NC 2025 | DeepSCFold: 基于序列衍生结构互补性的高精度蛋白质复合物结构建模
DeepSCFold提出了一种基于序列衍生结构互补性的蛋白质复合物建模思路,旨在突破传统方法对链间共进化信号的依赖。在许多真实体系中,如抗体–抗原或病毒–宿主复合物,共进化关系往往缺失,导致AlphaFold-Multimer等模型在界面预测上受限。DeepSCFold通过预测序列间的结构相似性与相互作用概率,构建更具结构感知的成对MSA,从而为复合物建模提供关键互作线索。这一策略使其在CASP15和SAbDab等基准任务中均展现显著优势,无论是整体拓扑还是界面几何,均优于现有最先进方法。特别是在抗体–抗原体系中,DeepSCFold对界面的准确捕捉展现出强大的泛化能力,为难以依赖传统共进化方法的体系提供了新的解决思路。这一工作展示了序列推断结构特征在复合物建模中的潜力,并为未来扩展至更广泛的生物大分子互作奠定基础。

获取详情及资源:
- 📄 论文: https://doi.org/10.1038/s41467-025-65090-7
- 💻 代码: https://github.com/iobio-zjut/DeepSCFold
- 🏠 课题组: http://zhanglab-bioinf.com/index.html
0 摘要
在活体系统中,蛋白质通过相互作用形成复合物,从而执行生命活动所需的关键功能。解析蛋白质复合物结构对于理解并掌握生物学功能具有重要意义。尽管AlphaFold2在预测蛋白质单体结构方面取得了革命性突破,但要准确捕捉链间相互作用信号并构建可靠的复合物结构仍然是一项艰巨任务。
此项工作报道了DeepSCFold这一用于提升蛋白质复合物结构建模能力的流程。DeepSCFold通过基于序列的深度学习模型预测蛋白–蛋白结构相似性与相互作用概率,为识别相互作用伙伴以及构建用于复合物结构预测的深度配对多序列比对(MSA)奠定基础。基准测试显示,与现有最先进方法相比,DeepSCFold能够显著提升复合物结构预测的精度。在CASP15的多聚体任务中,DeepSCFold相较AlphaFold-Multimer与AlphaFold3在TM-score上分别提高了11.6%与10.3%。
进一步将其应用于SAbDab数据库中的抗体–抗原复合物时,DeepSCFold在抗体–抗原结合界面的预测成功率上分别较AlphaFold-Multimer与AlphaFold3提高了24.7%与12.4%。这些结果表明,DeepSCFold能够通过序列衍生的结构感知信息有效捕捉内在且保守的蛋白–蛋白相互作用模式,而不是仅依赖序列层面的共进化信号。
1 引言
在细胞体系中,相互作用的蛋白质通过组装成功能性多蛋白复合物(即多聚体或组装体)来执行信号转导、运输与代谢等多种关键生命过程。为了深入理解这些功能,解析相关复合物的结构十分重要。然而,利用X射线晶体学、核磁共振(NMR)以及冷冻电镜(cryo-EM)等现有实验手段往往难以轻松获得这类结构,因此计算方法已成为结构生物学中不可或缺的重要补充。相比预测单体蛋白质的三级结构,预测复合物的四级结构更加困难,因为这要求同时准确建模多条蛋白质链之间的链内与链间残基相互作用。
传统的复合物结构预测依赖基于模板的同源建模或基于对接的方法。模板建模在有高质量模板时较为有效,但多数目标复合物往往难以获得合适的模板,从而限制了应用范围。在蛋白–蛋白对接方法中,工具通常依据“锁钥”与“诱导契合”原理,将单体结构组装成复合物,并通过能量最小化寻找满足空间形状互补性的最优结合模式。然而,对接方法在构象采样、能量函数精度以及界面区域的柔性方面仍面临挑战,使得其预测性能受限。
随着AlphaFold2在单体结构预测中取得显著突破,深度学习方法也被广泛用于预测蛋白质多聚体结构,包括AlphaFold-Multimer、DMFold-Multimer、MULTICOM3以及最新的AlphaFold3。其中,AlphaFold-Multimer作为针对复合物预测进行扩展的版本显著提高了预测精度,但其在多聚体任务中的准确性仍明显低于AlphaFold2在单体预测中的表现。为此,研究者基于AlphaFold-Multimer构建了多种改进策略,如在构建多序列比对(MSA)时进行多样化采样、使用多种随机种子、增加recycling次数以及加大网络dropout等。在CASP15中的结果表明,这些方法相较基础版AlphaFold-Multimer表现更佳,其中包括Zheng团队(DMFold)、Venclovas团队、Wallner团队、Yang-Multimer团队与MULTICOM3等。
在复合物结构预测中,MSA的质量至关重要,其隐含的共进化信息通常对于在蛋白构象空间中定位近似全局最优状态具有核心作用。在复合物情境下,链间结合模式的准确捕捉更依赖配对MSA的提供。通过对不同链的单体MSA进行系统性配对,可构建复合物的成对MSA,进而捕捉相互作用伙伴之间的链间共进化信号,为理解复合物内部的动态行为与稳定性提供信息。然而,主流序列搜索工具如HHblits、JackHMMER与MMseqs主要用于构建单体MSA,难以直接用于成对MSA的构建,从而可能影响复合物预测的准确性与泛化能力,尤其在如抗体–抗原等高度柔性或紧密交织的体系中表现尤为明显。
近期已有多种用于构建成对MSA的方法被提出,如DeepMSA2、MULTICOM3、DiffPALM与ESMPair等。DeepMSA2通过在基因组与宏基因组数据库中进行迭代比对搜索来改进单体MSA并构建配对MSA;MULTICOM3通过拼接亚基MSA并结合多来源的潜在相互作用信息生成多样化的成对MSA;ESMPair利用ESM-MSA-1b对单体MSA进行排序,再结合物种信息构建配对MSA;DiffPALM则利用MSA transformer估计氨基酸概率并生成排列矩阵以进行链间配对。尽管这些方法依赖序列相似性或物种相关信息来提取链间共进化信号,但对于缺乏此类信号的复合物体系仍可能受限。例如病毒–宿主与抗体–抗原体系通常难以出现清晰的链间共进化,宿主与病原体之间也往往缺乏可对应的同源序列。
蛋白质结构由于直接参与生物过程,通常比序列更具功能保守性,在蛋白–蛋白相互作用层面这一特征尤其显著,界面结构的保守性往往高于序列特征。大量实验证据表明,自然界中蛋白相互作用模式的类型相当有限,不同PPI间常常出现相似的结构结合模式。因此,利用深度学习模型直接从单体序列中提取空间构象互补性信息有望提供结构层面的洞见,从而提升复合物结构预测的精度。
基于此,该研究提出DeepSCFold,将蛋白序列嵌入与理化及统计特征相结合,通过深度学习框架系统地捕捉链间结构互补性,并准确预测蛋白质复合物的四级结构。在CASP15复合物数据集上的基准评测显示,DeepSCFold在整体结构与界面局部精度上均优于现有方法。为了进一步验证其稳健性,还在SAbDab数据库中选取缺乏链间共进化信号的抗体–抗原体系进行了评测,结果显示基于结构互补性的配对MSA能够为复合物提供可靠的链间相互作用信号,有效弥补共进化信息的缺失。

**图1|展示了DeepSCFold的整体流程。**a部分中,DeepSCFold以蛋白质复合物的多条查询序列作为输入,对每条序列分别在蛋白质数据库中进行搜索,构建对应的单体多序列比对(MSA)。随后,通过深度学习模型评估MSA中每条同源序列与查询序列之间的结构相似性,以便对同源序列进行排序与筛选。来自不同单体MSA的同源序列将通过两类策略进行配对:一类依赖物种信息或人工整理的注释,另一类利用基于相互作用概率预测模型生成的概率矩阵,对潜在同源序列对进行评分并据此进行配对。构建完成的成对MSA将输入AlphaFold-Multimer生成候选复合物结构。所得模型随后进行质量评估,排名最高的模型被用作模板进行一次精修迭代,最终获得预测的复合物结构。b部分展示了深度学习模型的架构:一对序列的特征首先输入多尺度保留模块(d)以生成序列表征,再通过交叉注意力模块(e)生成配对表征,随后进入下采样模块(c)得到最终预测分数(pSS或pIA)。对应的源数据可在附带的Source Data文件中获得。
2 结果
2.1 DeepSCFold的方法框架
DeepSCFold是一套专为高精度蛋白质复合物结构预测而开发的计算流程,其核心在于通过整合两类关键信息来构建成对多序列比对(pMSA):一是评估单体查询序列与其在各自MSA中的同源序列之间的结构相似性,二是识别来自不同单体MSA中的序列之间可能存在的相互作用模式。依托这一双重策略,方法能够系统性地产生高质量的pMSA,从而为后续的精确复合物建模提供稳固基础。
DeepSCFold的成对MSA构建依赖两个基于序列的深度学习模型:其一用于预测蛋白–蛋白结构相似性(pSS-score),仅基于序列信息推断不同链可能呈现的结构模式;其二用于预测相互作用概率(pIA-score),同样完全依赖序列层级特征判断序列是否可能形成链间互作。这两类模型无需任何已知结构输入,因而能够仅从序列层面对潜在的结构与相互作用性质做出推断,使DeepSCFold具备从纯序列数据建模复杂相互作用的能力。
图1展示了整体流程。从输入的复合物序列开始,DeepSCFold首先利用多个序列数据库(UniRef30、UniRef90、UniProt、Metaclust、BFD、MGnify以及ColabFold数据库)生成各单体的MSA。随后,预测得到的pSS-score被用作补充传统序列相似性的指标,用于优化单体MSA中同源序列的排序与筛选。接着,DeepSCFold基于新构建的深度学习模型对来自不同亚基MSA的潜在同源序列组合预测pIA-score,即序列对之间的相互作用概率,并利用这些概率系统性地拼接单体同源序列,构建成对MSA,从而捕捉具有生物学意义的互作模式。
此外,方法还整合了物种注释、UniProt登录号以及PDB中已有的实验结构等多源生物学信息,以构建更具生物相关性的补充型pMSA。最终,DeepSCFold利用上述一系列pMSA通过AlphaFold-Multimer进行复合物结构预测,并通过内部开发的复合物质量评估方法DeepUMQA-X筛选top-1模型,再将其作为模板进行一次AlphaFold-Multimer推理生成最终结构。
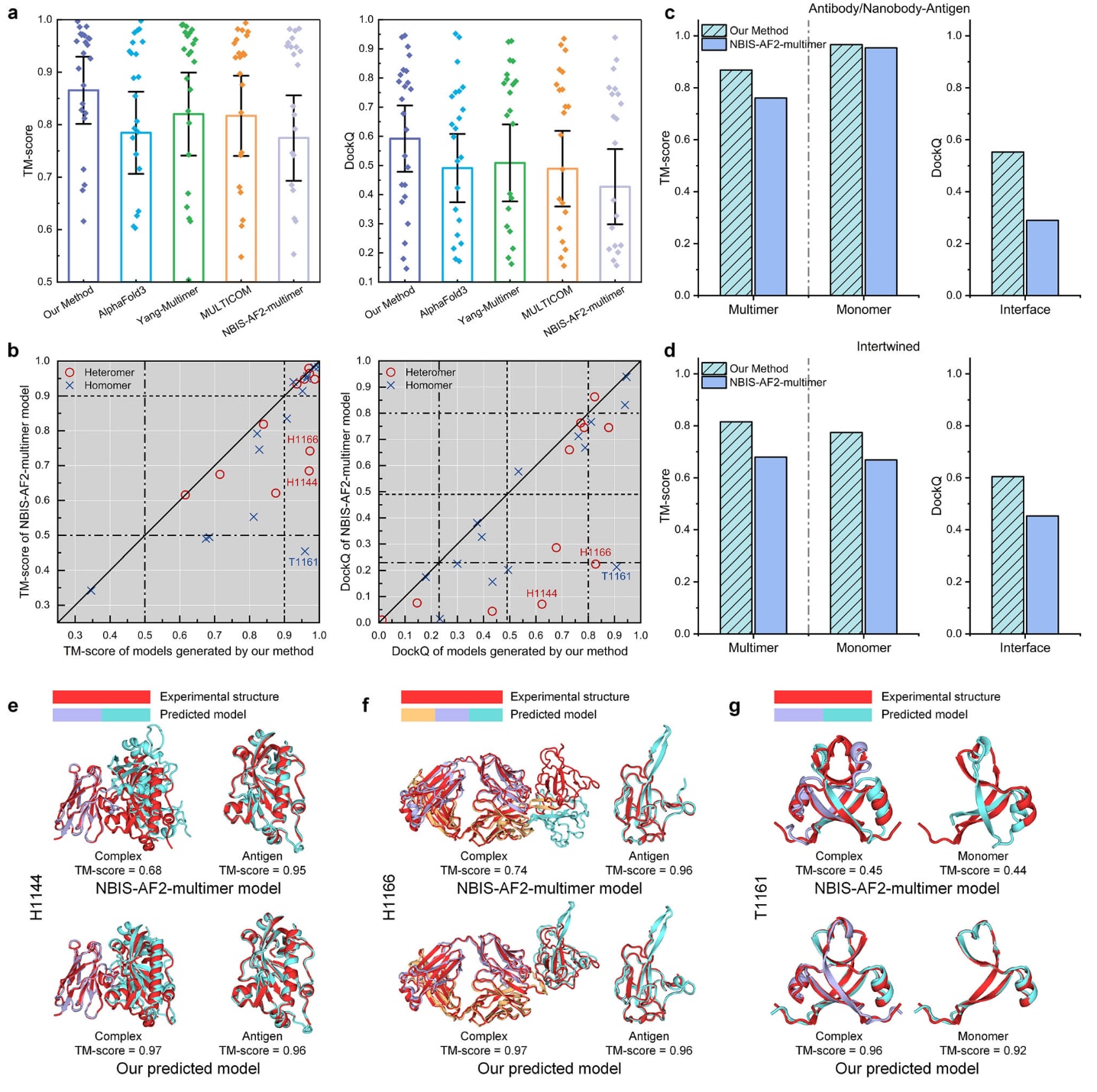
图2 | DeepSCFold在CASP15数据集上的蛋白质复合物结构预测性能(n=25个蛋白目标)。 a展示了DeepSCFold、Yang-Multimer、MULTICOM、NBIS-AF2-multimer与AlphaFold3生成的复合物模型的平均TM-score与DockQ,数据以平均值±95%置信区间呈现,n=25。b给出了DeepSCFold与NBIS-AF2-multimer在每个测试案例上的TM-score与DockQ的逐对比较,n=25。圆点表示异源复合物,叉号表示同源复合物,图中标注了三个代表性案例,其详细分析见(e、f与g)。c与d展示了基准集中抗体/纳米抗体–抗原复合物(n=8)与缠绕复合物(n=8)的性能对比,包括多聚体与单体的平均TM-score以及界面DockQ。e–g展示了DeepSCFold与NBIS-AF2-multimer在纳米抗体–抗原复合物H1144(e)、抗体–抗原复合物H1166(f)以及缠绕复合物T1161(g)上的建模示例,实验结构以红色表示,预测模型中的单体以不同颜色区分。H1144与H1166的复合物通过抗体链进行对齐。相关源数据可在Source Data文件中获取。
2.2 DeepSCFold在蛋白质复合物结构预测中的性能提升
为了评估DeepSCFold在蛋白质复合物结构预测中的性能,研究对CASP15竞赛中的多聚体目标进行了系统性测试。每个目标的结构预测均基于截至2022年5月的蛋白质序列数据库生成,从而确保评测在时间上不带偏倚。随后将预测结果与多种先进方法进行比较,包括AlphaFold3、Yang-Multimer、MULTICOM与NBIS-AF2-multimer。AlphaFold3的预测模型由其在线服务器生成,其余三类方法的结果取自CASP15官方网站。
为评估预测模型的准确性,采用两类互补指标:TM-score用于衡量与实验结构的整体拓扑一致性,而DockQ专注评估蛋白–蛋白界面的对接质量。指标的详细说明收录于补充说明中。图2a展示了预测复合物的TM-score与DockQ,详细结果见补充表格。DeepSCFold的平均TM-score达到0.87,DockQ为0.59,显著优于当前最先进方法,包括AlphaFold3(TM-score=0.78,DockQ=0.49)、Yang-Multimer(TM-score=0.82,DockQ=0.51)、MULTICOM(TM-score=0.82,DockQ=0.49)与NBIS-AF2-multimer(TM-score=0.78,DockQ=0.43)。两个指标均表现提升,说明DeepSCFold能更准确地建模复合物的整体拓扑与界面构象。DeepSCFold所使用的序列衍生结构互补性信息很可能在提升全局精度与界面预测质量方面发挥关键作用。
由于NBIS-AF2-multimer是CASP15中AlphaFold-Multimer的官方实现,研究进一步对两者进行了直接比较,以检验输入策略的改进是否有效提升预测性能。图2b显示了两者在各个测试目标上的TM-score与DockQ对比结果。DeepSCFold在92%的案例中获得更高TM-score,在88%的案例中获得更高DockQ。研究还依据不同阈值对结果进行分类,以便进行更细致的性能对照。在多种复杂系统中提升尤为明显,包括高度缠绕的复合物(T1161)、纳米抗体–抗原(H1144)与抗体–抗原体系(H1166)。
为进一步揭示提升的来源,研究对相关体系进行了深入分析,见图2c、d。图2c展示了抗体/纳米抗体–抗原复合物的平均多聚体TM-score、单体TM-score与界面DockQ。两种方法在单体预测上均表现稳定,DeepSCFold为0.97,NIBS-AF2-multimer为0.95。然而关键差异在界面预测中显现:DeepSCFold的DockQ为0.55,显著高于对方的0.29,直接推动了其多聚体TM-score的提升(0.87对0.76)。图2e展示了H1144案例,NIBS-AF2-multimer的单体质量虽高(TM-score=0.95),但复合物的链间取向严重错误导致TM-score降至0.68。而DeepSCFold提供更准确的链间信号,使复合物TM-score达到0.97。图2f展示了H1166抗体–抗原体系的类似情形。抗体/纳米抗体–抗原体系属于跨物种系统,单体MSA缺乏物种内的成对共进化信号,NIBS-AF2-multimer得到的MSA可能不理想,从而影响界面预测。而DeepSCFold通过序列推断的结构互补性可弥补此类缺陷,从纯序列中探索潜在互作信号。
图2d展示了八个高度缠绕复合物的对比分析,此类体系界面紧密交织,预测难度极高。DeepSCFold在所有指标上均显著优于对方:多聚体TM-score从0.68提升至0.82,单体TM-score从0.67提升至0.77,界面DockQ从0.45提高至0.60。图2g展示了代表性案例T1161,NIBS-AF2-multimer的单体TM-score仅0.44,导致复合物TM-score只有0.45;而DeepSCFold的单体TM-score达到0.92,复合物TM-score达0.96。结果说明,从序列推断的结构信息不仅改进链间距离与取向的确定,也补充了基于序列相似性生成的单体MSA中的共进化信号,使得对复杂体系的预测更加准确可靠。
关于CASP15的更详细分析见补充说明与附录图表。CASP16中出现了多种先进方法,包括AF3-server、Zheng-Multimer、Yang-Multimer与MULTICOM_GATE,代表当前复合物预测领域的最前沿。DeepSCFold也以两个组名参加CASP16并取得具有竞争力的结果,相关性能评估与分析见补充资料。
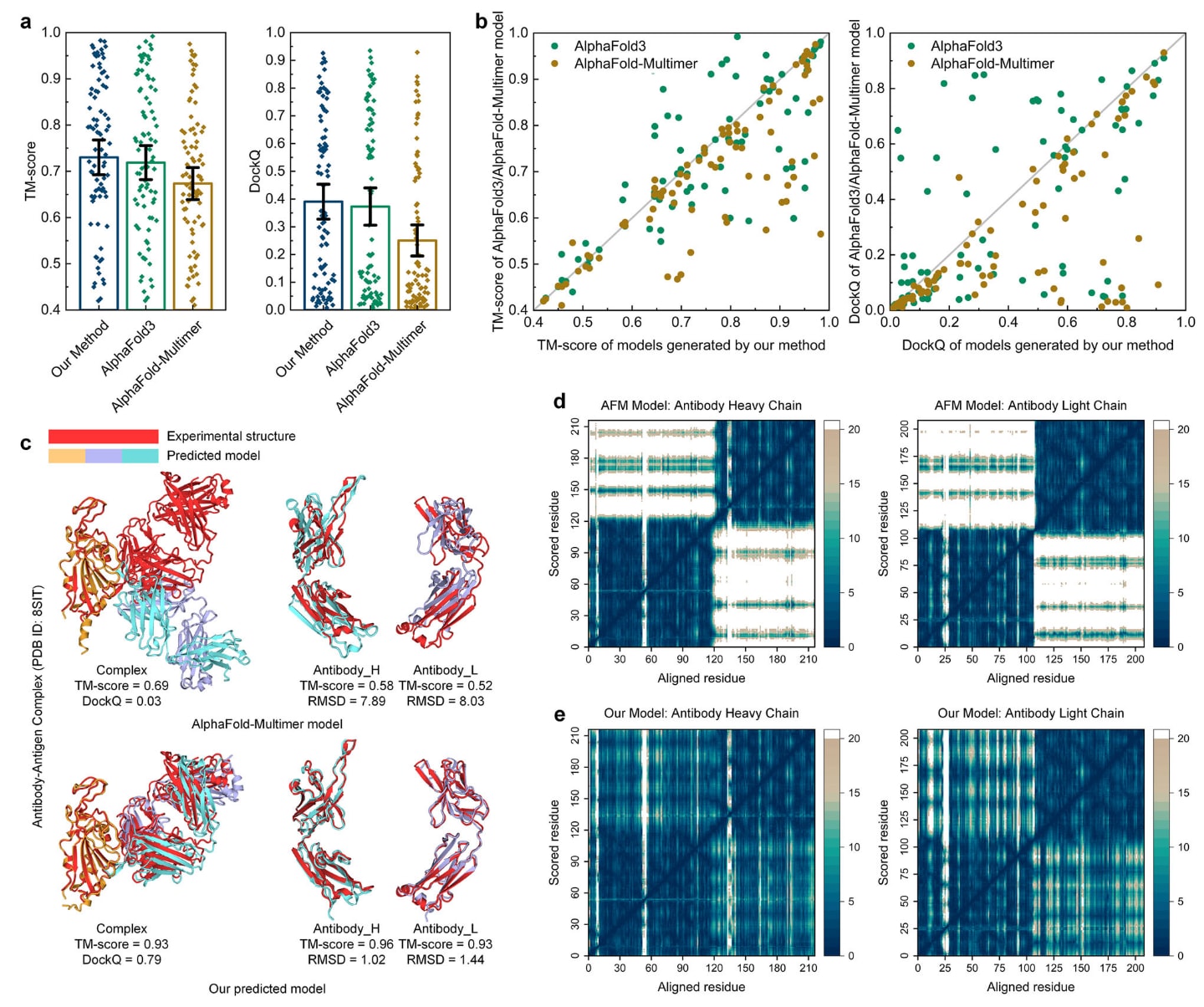
图3 | 抗体–抗原复合物结构预测结果(n=89个蛋白目标)。 a展示了DeepSCFold、AlphaFold-Multimer与AlphaFold3预测模型的复合物结构平均TM-score以及抗体–抗原界面的DockQ,数据以平均值±95%置信区间呈现,n=89。b给出了DeepSCFold、AlphaFold-Multimer与AlphaFold3在每个测试案例中的TM-score与DockQ的逐对比较,n=89。c展示了一个抗体–抗原复合物(PDB ID:8SIT)的预测示例,实验结构以红色表示,预测模型的单体以不同颜色区分。d与e分别展示了AlphaFold-Multimer(d)与DeepSCFold(e)对抗体重链与轻链的预测对齐误差(PAE)图。相关源数据可在Source Data文件中获取。
2.3 利用DeepSCFold进行抗体–抗原复合物的结构建模
鉴于抗体与抗原序列之间通常缺乏或几乎不存在共进化信号,研究特别针对抗体–抗原复合物建模进行了深入分析,重点评估复合物成对多序列比对(MSA)对建模性能的影响。DeepSCFold在SAbDab中筛选的抗体–抗原数据集上进行了评测,该数据集覆盖2024年1月25日至2024年6月1日期间的条目。这样的时间划分确保了模型训练数据与结构建模所用数据库之间不存在重叠,从而保证评估的独立性与公正性。
为实现公平比较,DeepSCFold与AlphaFold-Multimer均使用相同的模板信息与单体MSA。DeepSCFold所使用的成对MSA由单体MSA中各亚基独立推导而来。此外,研究还将最新的AlphaFold3纳入比较,其结果直接来自其在线服务器。
图3a展示了预测所得抗体–抗原复合物结构的TM-score与界面DockQ,与PDB中的实验结构进行对照。界面DockQ仅用于评估抗体链与抗原链之间的互作,不包含抗体重链与轻链之间的界面。详细结果见补充表格。DeepSCFold的复合物TM-score平均为0.73,界面DockQ为0.39,优于AlphaFold-Multimer(TM-score=0.67,DockQ=0.25)与AlphaFold3(TM-score=0.72,DockQ=0.37)。图3b展示了DeepSCFold与两类方法在各测试目标上的逐对比较。DeepSCFold在59.6%的案例中获得更高TM-score,在60.7%的案例中获得更高DockQ,界面预测成功率(DockQ>0.23)相比AlphaFold3提升12.4%。结果显示两类方法具有互补性,联合使用可能产生协同增强效果。相较AlphaFold-Multimer,DeepSCFold在77.5%的案例中获得更高TM-score,在92.1%的案例中获得更高DockQ。值得注意的是,AlphaFold-Multimer在56个目标中生成的复合物模型界面DockQ低于0.23,而DeepSCFold在其中39.3%的案例中成功获得DockQ>0.23的模型,表明基于结构互补性构建的成对MSA能够提供重要的链间信号,提升界面预测。
一个典型案例是SARS-CoV-2刺突蛋白受体结合域与广谱中和抗体CC84.24 Fab的复合物(PDB ID:8SIT)。AlphaFold-Multimer预测的模型存在严重的链间取向错误,复合物TM-score仅0.63,界面DockQ仅0.03(见图3c)。其单体质量也较差,抗体重链TM-score为0.58(RMSD=7.89 Å),轻链TM-score为0.52(RMSD=8.03 Å)。进一步通过预测对齐误差(PAE)评估各链结构的域间精度。AlphaFold-Multimer的PAE图如图3d所示,虽两个域内部误差较低(TM-score均高于0.9),但域间取向存在较高PAE,表明其不能正确预测域间关系。相反,DeepSCFold不仅准确建模各域,也正确恢复域间及链间的相对取向,其PAE在整个区域保持较低水平(图3e)。
这些结果说明,对于抗体这类具高度柔性的蛋白,单体MSA能较好地捕捉域级别拓扑,但要正确确定域间与链间的取向往往需要依赖复合物级别的MSA,以获取关键的互作信号。因此,在抗体–抗原的生物学相关构象预测中,整合结构互补性与链间互作信号至关重要。
此外,研究还构建了包含34个病毒–人类实验解析复合物的基准集,并将DeepSCFold与AlphaFold3及AlphaFold-Multimer进行对比。结果显示DeepSCFold能更有效捕捉跨物种互作的结构模式。详细的实验结果见补充说明,并附补充图表与数据。此外,补充说明中的其他部分(附补充图与表)还对结构预测中涉及的各组成模块进行了系统分析。
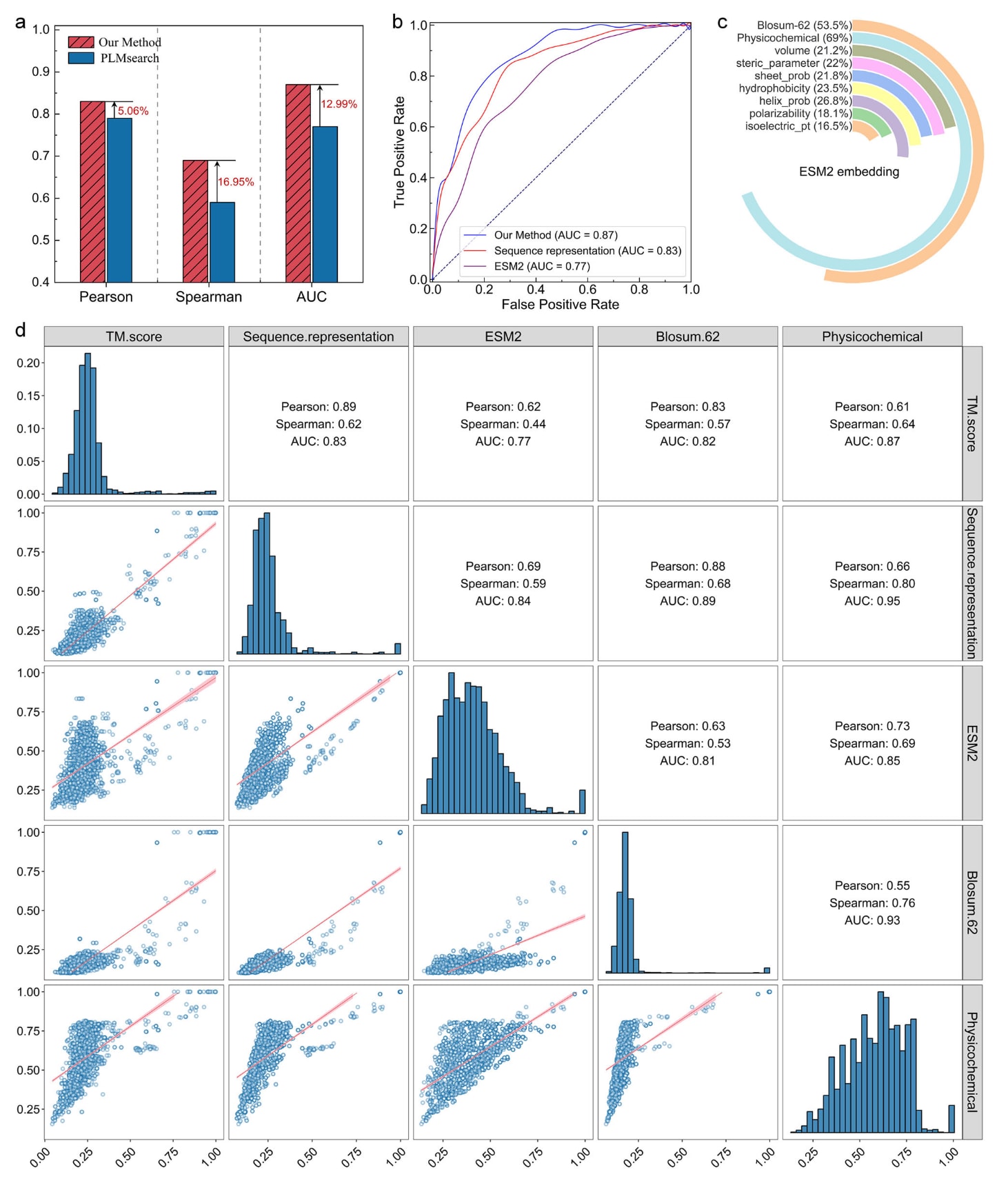
图4 | 蛋白–蛋白结构相似性预测的性能对比与特征分析(n=2701个蛋白对)。 a展示了该方法与PLMsearch在Pearson相关、Spearman相关以及ROC AUC指标下的性能比较,数据以平均值呈现,n=2701。b显示了该方法、ESM2以及序列表征在分类任务中的ROC曲线,n=2701。c展示了ESM2嵌入与额外序列衍生特征之间的相关性分析,n=2701。d展示了各特征的分布及其两两相关性,n=2701。对角线直方图表示各特征的分布,散点图显示特征之间的对应关系,右上三角给出了Pearson与Spearman相关系数及AUC。相关源数据可在Source Data文件中获取。
2.4 基于序列的结构相似性预测性能
DeepSCFold的核心组成部分之一是直接从序列数据预测蛋白–蛋白结构相似性。由同源序列比对推断的共进化信号对蛋白质结构预测十分关键,因为这些信号揭示了残基–残基之间的相互作用及其受进化约束的方式,从而影响折叠与复合物形成。由于序列数据规模庞大且成本低廉,基于序列的搜索方法通常比基于结构的方法更加高效便利,尤其适用于海量序列场景,例如宏基因组序列与抗体变体序列。然而,在系统发育距离较远的案例中,序列可能已经高度分化,导致难以判断它们之间的关联性,这一困难突显了亟需发展能够直接从序列推断蛋白–蛋白结构相似性的计算方法,以补充现有结构预测手段。
为提升检测能力同时保持序列搜索的通用性与效率,研究开发了一个基于深度学习的模型,能够直接从序列预测结构相似性,并作为DeepSCFold的重要组成模块(见方法部分)。其基本假设是:即便序列相似度很低,结构相似的蛋白质序列仍然可能携带足够的拓扑信息来弥补传统序列比对方法的不足,为构建高质量单体MSA提供重要基础,从而提升结构建模精度。对于系统发育距离较远的同源蛋白比对,这一策略尤为重要,有助于理解低序列相似度蛋白家族中的演化关系。
方法中预测的蛋白–蛋白结构相似性(pSS-score)采用TM-score刻画,这一指标广泛用于评价两个蛋白结构的全局相似性。为了全面客观地分析性能,研究构建了一个基于CASP15目标蛋白的基准集,并设置严格的时间切断以确保与训练数据完全独立。随后,将DeepSCFold与最新的PLMsearch进行对比,使用Pearson、Spearman与ROC(AUC)等统计指标进行评价。
图4a显示了pSS-score与真实TM-score二者之间的相关性。DeepSCFold的Pearson相关系数为0.83,相比PLMsearch的0.79提升了5.06%。Spearman相关系数为0.69,比PLMsearch的0.59提升了16.95%,说明模型更好地保持了结构相似性的排序关系。ROC分析中,DeepSCFold的AUC达到0.87,较PLMsearch的0.77提升12.99%。正样本被严格定义为真实TM-score处于数据集最高四分位的蛋白对,结果表明该方法具有更强的区分能力,能够更有效识别高可信度的结构相似序列对。
图4b进一步探讨性能提升的来源,将ESM2嵌入与该方法生成的序列表征进行比较。ESM2嵌入本身可达到0.77的AUC,说明蛋白语言模型确实捕捉了重要的生物信息。该方法在ESM嵌入的基础上进一步融合了序列衍生特征,如理化特性与氨基酸替换概率。仅使用多尺度保留模块提取的序列表征即可达到0.83的AUC,明显优于单独使用ESM2嵌入。这表明这些额外特征提供了重要补充信息。序列表征与最终模型(AUC=0.87)之间的性能差异则强调了分类模块的重要性,其有效整合各类特征,实现了更可靠的分类与排序能力。
图4c展示了ESM2嵌入与不同序列衍生特征之间的相关性,以评估它们的互补性。整体理化特征的相关性最高(69.0%),但单个特征例如疏水性(23.5%)、螺旋倾向(26.8%)与极化性(18.1%)等相关性较低,说明它们提供了ESM2未能完全覆盖的独特信息。氨基酸替换矩阵Blosum-62与ESM2嵌入的相关性为53.5%,显示替换概率中所包含的进化信息也具补充意义。
图4d展示了各类特征与真实TM-score之间的分布与相关性。所提出的序列表征与TM-score之间的Pearson相关高达0.89,Spearman相关为0.62,表明融合多源生物信息能够有效提升结构相似性预测。ESM2嵌入的相关性较低(Pearson=0.62,Spearman=0.44),但仍提供有价值的生物学参考。Blosum-62与TM-score的相关性为Pearson=0.83,Spearman=0.57,进一步体现进化信息的重要性。理化特征也表现出较高的相关性(Spearman=0.64,AUC=0.87)。不同特征之间的相关性存在差异,说明它们虽然部分重叠但各自携带独特信息。
综上所示,整合多种序列特征是必要的,因为这种组合能更全面地反映序列所蕴含的生物信息,显著提升序列–结构关系的预测能力。
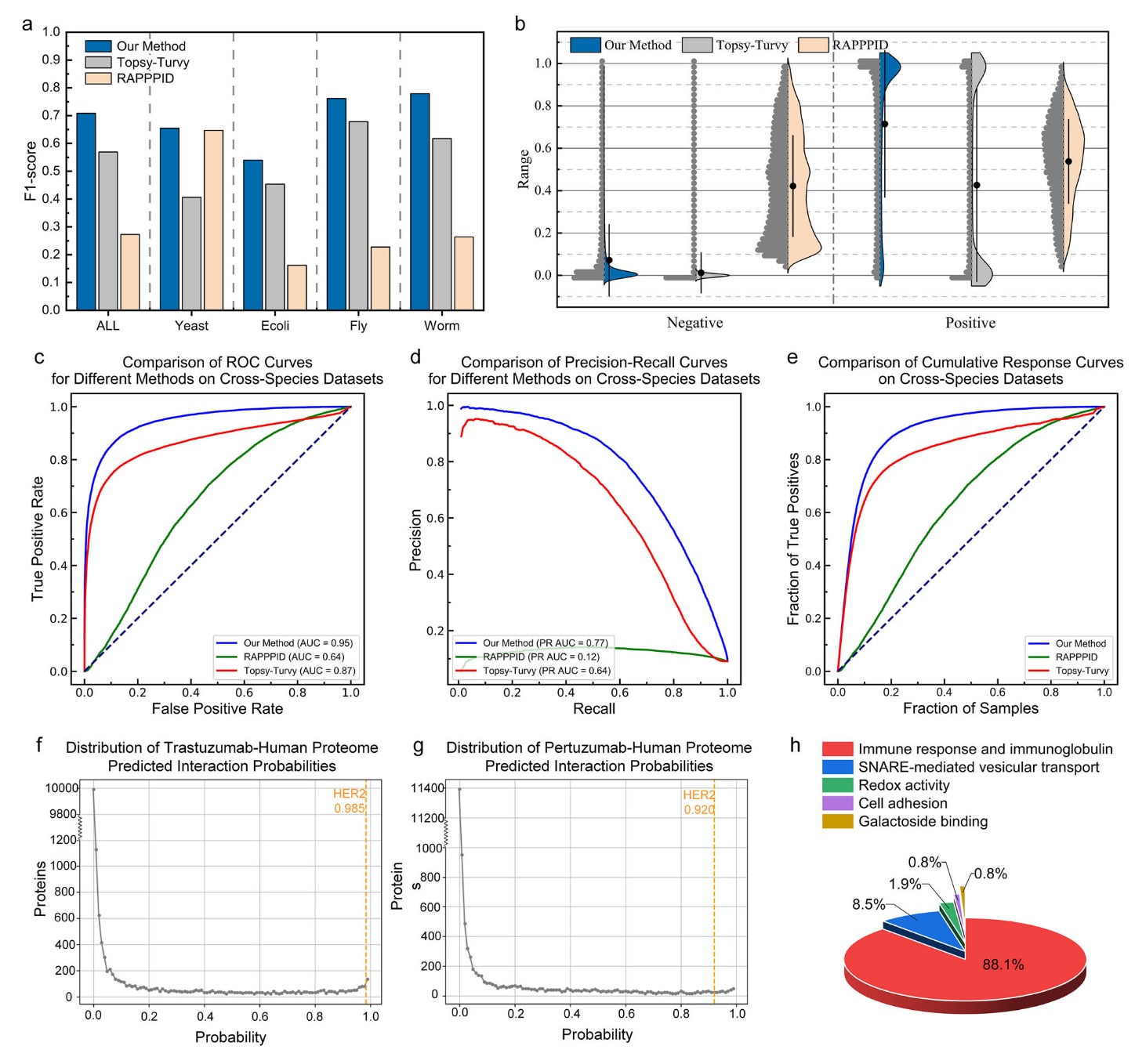
图5 | 蛋白–蛋白相互作用(PPI)预测的比较分析与实际应用,n=17,000个阳性样本与170,000个阴性样本。 a展示了该方法与当前最先进方法Topsy-Turvy与RAPPPID在不同物种中的F1-score,包括酵母(阳性5000/阴性50,000)、大肠杆菌(阳性2000/阴性20,000)、果蝇(阳性5000/阴性50,000)、线虫(阳性5000/阴性50,000)以及整体数据集(ALL,阳性5000/阴性50,000)。b小提琴图展示了不同方法在阳性与阴性蛋白对上的相互作用概率分布,数据以平均值±标准差呈现,n=17,000阳性与170,000阴性。c–e展示了该方法与Topsy-Turvy、RAPPPID的ROC曲线、PR曲线与累计响应曲线(CRC)对比分析,n=17,000阳性与170,000阴性。f与g展示了Trastuzumab(f)与Pertuzumab(g)在人体蛋白组中的相互作用概率分布(n=17,286个来自STRING数据库的蛋白),其中HER2显示出极高的相互作用分值(Trastuzumab:0.985,Pertuzumab:0.920)。h展示了Trastuzumab与Pertuzumab预测高相互作用蛋白的聚类结果,n=270个人类蛋白。相关源数据可在Source Data文件中获取。
2.5 基于序列的蛋白–蛋白相互作用概率预测性能
DeepSCFold的另一项关键能力是通过序列数据直接预测蛋白–蛋白相互作用(PPI)的概率。许多现有的成对MSA构建方法依赖实验数据与人工注释信息(例如物种注释),而这一依赖限制了它们在研究较少或未知物种时的适用性。如果能够直接预测任意两条序列之间的相互作用概率,就能够大幅扩展可用于构建MSA的数据来源,使大量未注释的序列数据库(如BFD与MGnify宏基因组数据库)可直接用于复合物结构预测。
为评估相互作用预测性能,研究基于人类蛋白对训练模型,并分别在四个物种(酵母、大肠杆菌、果蝇与线虫)上与当前先进的PPI预测方法Topsy-Turvy与RAPPPID进行比较。图5a展示了这些物种(以及汇总数据集ALL)上的F1-score。该方法在所有物种上都获得了更高的F1-score,表明其在精确率与召回率之间取得良好平衡,并具有稳健的跨物种泛化能力。尤其在果蝇与线虫上表现更为优异,这两个体系具有复杂的发育路径和信号网络,结果说明DeepSCFold能有效捕捉潜在共进化信号。详细结果见补充表格。
图5b的小提琴图展示了各方法在阳性与阴性蛋白对上的相互作用概率分布。该方法在阴性样本中呈现接近零的窄分布,说明其能够精确识别非互作蛋白并保持较低的假阴性率。在阳性样本中,DeepSCFold的预测大部分集中在0.8以上,显示了高度的预测置信度。相比之下,Topsy-Turvy的分布被分为两个区间(高置信与低置信),而RAPPPID的分布更为混杂,区分能力不足。整体而言,DeepSCFold在区分互作与非互作蛋白方面具有更高的判别力。
为了进一步评估DeepSCFold的稳健性,研究采用了三类标准指标:ROC曲线、PR曲线与累积响应曲线。图5c显示ROC结果,该方法的AUC达到0.95,显著优于RAPPPID(0.64)与Topsy-Turvy(0.87),说明其能在不同阈值下稳定区分真阳性与真阴性。PR曲线(图5d)特别适用于类别不平衡的数据集(互作蛋白对较少),DeepSCFold的PR AUC为0.77,远高于RAPPPID(0.12)与Topsy-Turvy(0.64),体现了其在保持高精确率的同时也维持较高召回率。图5e的累积响应曲线表明该方法在检测到大量真阳性所需的样本量更少,能更早富集真正的互作蛋白,适合用于探索性研究中的快速筛选。
上述结果在不同物种上的对比见补充图。关于特征贡献的详细分析见补充说明及附表附图。
为展示DeepSCFold的实际应用价值,研究进一步预测了两种广泛研究的抗体(Trastuzumab与Pertuzumab)与全人类蛋白组(17,286种蛋白)的相互作用概率,数据来自STRING数据库。图5f与图5g显示两种抗体对HER2的预测相互作用概率极高(Trastuzumab为0.985,Pertuzumab为0.920),符合其已知的生物学特异性。结果分布尖锐集中,说明该方法具有用于大规模互作筛选的潜力。
进一步地,研究从预测结果中筛选相互作用概率高于抗体与HER2之间的蛋白,获得共270种可能具有强互作关系的人类蛋白。为评估这些预测是否仅为噪声,研究利用STRING对其进行聚类分析,结果分为五簇。图5h显示,第一簇(红色)包含229种蛋白,主要富集于适应性免疫反应和免疫球蛋白相关结构域,这些结构与抗体框架具有相似性,因此可能获得高互作分值。其他簇分别富集于囊泡运输、氧化还原活性、细胞黏附及糖苷结合等功能。结果说明DeepSCFold倾向为具有结构或功能相关性的蛋白赋予更高互作概率,即使这些蛋白不是抗体的主要靶点,模型的预测仍呈现生物学意义,而非随机噪声。
3 讨论
准确捕捉链间取向并确定蛋白质复合物结构仍然是一项重大挑战,复合物结构预测的准确性普遍低于单链建模。此项工作提出了DeepSCFold,一种自动化复合物预测流程,利用深度学习网络捕捉潜在的结构互补性并构建复合物MSA。结果显示,DeepSCFold能够提供有效的互作信号,从而提升AlphaFold-Multimer的结构预测精度。
已有研究指出,自然界中蛋白–蛋白相互作用的方式可能是有限的,几乎所有已知互作都能找到相似的结合模式。为了获取反映结合模式的互作信号,DeepSCFold使用深度学习模型预测蛋白结构相似性与相互作用概率,并将其用于构建复合物MSA。值得注意的是,现有基于物种注释的序列拼接机制局限于基因组序列,无法充分利用宏基因组数据库中大量信息丰富的同源序列来辅助多链结构组装。DeepSCFold避开了这一局限,为单体MSA的拼接提供了一种可行途径。
通过将构建的复合物MSA与先进的AlphaFold-Multimer建模方法结合,DeepSCFold在CASP15复合物数据集上的实验结果显示了结构预测精度的提升。抗体–抗原体系被广泛认为缺乏链间共进化信号,因为宿主与病原体来自不同生物界,不存在可对应的同源序列。在此类体系中,DeepSCFold在相同单体MSA与模板条件下,在界面预测成功率(DockQ>0.23)方面较AlphaFold-Multimer提高了24.7%。这一提升进一步支持了模型的基本假设:性能改善源于DeepSCFold能够通过结构感知的复合物MSA捕捉形状与理化互补性,从而克服抗体–抗原体系中共进化信息缺失的固有局限。
尽管DeepSCFold在复合物预测中表现提升,仍然存在一些挑战。例如,使用者需在运行前提供复合物的化学计量信息(如各链的拷贝数),这在某些应用场景中可能限制其实用性。准确的模型质量评估(MQA)对于区分可靠预测与错误预测至关重要,尤其在缺乏实验结构进行验证的情况下。MQA还能够提供模型的可信度评分,帮助使用者判断结果的可靠性。此外,将DeepSCFold拓展至蛋白–DNA、蛋白–RNA以及蛋白–小分子等更广泛的体系,也是后续研究的重要方向。