。在此基准上,AEV-PLIG的加权平均PCC与Kτ分别为0.41与0.26,明显低于CASF-2016。其他ML方法同样表现一般,而FEP+的加权平均PCC与Kτ分别为0.68与0.49,保持领先。FEP基准通常具有较窄的实验亲和力范围,使得获得高相关性更加困难。然而,在具有较多配体的系列中,AEV-PLIG的表现明显改善。总体而言,这些结果展示了CASF-2016性能与更真实场景之间存在令人担忧的差距,强调评估ML模型需依赖多样化、贴近实际的基准才能真实判断其能力。
虽然AEV-PLIG在多数基准中表现领先,但其并非最终最优模型。重要的是通过这些比较揭示不同表示方法与架构的优势与局限,从而推动更可靠的蛋白–配体结合亲和力预测方法的发展。
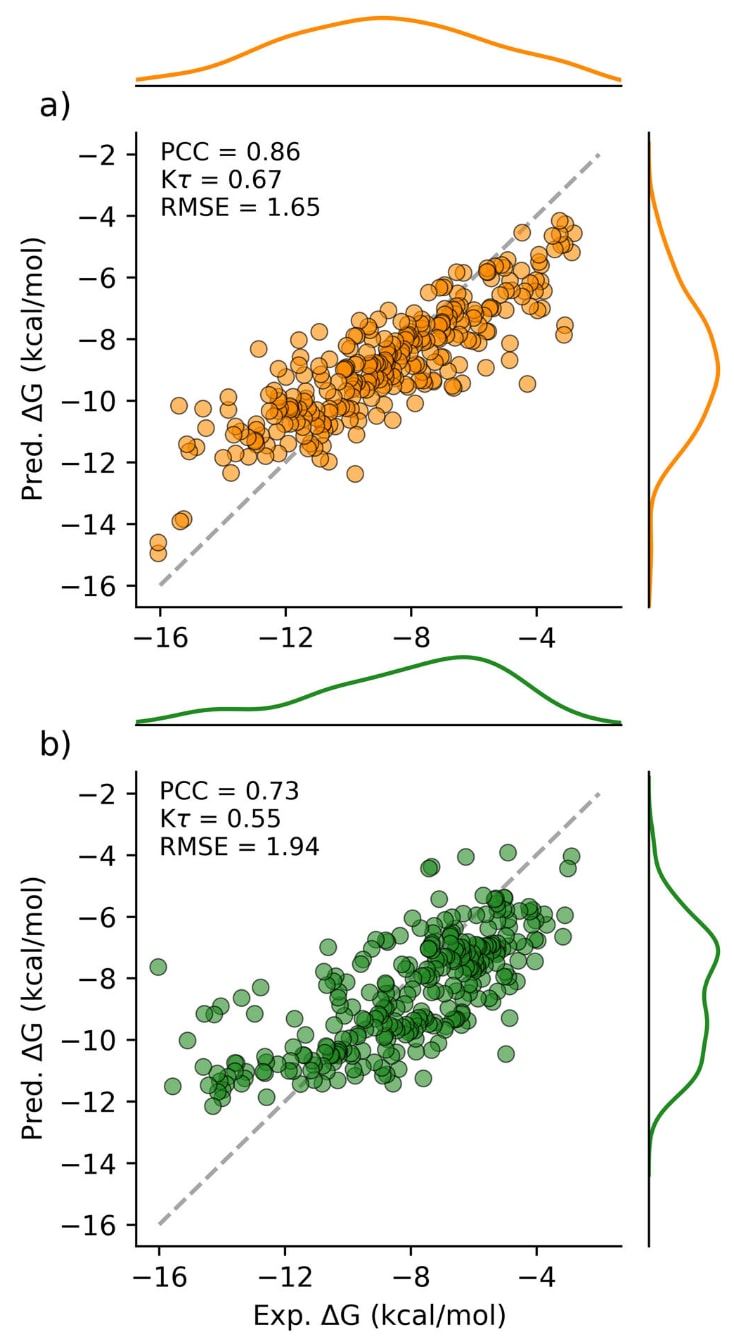
图2 | AEV-PLIG模型预测值与实验结合自由能的散点图。 a为使用PDBbind v2020训练并在CASF-2016基准上评估的模型,b为使用Refined2020+训练并在OOD Test基准上评估的模型。RMSE以kcal/mol表示。
2.2 增强训练数据能提升AEV-PLIG的性能
影响ML模型表现的核心问题之一在于缺乏多样且高质量的训练数据。与其他领域相比,这一困难在生化应用中尤为突出,因为获取新实验数据往往依赖高昂且耗时的实验测定,例如X射线晶体学或等温滴定量热。基于序列的亲和力预测方法尝试绕过结构依赖,通过蛋白与配体的非结构表征,在更大规模的亲和力测定数据(如来自ChEMBL)上训练,在目标蛋白序列已知但结构未知时尤其有用。然而,这类模型的表现仍未达到结构驱动方法的水准,并且在某些情况下仍需先验结构信息,例如配体结合口袋的位置。
与此并行,Li等人提出了一种基于模板的建模策略,为PDBbind中的蛋白靶点生成具有已知亲和力的配体三维结构。该策略最终构建了一个包含69816个额外复合物的增强数据集,称为BindingNet。这些复合物包含模型结构与实验亲和力数据。关键在于,这些化合物须与PDBbind v2019中所包含的蛋白靶点结合,并与其对应配体具有足够结构相似性,因此增强数据显著扩大了配体的结构覆盖范围。BindingNet还包含超过五万条活性悬崖信息,覆盖455个靶点。
类似地,Gilson等人亦在BindingDB项目中生成了增强数据。BindingNet Docked Congeneric Series(BindingDB-DCS)使用Surflex对接程序建模蛋白–配体复合物,其思路与BindingNet一致,依赖于一个同系配体的晶体结构作为模板生成对接构象。该数据集最终包含8822个复合物,来自1322个同系系列,从而进一步提升了配体覆盖度。
基于这些增强数据,该研究以PDBbind v2020为基础,加入BindingNet与BindingDB-DCS共同训练AEV-PLIG模型,并在上述各类基准上评估其性能。在CASF-2016、OOD Test及0 Ligand Bias三个基准上,增强数据未带来显著改善。然而,在FEP基准上,增强数据带来了实质性提升,无论对单个体系或平均表现均显著提高,PCC由0.41增至0.59,Kτ由0.26增至0.42。通过对加权平均PCC与Kτ的差异进行假设检验,确认该提升具有统计学意义(P < 0.0001)。
进一步分析增强数据在不同基准中的差异作用,可从数据本身的特性解释。FEP基准中每个蛋白对应多个配体系列,而CASF-2016与OOD Test由独立复合物组成,0 Ligand Bias由高度相似的配体与多个不同蛋白构成。增强数据恰恰与FEP基准的结构分布最为一致,具备较高的同一蛋白的配体覆盖率,而OOD Test与0 Ligand Bias则刻意包含与训练集不相似的蛋白或极低的配体覆盖度。因此,训练集中存在与测试体系结构相似的样本会提高预测效果。
基于此,研究者进一步研究了配体相似性对模型性能的影响,通过从训练集中移除与测试配体Tanimoto相似度(Ts)大于0.9、0.8、0.7或0.6的样本。结果显示模型性能确实受类似配体的存在影响,但影响幅度较为温和,例如将阈值从0.9收紧至0.6后,仅降低约0.05的相关性单位。即便采用严格阈值Ts ≤ 0.6,在FEP基准上,加入BindingNet和BindingDB-DCS仍比仅使用PDBbind v2020取得更好的平均表现。
进一步通过逐步增加增强数据量训练模型,结果显示模型在FEP基准上的性能随数据量增加而提升,并未在测试范围内饱和,暗示未来加入更多增强数据仍具潜力。
随后,将AEV-PLIG与FEP+在FEP基准上进行比较。总体来看,使用增强数据训练的AEV-PLIG模型的平均PCC与Kτ仍略低于FEP+,两者差异具有统计显著性(PCC P=0.012,Kτ P=0.014)。为分析单个靶点的表现,该研究进一步选取包含至少25个配体的14个靶点系列进行比较。整体趋势与综合结果一致:在14个靶点中,AEV-PLIG与FEP+在8个靶点上PCC表现相近,在11个靶点上Kτ表现相近。FEP+在五个靶点上具有更佳相关性,在两个靶点上具有更佳排序能力,而AEV-PLIG在BACE1系列上表现优于FEP+。对应的散点图见Fig. S6。
考虑到FEP+通过MD模拟采样大量蛋白与配体构象,其在精度上的优势是预期之内的;然而ML模型在速度、便利性与灵活性上具有无可比拟的优势。以该研究为例,AEV-PLIG训练完成后可在33 ms内基于三维结构输出一次预测,而FEP+执行四个配体扰动需要约24小时与8块显卡,这意味着AEV-PLIG至少快约40万倍。此外,即便从零开始使用约95000个增强数据训练整个AEV-PLIG模型,也仅需单卡28小时。训练完成后,AEV-PLIG可在几乎无需系统预处理的情况下直接用于任意体系,而FEP+通常需要复杂且体系特定的准备流程。
因此,ML方法,尤其是AEV-PLIG,可作为MD方法的有力补充,在许多情景下提供具有竞争力的预测,同时显著减少计算成本。FEP方法在精度上仍具优势,而AEV-PLIG在需要大规模快速筛选时具有显著价值,例如对成千上万个库内化合物或生成模型采样的分子进行快速排序。
表 1 | AEV-PLIG与其他先进结合亲和力预测方法在五个不同基准上的性能指标。
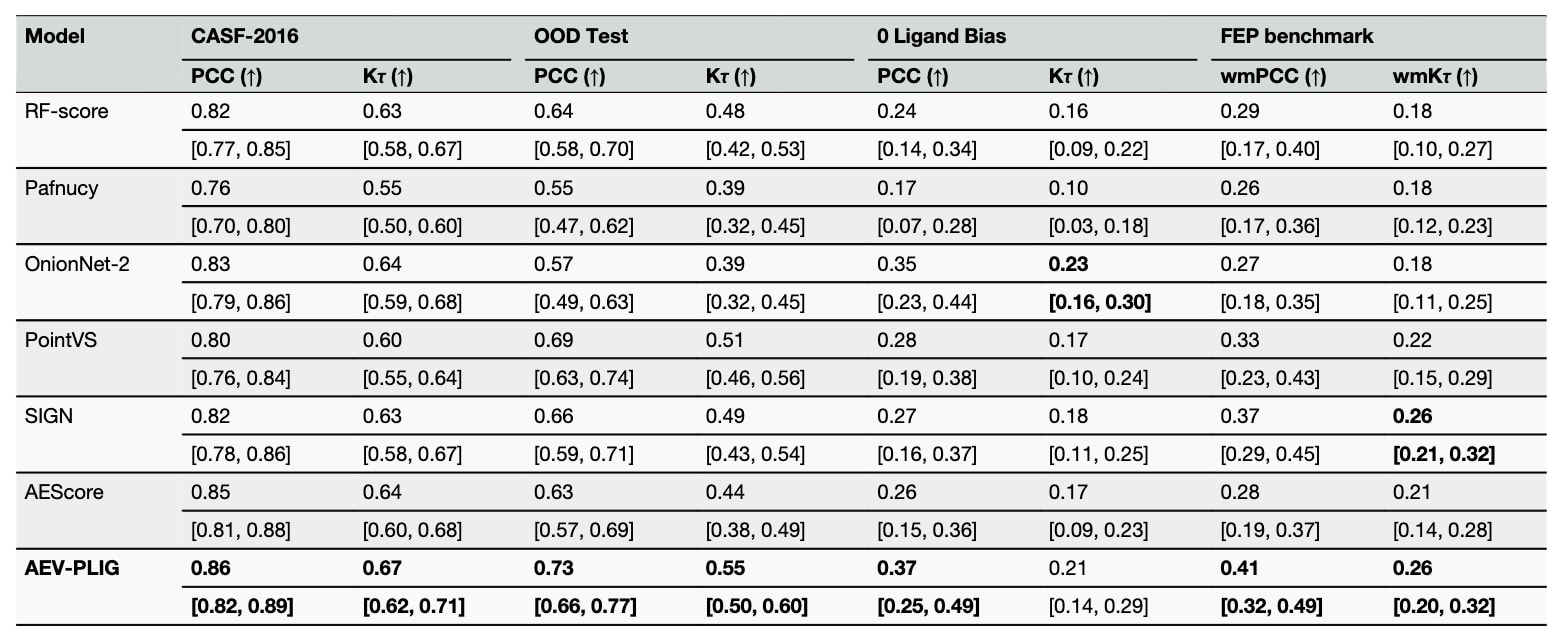
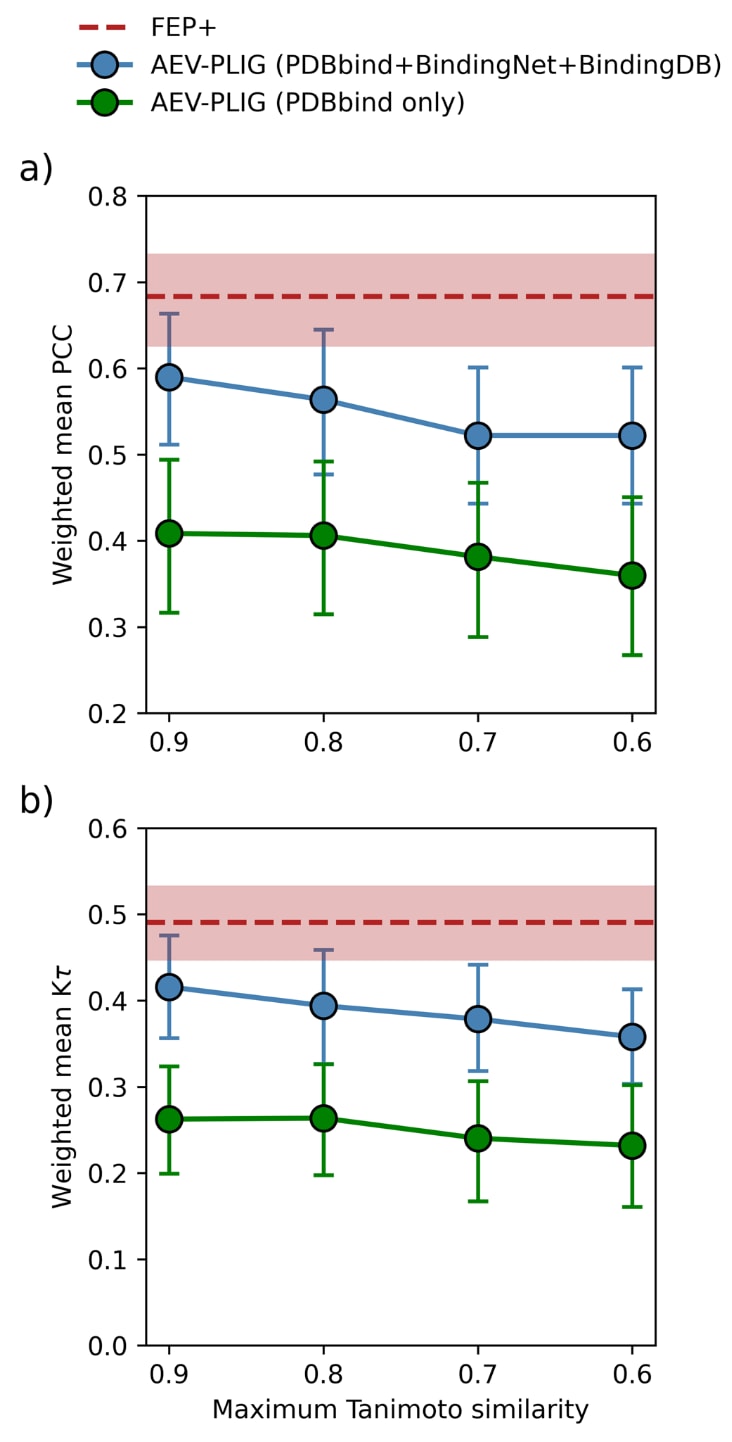
图3 | AEV-PLIG模型在FEP基准上的表现。 a为加权平均PCC,b为加权平均Kτ,纵轴为模型性能,横轴为训练集与测试集之间的配体Tanimoto相似度(Ts)上限。模型分别基于仅使用PDBbind v2020(绿色)或使用PDBbind v2020结合BindingNet与BindingDB-DCS进行训练。FEP+在FEP基准上的表现以红色显示。Tanimoto相似度基于ECFP6指纹计算,误差条表示95% BCa自举置信区间。
3 结论
该研究探讨了多种策略,以提升machine learning评分函数在药物发现场景中的适用性,并提供更为真实的性能评估。研究还提出一种新的结合亲和力预测方法AEV-PLIG,将原子环境向量与蛋白–配体相互作用图结合,通过注意力图神经网络架构捕获决定结合亲和力的复杂相互作用特征。在多个基准上对AEV-PLIG与RFScore、Pafnucy、OnionNet-2、PointVS、SIGN及AEScore进行了系统评估,所涉及的基准包括用于检验分布外表现的OOD Test、用于贴近真实药物研发体系的FEP基准,以及用于惩罚记忆行为的0 Ligand Bias。结果显示,与广泛使用的CASF-2016相比,这些更严格或更贴近真实的基准均导致不同程度的性能下降。尽管AEV-PLIG整体表现良好,但没有任何单一模型能够在所有基准上显著优于其他模型,这说明新模型与新特征的评估必须依赖多样化的测试体系。
针对生化数据普遍稀缺的问题,该研究探索使用增强数据进行训练,即保留实验亲和力数据,但使用模板对齐或分子对接生成三维蛋白–配体结构。增强数据显著提升了AEV-PLIG在药物研发典型体系中的排序能力,这些体系通常以一系列结构高度相似的小分子为对象,通过细微结构修饰调节其对靶点的结合亲和力。分析还表明,未来加入更多增强数据可能进一步提升性能。随着高精度蛋白–配体结构预测模型的出现,例如AlphaFold 3、Umol与NeuralPlexer,这类增强数据的获取或将变得更加容易。基于此,该研究认为更丰富的结构数据可能在未来促进ML方法达到严谨模拟方法(如FEP)在预测结合自由能方面的精度。
总体而言,该研究显示,AEV-PLIG等ML模型正逐步缩小与炼金术FEP+等“金标准”方法之间的差距,在使用增强数据训练后,在FEP基准中的许多同系系列上已达到接近的性能。然而,由于FEP方法仍在持续进步,这一差距是否会继续缩小仍需进一步观察。同时,ML模型如AEV-PLIG具有许多实际优势,例如预测速度比基于MD的工作流程快五个数量级,并且无需手动重新参数化,因而非常适合高通量虚拟筛选。整体来看,这些新方法为加速药物发现早期阶段提供了强大且互补的技术路径。
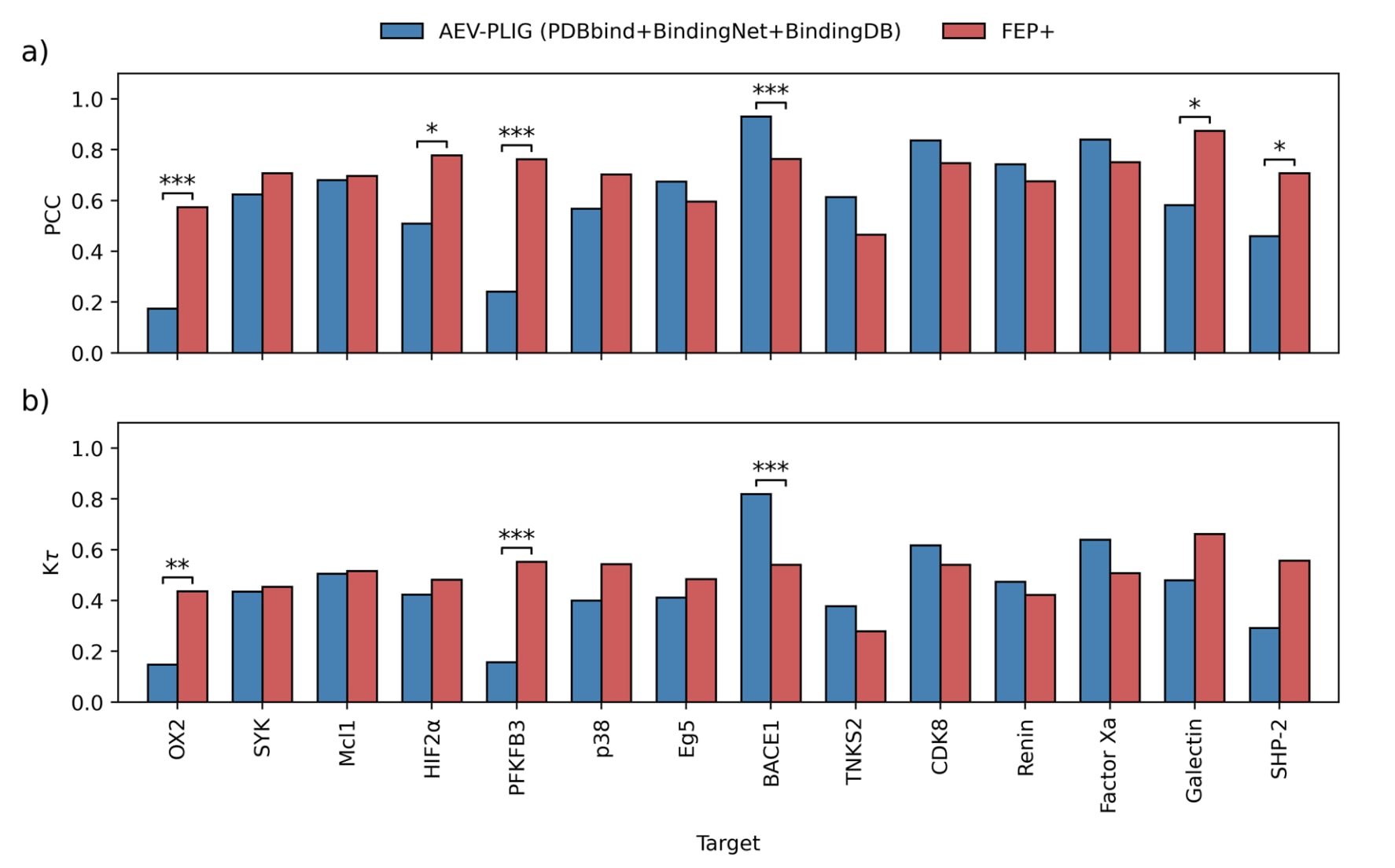
图 4 | FEP基准中包含25个及以上配体的靶点上,FEP+与AEV-PLIG模型的性能比较。 a为PCC,b为Kτ。AEV-PLIG模型基于PDBbind v2020、BindingNet与BindingDB-DCS数据训练(Ts ≤ 0.9)。为便于展示,未绘制误差条。星号表示两种方法之间存在统计学显著差异(*P < 0.05,**P < 0.01,***P < 0.001),其统计计算方式详见“Methods”。