Science 2023 | CLEAN: 利用对比学习的酶功能预测
今天介绍的研究发表于 Science ,题为“利用对比学习的酶功能预测”。随着DNA测序和宏基因组学的迅速发展,科学家获得了数以亿计的蛋白质序列,但其中仅有极少部分经过实验验证。如何准确注释这些序列的功能,尤其是研究不足或具有多重活性的酶,已成为生物信息学的一大挑战。传统基于序列相似性的注释工具(如BLASTp)在序列同源性较低时预测准确性显著下降,而现有的机器学习模型又受限于数据不平衡与标签稀缺问题。
为此,研究者提出了CLEAN(Contrastive Learning–Enabled Enzyme Annotation)模型,首次将对比学习引入酶功能预测。该方法通过学习酶序列的功能嵌入空间,使欧氏距离能够反映酶的功能相似性。相比多标签分类模型,CLEAN能同时利用正负样本信息,从而在稀有酶类别上保持稳健性能。通过在UniProt高质量数据上的训练与独立验证,CLEAN在多个基准测试中表现优异,显著超越BLASTp、DeepEC与ProteInfer等先进方法。
更为重要的是,CLEAN在实验中成功注释了36种未表征卤素化酶,纠正了错误标注,并识别出具有三重催化功能的多活性酶SsFlA。这项研究展示了对比学习在解决生物数据不平衡问题中的潜力,也为功能基因组学、酶工程与合成生物学开辟了新的方向。

获取详情及资源:
0 摘要
酶功能注释是生物信息学中的核心难题,虽然已有众多计算工具被开发出来,但大多数工具仍难以准确预测那些研究较少、功能未知或具有多重活性的蛋白质的功能注释,如酶委员会(Enzyme Commission,EC)编号。研究者提出了一种名为CLEAN(Contrastive Learning–Enabled Enzyme Annotation,对比学习驱动的酶注释)的机器学习算法,与当前最先进的工具BLASTp相比,该方法在准确性、可靠性和灵敏度上均有显著提升。对比学习框架使CLEAN能够自信地实现三项关键功能:(i)为研究不足的酶进行注释;(ii)纠正错误标注的酶;(iii)识别具有两种或以上EC编号的多功能酶。通过系统的计算实验与体外实验,研究者验证了这些能力。该工具有望广泛应用于未知酶功能的预测,从而推动基因组学、合成生物学以及生物催化等多个领域的发展。
1 引言
DNA测序技术的发展,尤其是基因组学与宏基因组学工具的出现,促使研究者在生命各个分支的生物体中发现了大量蛋白质序列。例如,UniProt知识库已收录约1.9亿条蛋白质序列。然而,其中仅不到0.3%(约50万条)经过人工审核,而在这些经审核的序列中,只有不到19.4%具有明确的实验支持。因此,蛋白质功能注释在很大程度上依赖于计算方法。然而,大规模社区协作的蛋白质功能注释关键评估项目(CAFA)研究发现,现有计算工具自动注释的酶中约有40%被错误标注。由此可见,蛋白质功能注释依然是蛋白科学中极具挑战性的问题。尤其是研究较少的蛋白质与具有多重功能的“多活性”蛋白质,其注释的不平衡性严重阻碍了生物医学研究与药物发现的进展。
酶委员会(Enzyme Commission,EC)编号是目前最常用的酶分类体系,通过四位数字精确描述酶的催化功能。由于实验手段表征目标酶功能往往耗时且成本高昂,研究者开发了多种基于计算的酶功能注释方法,包括但不限于序列相似性、同源性、结构特征以及机器学习等方法。其中,基于序列比对的BLASTp工具是应用最广泛的代表。然而,BLASTp及类似比对方法仅依赖序列相似性进行功能推断,当序列相似性较低时,预测的可靠性显著下降。另一方面,现有的机器学习模型(如DeepEC与ProteInfer)多基于多标签分类框架,但受限于生物学领域常见的训练数据稀缺与不平衡问题,难以实现高精度预测。因此,亟需一种具有更高准确度与更全面EC覆盖率的稳健工具,以挖掘大量尚未表征的蛋白质潜能,拓展对蛋白质功能空间的理解。
为此,研究者开发了一种名为CLEAN(Contrastive Learning–Enabled Enzyme Annotation,对比学习驱动的酶注释)的机器学习模型用于酶功能预测。CLEAN以氨基酸序列为输入,输出按照可能性排序的酶功能列表(以EC编号为例),其训练数据来自UniProt中的高质量数据集。为了验证CLEAN的准确性与稳健性,研究者进行了大量计算实验,并进一步利用自建的36个未表征卤素化酶数据库进行EC编号注释与体外实验验证。结果表明,CLEAN在这些任务中均优于BLASTp及其他先进的机器学习模型,展现出在酶功能预测领域的显著优势。
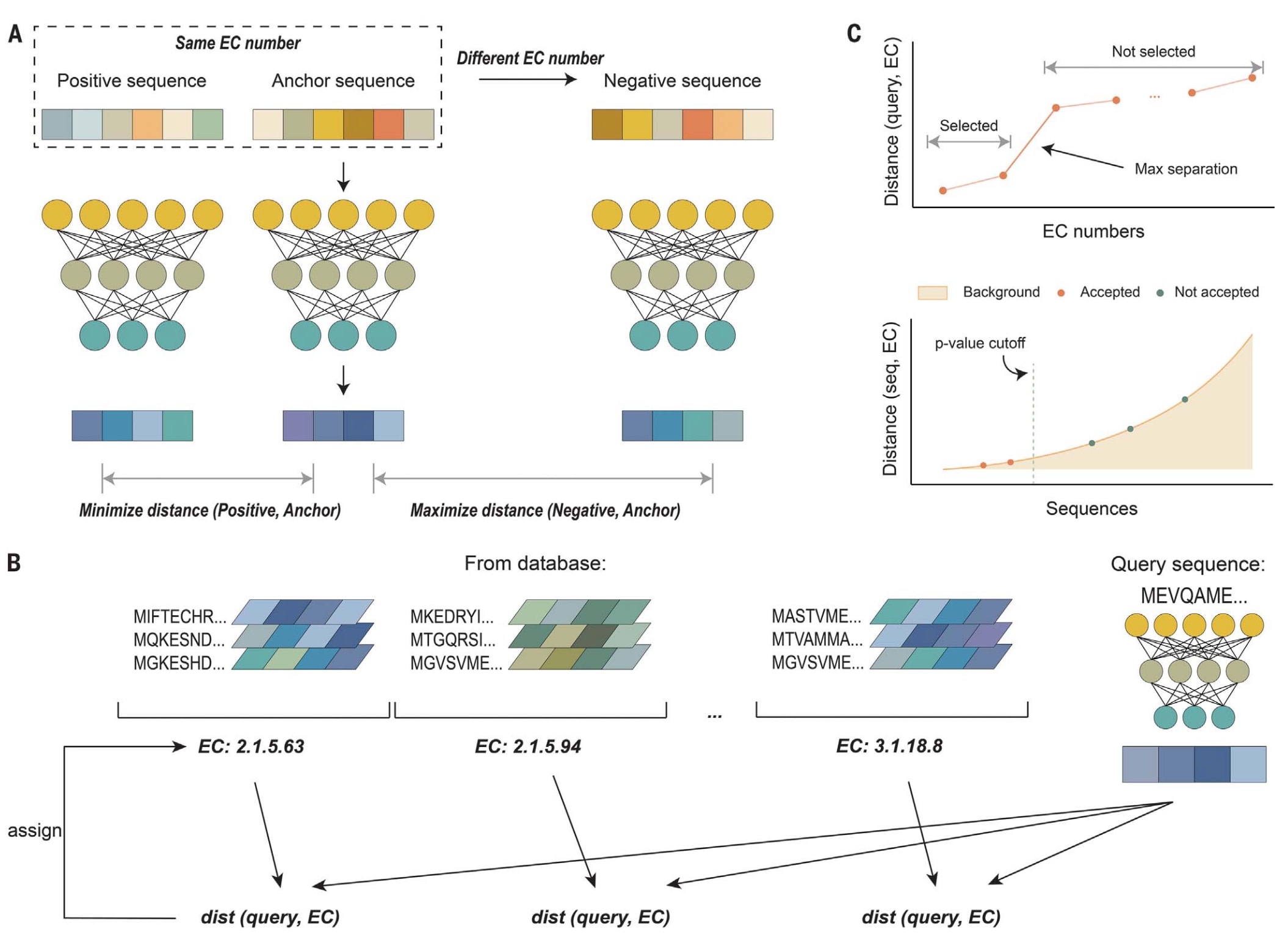
图1 | 展示了CLEAN基于对比学习的框架。 (A) 在训练阶段,正负样本的选取基于酶的EC编号。输入序列首先被嵌入并传入神经网络。图中一系列暖色方块表示由ESM-1b模型生成的输入序列嵌入表示;相对应地,经过监督式对比学习神经网络得到的序列嵌入则以冷色方块表示。(B) 每个EC编号的表示由该编号下所有酶的嵌入取平均获得。当进行EC编号预测时,查询序列的嵌入会与各EC编号的表示(以冷色平行四边形表示)进行比较,从而计算查询序列与每个EC编号之间的欧氏距离。该距离反映了查询序列与各EC编号之间的相似程度。(C) 当CLEAN作为分类模型使用时,采用了两种方法来提升预测结果的置信度:其一是最大分离法(上方),其二是基于P值的排序法(下方),通过这两种策略从排名结果中优先筛选出高可信度的EC编号预测。
2 模型开发与评估
与以往将EC编号预测任务视为多标签分类问题的机器学习算法不同,CLEAN采用了对比学习(contrastive learning)框架。该模型的训练目标是学习一种酶的嵌入空间,使欧氏距离能够反映功能相似性。所谓嵌入,即是将蛋白质序列以数值形式(向量或矩阵)表示,使机器能够读取,同时保留酶所包含的重要特征与信息。在CLEAN的任务中,具有相同EC编号的氨基酸序列在嵌入空间中的欧氏距离较小,而不同EC编号的序列距离较大。模型采用带监督的对比损失函数进行训练。
在训练过程中(见图1A),每个训练集中的参考序列(称为锚点,anchor)都会与一个具有相同EC编号的序列(正样本,positive)和一个不同EC编号的序列(负样本,negative)配对。为了提高训练效率并提供更具挑战性的负样本,模型优先选择那些与锚点嵌入距离较近的负样本,而非随机抽取。训练阶段使用来自语言模型ESM-1b的蛋白质表示作为输入,输入经过前馈神经网络后,输出层生成与功能相关的精炼嵌入表示。学习目标是通过对比损失函数使锚点与正样本的距离最小化,同时最大化锚点与负样本的距离。
在预测阶段,首先通过对训练集中属于同一EC编号的所有序列嵌入取平均,获得每个EC编号的聚类中心表示(见图1B)。随后,计算查询序列与所有EC编号聚类中心之间的两两欧氏距离。与查询序列距离显著较近的EC编号被视为输入蛋白质的预测功能。
模型的开发与评估基于通用蛋白质知识库UniProt。为从输出排名中筛选出高置信度的EC编号,CLEAN设计了两种选择策略(见图1C):(i)最大分离法,即选择在与查询序列的距离上与其他编号差异最大的EC编号;(ii)基于P值的方法,即选出与背景相比具有统计显著性的EC编号。
在训练集与测试集划分时,测试集中的任何酶与训练集中酶的序列同一性均不超过50%。在采用最大分离法的条件下,CLEAN取得了F1值为0.865的表现,该指标反映了模型在精确率与召回率之间的调和平均。即使在10%序列同一性聚类条件下,CLEAN依然保持0.67的F1值。此外,与未采用对比学习的ESM-1b基线模型相比,CLEAN的性能显著更高。
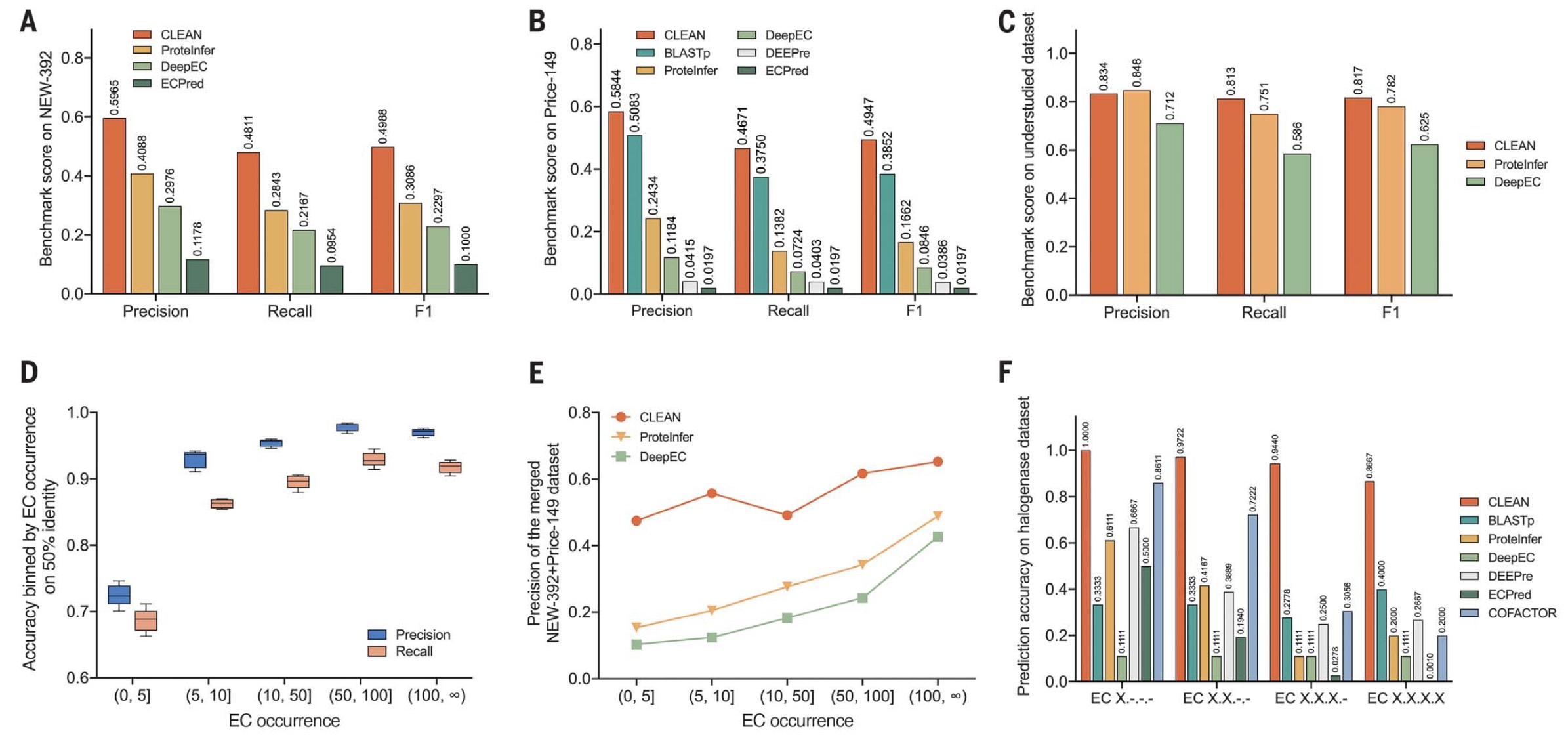
图2 | 展示了CLEAN与多种先进EC编号预测工具的定量比较结果。 (A) 在New-392数据库上评估了CLEAN在三项多标签准确性指标(精确率、召回率与F1得分)上的表现。对比模型包括ProteInfer、DeepEC、CatFam与ECPred四种表现最优的工具。结果显示,CLEAN在所有指标上均取得最高值。(B) 在Price-149数据库上比较了CLEAN与BLASTp、ProteInfer、DeepEC、DEEPre、CatFam及ECPred的表现。CLEAN在该实验中同样获得最高的F1分数,显著优于所有对比模型。(C) 针对稀有EC编号数据集,比较了CLEAN、ProteInfer与DeepEC三种模型的预测性能。CLEAN在处理低样本、分布不均的EC编号时依然保持最优表现。(D) 显示了CLEAN在与训练集序列同一性低于50%的测试集上的准确度分布图。采用SupconH损失函数训练后,精确率与召回率按EC编号在训练集中的出现次数分箱统计。例如区间(0,5]表示该EC编号在训练集中出现次数少于5次。箱线图展示了五折交叉验证的结果,表明CLEAN在低样本区间仍保持稳定性能。(E) 将Price-149与New-392数据集合并后,依据EC编号在CLEAN训练集中的出现次数分箱,对整体准确性进行评估。结果表明,CLEAN在预测少样本EC编号时依旧具有显著优势,而其他模型对高频EC编号表现出明显偏倚。(F) 展示了CLEAN在自建卤素化酶数据集上的预测准确率,并与六种常用工具(BLASTp、ProteInfer、DeepEC、DEEPre、ECPred与COFACTOR)进行比较。该数据集具有良好的多样性,涵盖11个不同EC编号。CLEAN在此数据集中同样表现最佳,进一步验证了其在实际酶功能预测中的广泛适用性与高可靠性。
3 CLEAN模型在EC编号注释工具中的基准测试
在模型训练完成后,研究者系统性地评估了CLEAN的预测性能,并将其与六种先进的EC编号注释工具进行了比较,包括ProteInfer、DeepEC、BLASTp、DEEPre、CatFam与ECPred。为了确保比较的公平性与严谨性,实验采用了两个独立的数据集,这些数据均未被用于任何模型的开发阶段。
第一个数据集New-392包含392条酶序列,覆盖177个不同的EC编号,数据来源于CLEAN训练完成后发布的Swiss-Prot版本(2022年4月)。这一预测场景模拟了实际应用中常见的情况,即以Swiss-Prot数据库作为标注知识库,而待预测序列的功能未知。总体而言,CLEAN在多标签准确率指标上均取得最高表现,包括精确率(0.597)与召回率(0.481),显著优于ProteInfer与DeepEC(见图2A)。在综合指标F1得分方面,CLEAN达到0.499,而ProteInfer与DeepEC分别为0.309与0.230。
第二个独立数据集Price-149来自Price等人的实验验证数据集。该数据集最初由ProteInfer整理,被认为是一个具有挑战性的集合,因为其中的许多序列在KEGG等数据库中被自动注释工具错误或不一致地标注。再次比较中,CLEAN在该数据集上同样取得最高的F1得分(0.495),明显优于BLASTp、ProteInfer和DeepEC(见图2B)。值得注意的是,在这一困难任务中,CLEAN的F1值是ProteInfer的约3倍(0.166),是DeepEC的近5.8倍(0.085)。
对New-392与Price-149数据集的评估结果充分表明,CLEAN在预测新发现蛋白质的功能时,尤其是在面对未知酶功能的情况下,具有比以往机器学习模型更高的精确性与可靠性。
4 理解CLEAN在注释研究不足EC编号中的性能优势
研究进一步探讨了CLEAN在预测研究不足(understudied)的EC编号时优于其他机器学习模型的原因。为此,研究者构建了一个验证数据集,专门收录来自稀有EC编号的酶,以验证这样的假设:相较于传统的多标签分类框架,对比学习能够更好地应对EC编号分布极度不平衡的特性——即部分EC编号下有上千个酶样本,而另一些仅有极少数(少于五个)。该验证数据集包含三千余个样本,涵盖一千多个不同EC编号,每种EC编号的出现次数均不超过五次。
在评估过程中,ProteInfer与DeepEC均使用其公开的预训练模型进行测试,因此本验证数据集中的部分样本曾出现在这两个模型的训练阶段。换言之,ProteInfer与DeepEC在此实验中具有先验优势,因而取得了F1分数介于0.625至0.782之间的结果。然而,即便在这种不利条件下,CLEAN依然表现出更高的性能,达到了0.817的F1分数(见图2C)。
研究还进一步分析了CLEAN性能与训练集中EC编号出现次数之间的关系。即使在50%序列同一性聚类的低相似度条件下,当训练样本极为稀少时,CLEAN的性能下降仍然有限(见图2D)。随后,将前述两个独立数据集(New-392与Price-149)合并重新分析,以EC编号在训练集中的出现次数为分层依据考察模型的准确率。结果显示,ProteInfer与DeepEC在分类框架的限制下,表现出对常见EC编号的偏向性;相反,CLEAN在预测罕见或研究不足的功能时依然保持优势,且准确率几乎不受EC编号出现频率的影响(见图2E)。
这一结果揭示了分类模型在不平衡数据集上的根本问题:当稀有EC编号的正样本过少时,模型几乎无法有效学习。相比之下,CLEAN能够通过对比学习同时利用正样本与负样本的信息,因此在少样本条件下依然具有较强的学习能力。为进一步验证这一假设,研究者引入了Supcon-Hard(SupconH)损失函数,这种损失函数较三元组损失(triplet loss)采样更多的负样本,从而增强模型的判别能力(见补充文本第2节及图S2)。
此外,研究者还设计了一种用于量化预测置信度的方法。具体而言,通过拟合酶序列嵌入与EC编号嵌入之间欧氏距离分布的双峰高斯混合模型(GMM),可对每个预测结果的可信度进行估计。置信度的引入不仅帮助研究者定量解读CLEAN的预测结果,还能有效防止过度预测:当模型置信度较低时,CLEAN将仅报告至EC编号的第三级别,以确保注释的稳健性(见图S11–S14及补充文本第3节)。
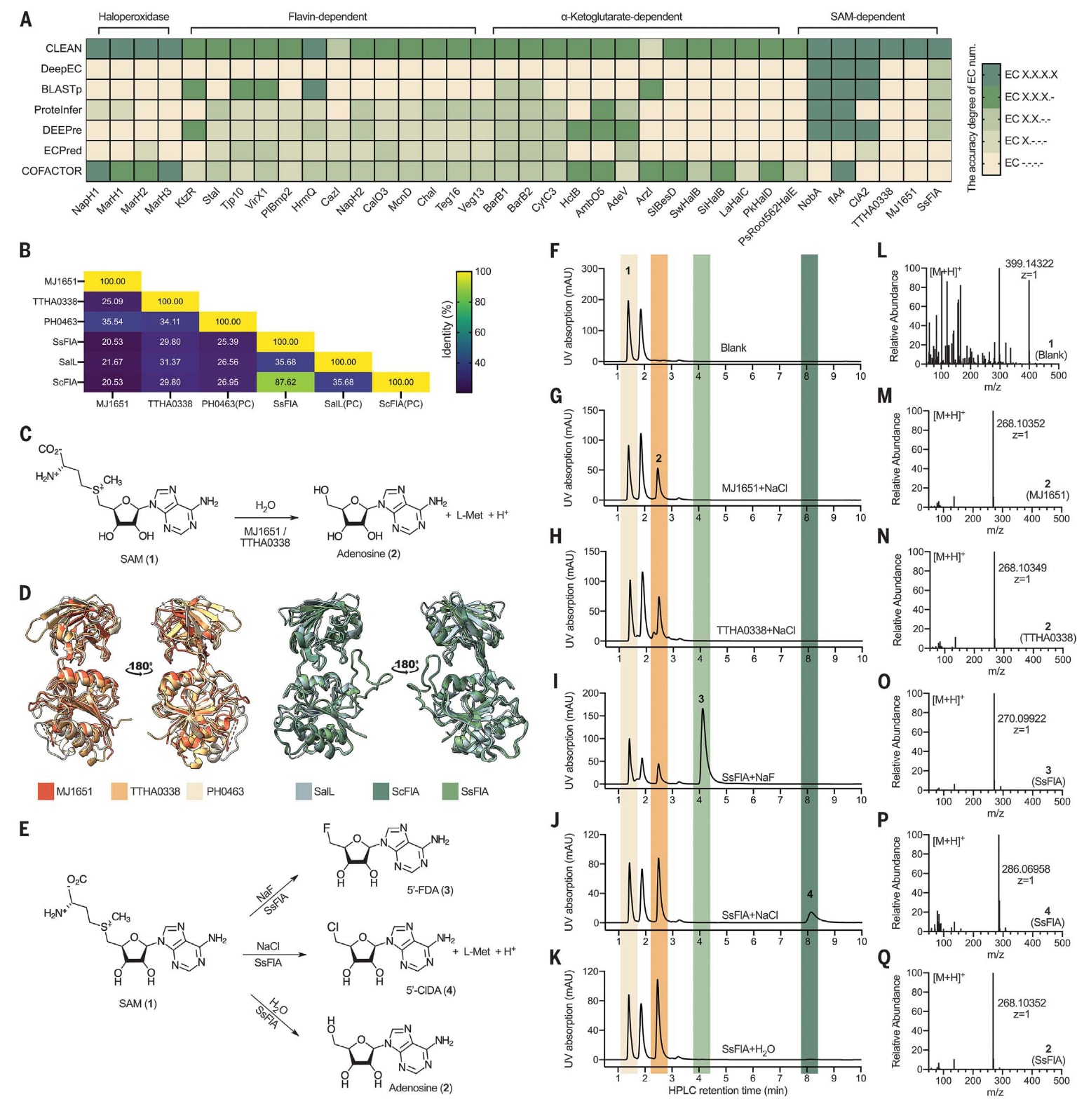
图3 | 展示了CLEAN在未表征卤素化酶(halogenases)中的实验验证结果。 (A) 显示了36种已鉴定卤素化酶的EC编号预测准确度热图。色块的深浅反映了模型预测与实际功能的一致程度。(B) 展示了这些未表征蛋白质与阳性对照(PC)酶之间的序列同一性热图,颜色条采用“viridis”色标,颜色深浅代表同源性百分比。(C) SAM羟基腺苷转移酶MJ1651–TTHA0338催化反应示意图。(D) 未表征蛋白MJ1651 [PDB ID: 2F4N]、TTHA0338 [PDB ID: 2CW5]与阳性对照酶PH0463 [PDB ID: 1WU8]的三维结构叠合图。同时也展示了SsFlA [PDB ID: 5B6I]、SalL [PDB ID: 2Q6O]与ScFlA [PDB ID: 1RQR]的结构叠合结果。可以看出,这些SAM结合型酶的三维结构高度相似,但CLEAN仍能准确区分其功能。每个晶体结构中均选取链A用于叠合。(E) 展示了SAM在SsFlA催化下与卤化物离子或H₂O发生亲核取代反应的过程。(F–K) 展示了SAM与NaCl/NaF/H₂O反应混合物的高效液相色谱(HPLC)分析结果。分别包括空白对照(F)、纯化的MJ1651(G)、纯化的TTHA0338(H)以及纯化的SsFlA[(I)–(K)]。图中标注了底物SAM(1)、产物腺苷(2)、5′-氟-5′-脱氧腺苷(5′-FDA)(3)与5′-氯-5′-脱氧腺苷(5′-ClDA)(4)的峰位,分别以浅黄、橙色、绿色与深绿色标识,并在相同保留时间处对齐。UV表示紫外检测信号,mAU为毫吸光单位。(L–Q) 展示了反应混合物中产物的质谱结果:空白体系中的底物1(L)、MJ1651催化反应产物腺苷(2)(M)、TTHA0338催化反应产物腺苷(2)(N)、5′-FDA(3)(O)、5′-ClDA(4)(P)以及腺苷(2)(Q)。m/z表示质荷比。这些实验验证表明,尽管未表征的SAM结合酶在结构上高度相似,CLEAN仍能精确区分其催化功能,进一步证实了其在功能注释上的可靠性与灵敏度。
5 实验验证
为了验证CLEAN在EC编号预测方面的准确性,研究以卤素化酶(halogenases)作为概念验证对象进行了实验测试。卤素化酶因其优异的催化选择性,近年来被广泛用于C–H键的生物催化官能化反应。通常,由卤素化酶生成的含卤小分子具有良好的生物活性与理化性质,因此在医药与农用化学领域均具有广阔的应用前景。迄今为止,从UniProt数据库中共鉴定出36种未完整注释的卤素化酶,涵盖四大类:卤过氧化物酶、黄素依赖型、α-酮戊二酸(α-KG)依赖型以及S-腺苷-L-蛋氨酸(SAM)依赖型酶(见图3A与表S2)。这些酶在UniProt中多被标注为“未表征”或“假定蛋白”,部分在文献中亦存在注释冲突。由于卤素化酶家族整体研究较少,且数据库中相关序列有限,该数据集对模型的预测能力构成了极大挑战。
在经过专家整理与后续实验验证后,36种卤素化酶均被CLEAN准确地分配了EC编号。总体上,CLEAN的预测准确率在86.7%至100%之间(见图2F与图3A),远高于六种常用计算工具的结果(例如DeepEC约为11.1%,ProteInfer在不同EC位数层级下为11.1%至61.1%)。这些结果表明,CLEAN不仅能区分相似的生物催化反应类型,还能精准识别功能细微差异。
在这36种酶中,MJ1651、TTHA0338与SsFlA三种酶的功能描述在文献与UniProt数据库中存在不一致。CLEAN为这三者预测了新的EC编号,提示它们可能具有不同或额外的催化功能。为验证这一预测,研究团队进行了体外实验。通过高效液相色谱–质谱(HPLC–MS)与酶动力学分析发现,MJ1651确实为SAM水解酶(EC 3.13.1.8),与CLEAN预测结果一致,而非UniProt及其他计算工具所误标的氯化酶(EC 2.5.1.94)或氟化酶(EC 2.5.1.63)(见图3C、D、F、G、M及图S3–S5A、S7、表S3)。此外,CLEAN还正确注释了TTHA0338,该酶隶属于无已知功能的DUF62 Pfam家族,实验验证其确为SAM水解酶(见图3C、D、H、N及图S5B、S7、表S3)。除BLASTp正确预测TTHA0338外,其余六种计算工具均未能识别MJ1651与TTHA0338的真实功能。这表明CLEAN能够有效纠正错误标注,并准确识别研究不足的催化功能。
此外,CLEAN还成功识别出具有三重功能的多活性酶SsFlA,其被同时注释为EC 2.5.1.63、EC 2.5.1.94与EC 3.13.1.8(见图3E、I–K、O–Q)。这一结果显示CLEAN能够有效回溯并识别酶的多功能性。令人瞩目的是,CLEAN能够区分结构高度同源的SAM结合蛋白,其序列同一性仅为20.5%至35.7%,唯有SsFlA与ScFlA例外,二者的序列同一性达87.6%(见图3B与图S6)。通常而言,在此序列同一性范围内预测功能极具挑战性,这进一步说明CLEAN在处理结构相似但功能不同的酶时,性能优于结构比对方法(如COFACTOR)。
6 讨论
通过系统的计算与体外实验验证,研究表明CLEAN在预测性能上明显优于六种先进工具(ProteInfer、BLASTp、DeepEC、DEEPre、COFACTOR与ECPred)。在未表征卤素化酶数据集上的综合分析显示,CLEAN能够识别假定蛋白并纠正错误标注,而多数基于序列、结构或机器学习的注释方法在此类任务中表现不佳甚至无法给出预测。识别酶的多功能性对于改造与优化酶性能至关重要,CLEAN在这方面展现出独特优势(例如SsFlA的三重功能预测)。
与传统分类模型不同,对比学习更适用于具有稀疏与偏倚特征的生物学数据。CLEAN有望成为预测酶催化功能的强大工具,并在功能基因组学、酶学、酶工程、合成生物学、代谢工程及逆向生物合成等领域发挥重要作用。此外,CLEAN基于语言模型表示并结合对比学习的设计框架,可灵活扩展至其他生物功能预测任务,如功能分类体系(FunCat)与基因本体论(GO)注释。其简洁友好的特性使其既可独立高通量运行,也能作为组件集成入其他计算平台。CLEAN在研究不足蛋白预测中的卓越表现将进一步丰富生物信息学工具体系,为未来的酶功能机制研究奠定基础。