NMI 2025 | MSGo: 未知化学物揭示中的伪数据分子结构生成器

获取详情及资源:
0 摘要
将质谱数据转译为化学结构是外暴露组学中的核心难题,使得快速追踪存在于人体与环境中的数百万种化学物变得十分困难。相较于代谢组学,更大分子空间的化学物在模型开发上面临数据稀缺、模型复杂性高以及查询策略难以制定等关键问题。该研究提出一种可直接由质谱生成分子结构的生成器MSGo,用于发现外暴露组中的未知多氟化学物。该方法通过变换器神经网络并仅利用虚拟质谱进行训练,在验证集中能够正确识别48%的结构;在从文献报道的废水样本中挖掘新的多氟化学物方面,其表现也优于专家。面向概率的虚拟质谱掩蔽策略是MSGo取得高性能的关键。借助MSGo等自动化工具,在实验质谱数据有限的情况下快速发现化学物,将成为应对当前未知多氟化学物危机的重要途径。
1 引言
外暴露组学旨在测量并理解一生中所有环境暴露对健康的影响。然而,未知化学物对健康的具体作用仍缺乏清晰界定,其对全球疾病负担的贡献也长期被忽视。目前,化学结构的鉴定通常依赖将实验质谱与质谱库中的参考谱图比对,或利用PubChem、ChemSpider等数据库,通过计算工具对候选结构进行排序。然而,这些方法无法标注未知结构,仍需依靠经验进行人工判定。鉴于人类每天暴露于成千上万种未披露化学物及其复杂转化产物,迫切需要更快速的自动化方法来探索和发现未知化学物。
已有研究尝试通过基于分子式或反应规则穷举候选分子以扩展分子数据库,但这种方式会指数级增加同分异构体数量,或仅能在有限且既定的反应规则下推断少量转化产物。为解决这些问题,研究者提出了基于“谱图相似则结构相似”假设的间接结构标注策略,例如利用质谱相似性算法、分子网络可视化或基于碎裂谱预测分子指纹并按树状聚类。此外,人工智能方法也被用于识别结构关系或预测化学类别,但这些结构关系依旧无法给出未知化学物的具体结构。
基于自然语言处理的分子生成模型被广泛应用于药物设计,也为未知分子的结构生成提供了可能。一些研究利用生成算法解决局部分支问题,并与现有计算工具结合以辅助结构标注。例如,MSNovelist结合SIRIUS的同位素模式排序,是首个通过中间分子指纹实现从质谱到结构的流程;MassGenie、Spec2Mol与MS2Mol等方法则尝试直接从质谱生成结构,但受限于质谱数据不足及巨大化学结构空间,性能仍有限。
该研究提出的MSGo是一种可直接将质谱转译为分子结构的生成器,能够为外暴露组中的未知多氟化学物寻找全局最优结构。MSGo以数万条虚拟质谱为训练数据,并采用曾用于蛋白预测的变换器神经网络。变换器的注意力机制可同时分析整个谱图,捕捉不同离子峰之间的复杂关系,从而推断相应的子结构及其连接方式。通过概率导向的掩蔽策略去除虚拟谱图中低强度且易误导的碎片,大幅提升伪数据的可靠性,也是MSGo性能的关键。MSGo在验证集中能够正确识别48%的结构,并发现至少两类新的多氟化学物。此类化学物广泛存在于环境中,且已知会引发神经毒性、生殖障碍及代谢疾病,却因结构多样、信息不公开而难以鉴定。尽管部分多氟化学物已被禁用,但厂商仍在生产新的替代物,并以商业机密形式隐藏其结构,使未知多氟化学物危机持续加剧。因此,开发能够快速识别外暴露组未知化学物的工具,对人类健康与环境保护具有紧迫意义。
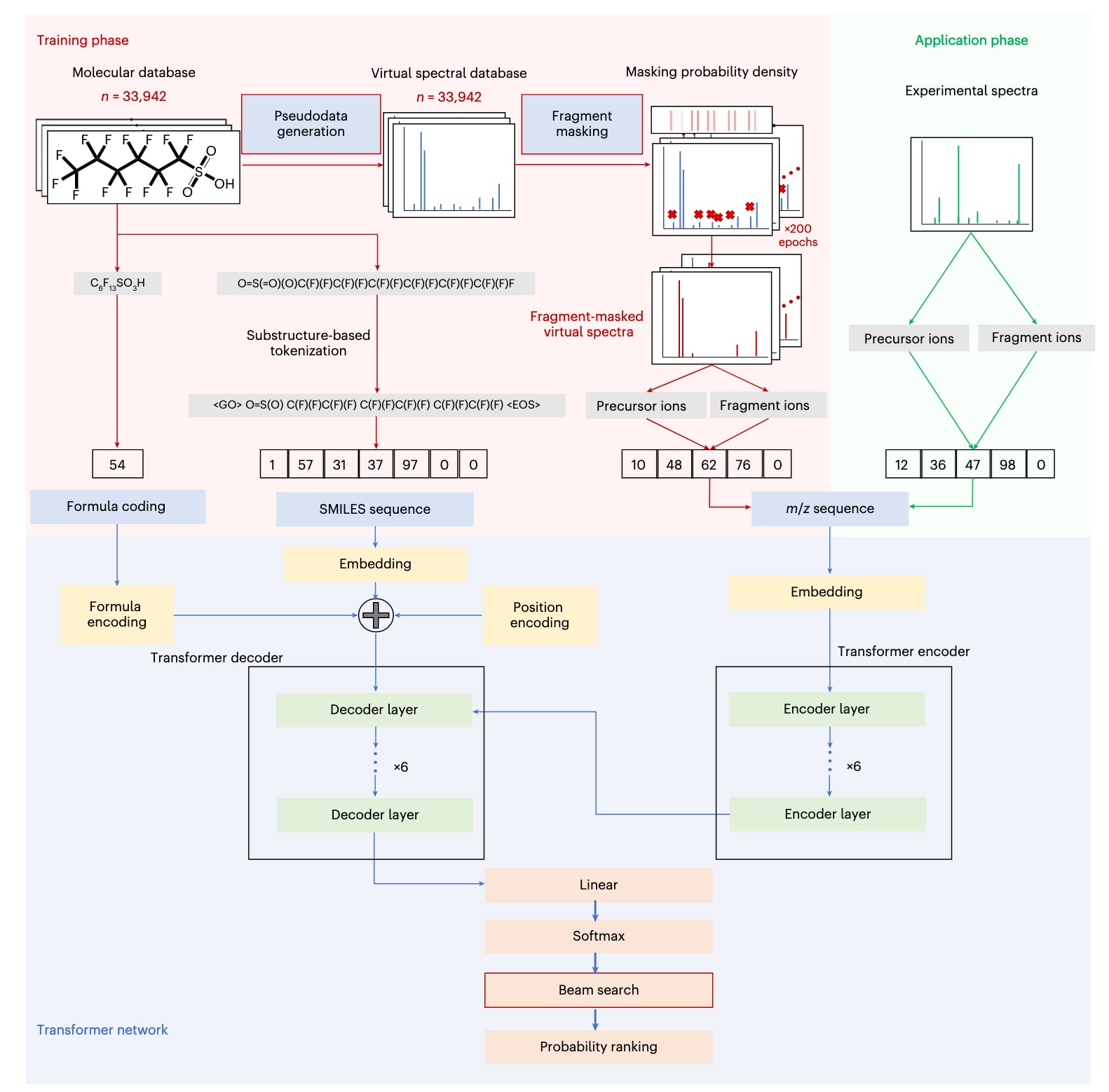
图1|MSGo的模型架构 在训练阶段,分子式、SMILES与质谱信号首先被转换为序列数据类型。SMILES通过基于LSTM的SMILES对编码算法转化为子结构序列。质谱的伪数据由CFM-ID生成,用于构建虚拟谱图库,并进一步通过掩蔽策略处理,以缩短其与实验数据的差距并扩大可用于训练的数据规模。在变换器神经网络中,编码器部分移除了位置编码,而在解码器部分加入了分子式编码。在应用阶段,质谱被处理为序列数据并输入变换器神经网络以生成SMILES。随后通过束搜索与概率排序从生成的候选结果中选出全局最优的SMILES结构。
2 训练MSGo
MSGo的构建、训练与验证需要规模足够大的结构—质谱成对数据。通过查阅文献与数据库,共收集33,942种多氟化学物,并以SMILES记录其结构,建立用于训练的分子结构数据库。然而,与大多数化学物类似,可用的质谱数据远少于结构数据,仅找到219种多氟化学物的300条实验质谱。由于这些数据不足以支持基于变换器神经网络的MSGo训练,因此采用CFM-ID v.3.0生成伪数据,构建虚拟质谱数据库。
实验质谱与虚拟质谱之间存在较大差异,因此如何缩短两者的距离是MSGo训练中的关键。利用100条实验质谱评估二者相似性时发现,相似度低于0.5的虚拟—实验配对占89%。为此,在将虚拟质谱输入嵌入层前,采用掩蔽策略去除低丰度的碎片离子。掩蔽后,相似度大于0.5的配对比例从11%提高至32%。这种概率导向的掩蔽策略与近期的光谱降噪方法类似,是一种稳健的降噪方案。在该策略下,每个epoch都会对同一化学物的虚拟质谱重新掩蔽,使模型能够从不同版本的虚拟光谱中不断学习与优化。碰撞能量与质量精度在训练过程中也经过调优。
MSGo基于注意力机制的改进变换器网络,从质谱生成分子结构。MS1提供精确质量信息,MS2提供碎片离子信息,并共同作为输入。由于MS2中碎片的顺序无意义,因此去除了输入阶段的位置信息编码。在输出阶段,模型采用基于子结构单元的生成策略,而非逐字符生成。通过SMILES对编码算法获取子结构序列,并将分子式及子结构序列作为监督信号输入输出嵌入层。相比逐字符生成,该策略更为准确。为检验生成SMILES的有效性,从300条实验谱生成3,000条候选SMILES,其中95.3%为有效结构,且每条测试谱均能生成有效SMILES,说明对编码方式保证了生成结果的语法有效性。
为了捕获全局最优结构,模型通过束搜索生成SMILES,再依据质量误差或分子式进行过滤,并最终依据生成概率进行排序。

图2|利用文献和数据库中的实验质谱验证MSGo的性能 a,MSGo在不同设置下于前K个候选结果中正确识别多氟化学物的比例,包括完整MSGo、去除分子式、去除掩蔽、同时去除分子式与掩蔽,以及进一步去除前体离子的版本。b,在束搜索中将num_beams设为1、10、50、100与500时,MSGo在前K个候选中正确识别多氟化学物的比例。c,利用100条随机抽取的实验质谱,比较MSGo、CFM-ID与SIRIUS在前K个候选中正确识别多氟化学物的数量。d,来自文献的全氟辛烷磺酰胺丙酸的实验质谱。e,MSGo基于该谱图生成的有效结构,其中发生异构化的子结构以红色标注。
3 MSGo的性能验证
MSGo的性能通过300条实验质谱进行了验证,其总体准确率为48.0%,在候选前十的范围内能够找回80.7%的正确结构。为了进一步评估各类输入特征对模型表现的贡献,对虚拟质谱、精确质量、分子式以及掩蔽策略逐一进行了测试。在仅使用虚拟质谱训练的情况下,模型的正确识别率仅为8.33%;当加入精确质量、分子式与掩蔽策略后,准确率显著提升至48.0%。相比精确质量和分子式,掩蔽策略贡献最大,因为它使虚拟质谱更接近实验谱,并扩大了训练数据的有效规模。
束搜索生成结构的数量(num_beams)同样影响性能。当num_beams=1时束搜索等同于贪婪搜索,随着num_beams增大,MSGo的准确率持续提升。为在计算成本与性能间取得平衡,num_beams最终设为500。随机抽取100条实验质谱,将MSGo与CFM-ID v.3.0和SIRIUS v.5.8比较时,MSGo在识别多氟化学物方面均表现更优。这部分原因在于数据库检索类方法难以处理数据库中缺失的含氟结构,而MSGo直接生成结构,因而具备更广的探索能力。与其他生成模型相比,MSGo在结构注释准确性及结构相似度方面也优于MSNovelist、MassGenie、Spec2Mol与MS2Mol,表明结合前向模型与掩蔽策略的伪数据训练方案可有效扩展至代谢组学之外,并适应外暴露组中更大的结构空间。
尽管训练用虚拟质谱均由CFM-ID生成,但在反向任务中,MSGo表现仍优于CFM-ID,进一步证明概率导向掩蔽策略的必要性。对300条实验质谱进一步分析显示,在43.3%的样本中,MSGo在前十候选中给出了至少三个有效结构,这些结构即便专家或CFM-ID也难以仅凭质谱直接区分。例如,全氟辛烷磺酰胺丙酸的多种支链异构体常需更多碎片信息与保留时间才能人工辨识,而MSGo已能将其纳入候选。值得注意的是,这些支链异构体并未出现在训练数据库中,说明模型并非记忆训练集,而是探索了更大的化学空间并提供更多结构多样性,适用于数据库之外的未知物发现。
MSGo在未知化学物鉴定上甚至优于专家。基于文献报道的废水数据,MSGo预测出17类共51种多氟化学物,与人工鉴定互补。在专家鉴定的15类、90种化学物中,MSGo对其中11类、31种实现首候选正确;在前十候选中则对12类、43种给出了有效结构。对于通过MZcloud数据库鉴定的化学物,MSGo的正确预测比例更高。更重要的是,MSGo还预测到专家未报告的6类新多氟化学物,其中二氯取代的全氟烷基羧酸(Cl2-PFCA)与氯、氢取代的PFOS(H,Cl-PFOS)此前从未在环境外暴露组中出现,且Cl2-PFCA不在训练数据中。这两个结构的碎片裂解途径与同位素分布均支持其合理性。尽管已知的合成路线常停留在单氯产物,但专利资料显示Cl2-PFCA可通过常见氯气制得,并可能用于表面活性剂或氟聚合物生产,提示其在环境中的存在具有合理性。
为了测试MSGo方法在其他化学类别中的可扩展性,进一步收集了3万余种脂质分子并生成其虚拟谱图进行同样方式的训练。使用实验谱图测试时,模型的准确率为27.7%,在前十候选中找回正确结构的比例为50.3%。在分子指纹相似性上,该脂质模型逐步接近MSGo的表现。鉴于脂质的结构空间比多氟化学物更大,若扩大训练分子的数量,其性能仍有提升空间。
此外,MSGo在真实样本含噪质谱中的表现也具有一定鲁棒性,即使谱图包含随机噪声与共洗脱分子的干扰,仍能生成正确的全氟醚羧酸与全氟二醚羧酸结构。尽管并非必须,若在前处理阶段使用解卷积软件进行去噪,可进一步提升MSGo的性能。
总体而言,MSGo能够直接从复杂样本的质谱生成分子结构,加速未知化学物的发现,为解决环境外暴露组中的未知化学物难题提供了一种有效途径。
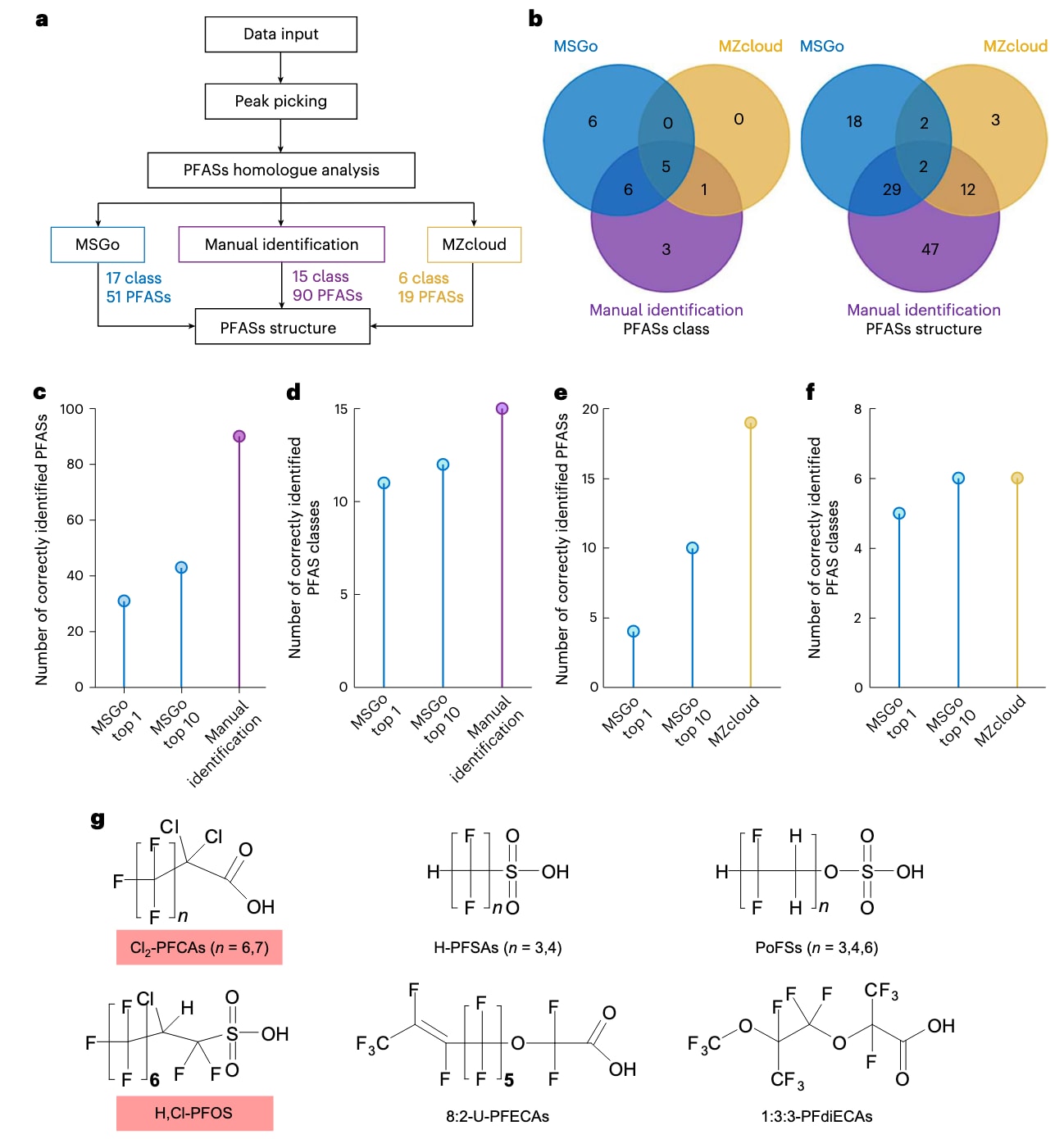
图3|利用环境样本中的质谱数据验证MSGo的性能 a,基于文献报道的数据,对比MSGo、MZcloud与专家的分析流程。b,MSGo、MZcloud与人工鉴定三者在PFAS类别及结构层面的维恩图对比。c,在专家鉴定的90种PFAS中,MSGo在前1与前10候选结果中正确识别的数量。d,在专家鉴定的15类PFAS中,MSGo在前1与前10候选结果中正确识别的类别数量。e,在MZcloud标注的20种PFAS中,MSGo在前1与前10候选结果中正确识别的数量。f,在MZcloud标注的6类PFAS中,MSGo在前1与前10候选结果中正确识别的类别数量。g,MSGo基于文献数据识别出的新PFAS结构,其中首次注释的Cl2-PFCA及氯、氢取代的PFOS(H,Cl-PFOS)以标注方式突出显示。图中还包括H-PFSAs(氢取代全氟烷基磺酸盐)、PoFSs(多氟烷基硫酸酯)、8:2-U-PFECAs(8:2不饱和全氟烷基醚羧酸)与1:3:3-PFdiECAs(1:3:3全氟烷基二醚羧酸)。
4 讨论
外暴露组学面临的核心问题之一,是如何快速追踪存在于人体与环境中的数百万种未知化学物。该研究提出一种基于伪数据训练的深度学习模型,可将质谱自动转译为化学结构,并预测外暴露组中的未知多氟化学物。近期CASMI 2022竞赛的结果显示,深度学习模型在小分子鉴定中的整体表现仍落后于传统机器学习方法,其主要瓶颈在于深度学习通常需要规模巨大的训练集,而目前可获得的参考质谱仅覆盖数万到十数万种化学物,远不足以揭示碎片离子与子结构之间的复杂对应关系。该研究通过使用多氟化学物的伪数据构建并训练MSGo,有效缓解训练集不足的问题。
然而,现有计算工具生成的伪数据并不如实验质谱可靠,直接使用虚拟质谱训练模型会导致性能不佳。研究表明,掩蔽策略的应用对模型性能至关重要。概率导向的掩蔽方法能够去除低强度且多为错误的碎片,同时保留噪声离子,使生成的虚拟质谱不再试图完全复制实验谱,而是为同一结构形成多个不同版本的虚拟谱图。变换器网络在这些包含不同噪声离子的虚拟谱中学习,从而更善于区分真实碎片与噪声,捕捉离子与子结构之间复杂的联系,并在一定程度上抵御仪器噪声。掩蔽策略在机制上实现了信息选择性削减以防止过拟合、通过噪声分布建模提升泛化能力以及促进虚拟与实验谱之间的域适配。此外,这一策略对谱图检索模型也具有潜在提升作用,例如CFM-ID若结合此类后处理策略,可能进一步改善性能。
除了掩蔽策略,MSGo框架还融合了子结构生成策略、束搜索优化以及碰撞能量与质量精度调控等关键创新,为实验谱图匮乏的化学类别提供了一种新的结构注释范式。就多氟化学物而言,MSGo在准确率上已显著优于现有生成模型。尽管目前版本的MSGo专用于多氟化学物,但通过调整伪数据规模及变换器参数,使其与不同化学类别的结构空间相匹配,该训练策略可推广至其他分子类型。对脂质的测试显示,模型的结构识别率可达27.7%,前十候选中找回正确结构的比例为50.3%;虽然训练数据规模仍不足以覆盖脂质更广阔的结构空间,但其性能已优于现有生成式模型,证明基于伪数据的训练在多种化学类别上具有可行性。
为了进一步拓展MSGo能够识别的化学结构类型,亟需积累更多实验质谱,尤其是当前缺乏谱图的结构类别。同时,MSGo有潜力与距离几何工具(如RDKit)及三维深度学习模型(如Uni-Mol)整合,用于三维相关的下游任务。未来,结合X射线晶体学、离子迁移谱与质谱,有望实现从谱图到分子三维构象的端到端预测。