Nature Methods 2024 | CellRank 2: 多视图单细胞数据中的统一细胞命运映射方法
在单细胞技术迅速演进的背景下,如何在多模态、多时间维度的数据中重建细胞命运变化,已成为理解发育、再生与重编程过程的核心难题。发表于 Nature Methods 2024 的 CellRank 2 提供了一套真正意义上的“统一框架”。它将细胞状态变化建模拆解为“核函数 + 估计器”的模块化结构,实现了对伪时间、RNA velocity、实验时间点、发育潜能以及代谢标记等多种视角的灵活组合,可扩展至百万级细胞。
CellRank 2 不仅在造血、内胚层发育等体系中稳定恢复初始/终末状态与命运概率,还能融合跨时间点与时间点内的转移信息,识别传统方法难以捕捉的关键祖细胞。此外,它首次在大规模单细胞背景下实现细胞特异的转录与降解速率估计,为解析发育与命运决策中的调控策略提供了新的量化工具。CellRank 2 的推出,标志着多视图单细胞命运映射进入了统一、可扩展、可解释的新阶段。

获取详情及资源:
- 📄 论文: https://doi.org/10.1038/s41592-024-02303-9
- 💻 代码: https://github.com/theislab/cellrank
- 🌐 网站: https://cellrank.org
0 摘要
单细胞RNA测序可通过表达相似性或RNA velocity来建模细胞状态动态与命运决策,从而重建状态变化轨迹;然而,这类轨迹推断方法并未整合宝贵的时间点信息,也无法利用其他数据模态,而能够处理这些不同数据视角的方法又往往无法结合,或无法扩展到大规模数据。在此背景下提出CellRank 2,这是一种可扩展且灵活的框架,能够以统一方式分析多视图单细胞数据中多达百万级别细胞的命运变化。CellRank 2在人体造血与内胚层发育等多类数据模态中,都能够一致地恢复终末状态与命运概率。该框架还允许同时整合实验时间点内与跨时间点的转移信息,并据此在咽内胚层发育过程中识别促进髓质胸腺上皮细胞形成的基因。此外,该方法还能基于代谢标记数据估计细胞特异性的转录与降解速率,并应用于肠类器官体系,以解析分化轨迹并定位潜在调控策略。
1 引言
单细胞检测技术以此前所未有的分辨率与规模揭示细胞异质性,使得复杂的分化轨迹能够通过计算方法重建。然而,这些轨迹推断方法大多基于一次性测量的单细胞RNA测序数据,难以整合理解细胞状态动态所需的其他信息,例如实验时间点、多模态测量、RNA velocity以及代谢标记。此前已有多种面向新兴数据模态的方法被提出,如用于RNA velocity的CellRank、利用实验时间点的Waddington optimal transport(WOT)以及用于代谢标记数据的dynamo,但它们均只处理单一模态,从而忽略了多模态数据在轨迹分析中的潜力。这种高度专门化限制了许多生物体系的解析。例如,成体造血体系违背了当前RNA velocity模型的核心假设,使CellRank无法应用于这一重要系统,也提出了一个问题:是否能基于其他信息源扩展算法以重建其分化动态。
为解决这一挑战,研究将轨迹推断拆分为两个部分:基于模态的细胞转移建模与独立于模态的轨迹推断,并据此开发出CellRank 2,一个稳健且模块化的框架,可用于分析百万级细胞的多视图数据。CellRank 2将原版CellRank进行了推广,能够充分利用伪时间、发育潜能等替代信息源,以及实验时间点、代谢标记等新数据模态,从而刻画复杂的细胞状态变化,并识别初始与终末状态、命运概率及谱系相关基因。与先前版本相比,新框架结构更模块化,适用模态更多,计算速度也显著提升。
研究展示了CellRank 2在多种应用中的灵活性:在造血体系中利用伪时间推断分化结构,在类胚体形成中推断发育潜能,以及在咽内胚层发育中同时整合实验时间点与时间点内信息。相比传统的跨时间点映射策略,该方法能更准确恢复终末状态,并允许沿时间轴连续分析基因表达变化,还能识别其他方法遗漏的潜在祖细胞。此外,研究还提出了一种用于代谢标记数据的新计算策略,能够在小鼠肠类器官中恢复细胞特异的转录与降解速率,由此揭示潜在调控机制。
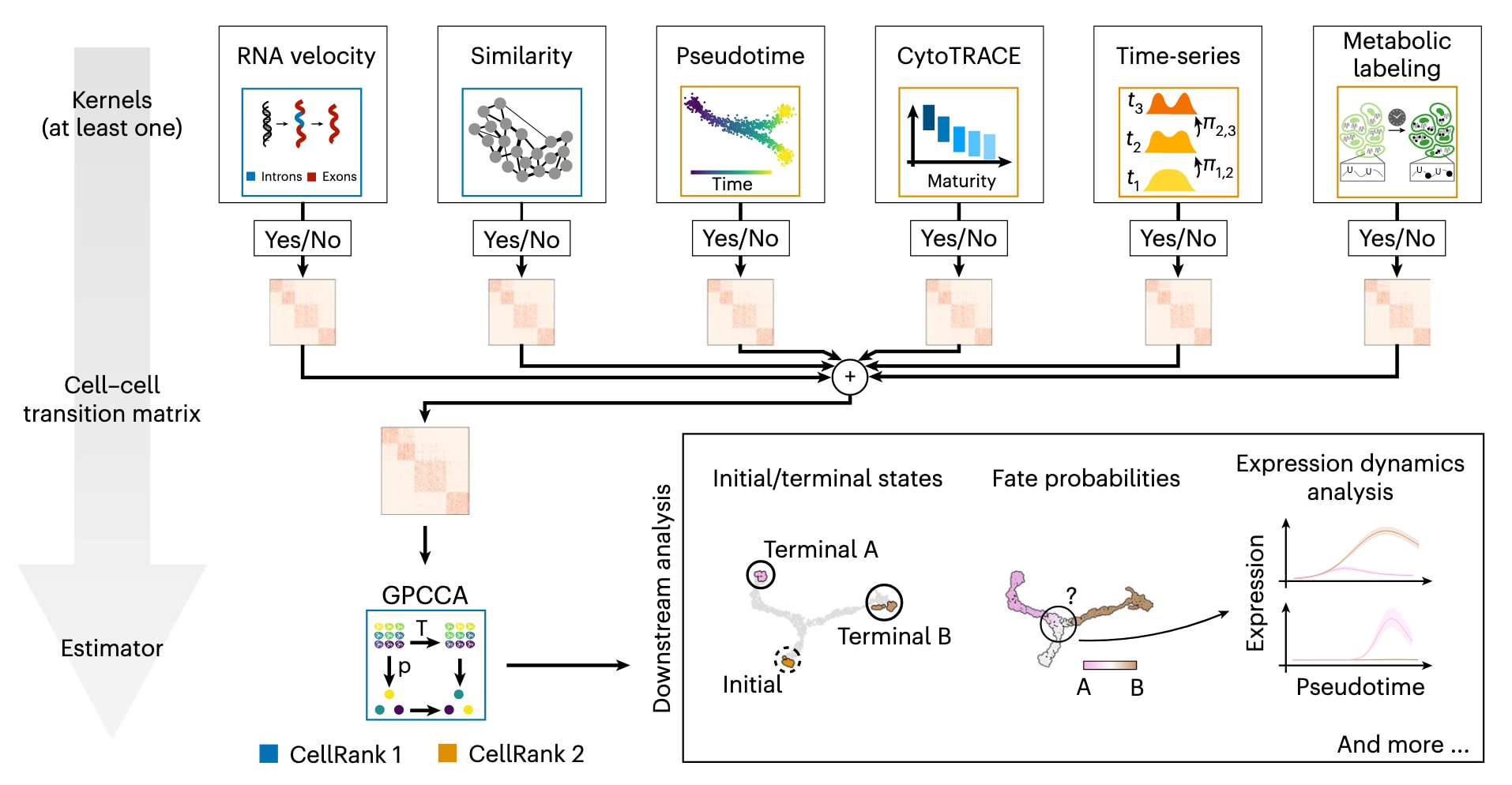
图1 | 展示了CellRank 2如何通过马尔可夫链提供一个统一框架,用于研究单细胞命运决策。 CellRank 2采用模块化设计。数据与问题特定的核函数用于计算细胞间的转移矩阵,从而诱导出一个马尔可夫链;这些核函数至少需要使用一种,但也可通过核组合方式加以整合。估计器对马尔可夫链进行分析,以推断初始与终末状态、命运概率以及与谱系相关的基因。结合命运概率与伪时间,可以研究谱系预示过程中基因表达的变化。图中以蓝色标出源自原始CellRank的功能,以橙色标出新增功能。
2 结果
2.1 状态变化轨迹的模块化框架
CellRank 2基于多视图单细胞数据来建模细胞状态动态,能够自动识别初始与终末状态,计算命运概率,描绘特定轨迹上的基因表达趋势,并筛选与谱系相关的基因。其稳健、可扩展且模块化的设计使其能够适用于多种生物情境。与原版CellRank类似,CellRank 2同样将体系描述为一个概率模型,其中每个细胞对应马尔可夫链中的一个状态,边表示细胞之间的转移概率;不同之处在于,该版本允许从多种生物学先验中推导转移概率。参考已有成功的轨迹推断方法,模型假设细胞沿表型流形以渐进且无记忆的方式转换,而无记忆性假设成立在于模型描述的是平均细胞行为。
为了实现广泛适用性,CellRank 2被划分为两大组件:用于基于多视图数据计算细胞间转移矩阵的“核函数”,以及用于分析转移矩阵以识别初始与终末状态、计算命运概率及执行其他下游任务的“估计器”。CellRank 2提供多种核函数,可基于基因表达、RNA velocity、伪时间、发育潜能、实验时间点以及代谢标记数据计算转移概率。根据数据特性与研究问题,既可单独使用某一核函数,也可组合多种核函数以构建多视图马尔可夫链。为便于获得动态概览,研究引入了基于随机游走的可视化方案。
在许多生物过程中,起始点可以可靠确定,细胞也可沿伪时间排序。基于这一优势,CellRank 2利用伪时间偏置最近邻图的边,使其更偏向成熟细胞状态,从而估计细胞之间的转移;发育潜能也可以类似方式使用。PseudotimeKernel与CytoTRACEKernel将既有概念推广至任意伪时间与图谱级数据。对于起始状态未知或发育时间尺度较长的体系,可通过多个实验时间点更好地恢复整体动态。为重建跨时间点与时间点内的分化过程,研究扩展了经典最优传输框架,特别是WOT,并通过RealTimeKernel纳入时间点内动态。另一方面,代谢标记提供实验手段,能够突破离散时间点的限制。基于此类数据,研究开发了推断动力学速率的方法,用以恢复细胞转换信息。
在阐述各类核函数与其应用后,研究强调它们可组合用于多视图建模,从而获得更全面的动态视角。一旦得到转移矩阵,估计器模块即可用于推断轨迹,包括初始与终末状态、命运概率与谱系相关基因。估计器与数据视图无关,因此适用于任何转移矩阵。为提升大规模数据上的计算效率,研究假设每个细胞仅能产生少量潜在后代,从而保证所有核函数生成的转移矩阵均为稀疏格式,并使CellRank 2计算命运概率的速度比原版提高约30倍。
这种模块化与稳健的设计使CellRank 2成为分析多视图单细胞数据中状态动态的灵活框架,能够快速适应新兴数据模态,包括谱系追踪与时空数据,并通过扩展核函数与估计器支持新的分析任务。
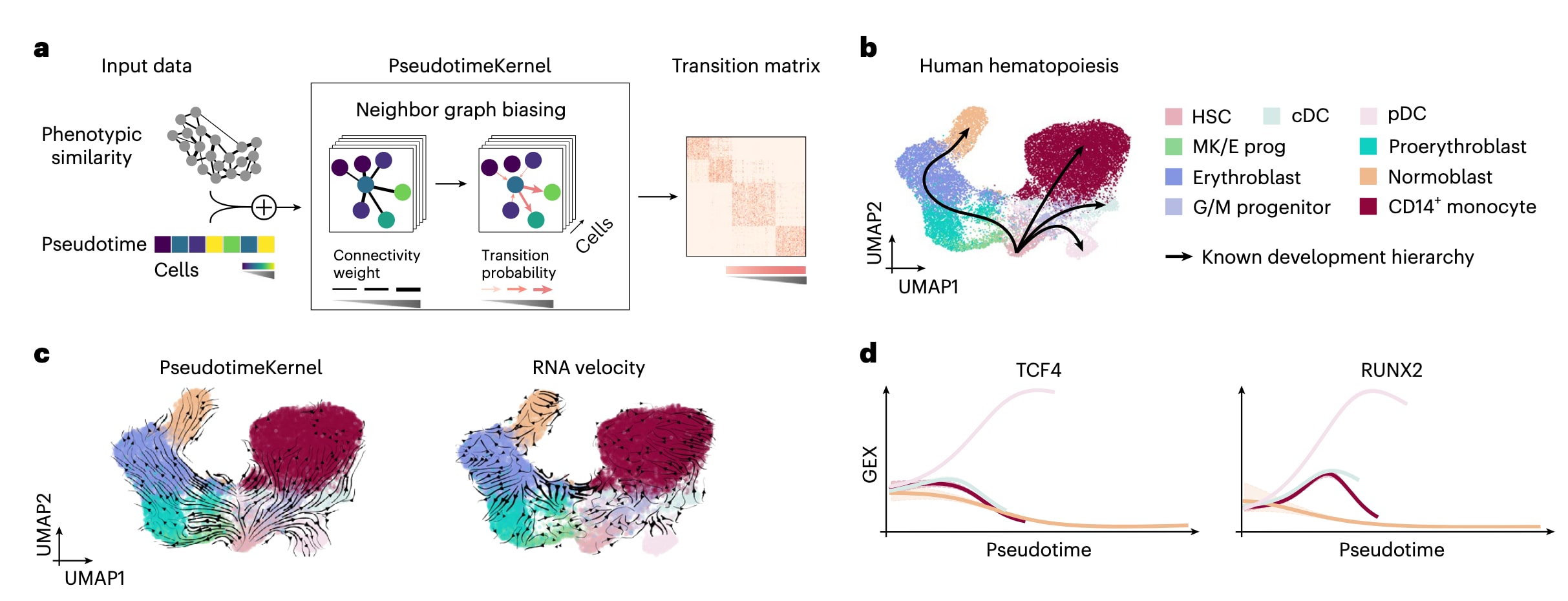
图2 | 展示了如何利用伪时间排序进行细胞命运映射。 a部分中,PseudotimeKernel将基于表型相似性的最近邻图中的边偏向于更高的伪时间方向,从而定义细胞间的转移概率。b与c部分为24,440个外周血单个核细胞的UMAP嵌入,按细胞类型着色(cDC;G/M progenitor,粒细胞/髓系祖细胞;HSC;MK/E prog,巨核/红系祖细胞;pDC)。b部分以黑色示意该体系已知的分化层级。c部分展示基于PseudotimeKernel所得的投影velocity场(左)以及基于RNA velocity的velocity场(右)。d部分展示利用命运概率与基因表达的相关性能够恢复pDC谱系的已知调控因子。图中按照此前工作所提出的方式,在伪时间(横轴)上拟合广义加性模型,显示基因表达(纵轴)的谱系特异趋势;每个细胞对各条谱系的贡献根据CellRank 2恢复的命运概率加权。颜色对应b部分中所示的不同谱系。
2.2 克服RNA velocity的局限性
在稳态的人类造血体系中,RNA velocity由于模型假设被违反而无法推断出正确的动态,即便伪时间仍能被可靠恢复。具体而言,传统RNA velocity模型假设的恒定速率在该体系中并不成立,而系统中关键基因的高噪声或低覆盖度进一步削弱了模型表现。考虑到在初始状态明确的体系中,传统伪时间方法表现稳定,研究由此提出以PseudotimeKernel避开RNA velocity的限制。该核函数利用伪时间计算细胞间的转移概率及相应向量场(图2a)。基于此前在Palantir中提出的概念思想,该方法被推广至任意预计算的伪时间,并采用软加权策略。
将PseudotimeKernel应用于人类造血体系,研究基于扩散伪时间(DPT)为红细胞母细胞、单核细胞与树突状细胞谱系计算转移概率。PseudotimeKernel能够正确恢复四个终末状态以及初始状态。为进一步可视化动态过程,研究将RNA velocity提出的流线投影方法拓展至任意基于邻接图的核函数(图2c)。通过将基因表达与谱系特异的命运概率相关联,方法识别出可能参与谱系承诺的候选基因;这一分析正确检测到已知参与浆细胞样树突状细胞谱系调控的转录因子RUNX2与TCF4。
相比之下,基于RNA velocity的分析未能恢复经典树突状细胞谱系,VelocityKernel赋予的命运概率也违背了已知的谱系层级,例如将原红细胞母细胞与红细胞母细胞误判为向单核细胞而非红细胞母系转移。这些偏差源于RNA velocity模型假设在此体系中无法满足。
为提供定量评估,研究计算了不同核函数跨边界正确性(CBC)得分的对数比,用以衡量它们在已知细胞状态转移中的表现。结果显示,PseudotimeKernel在大多数状态转移中显著优于基于velocity的方法(8个中有6个)。研究还引入终末状态识别(TSI)得分,用以衡量相较于最优策略(TSI=1)对已知终末状态的识别质量。伪时间方法同样优于RNA velocity方法(TSI=0.9 对 0.81)。
PseudotimeKernel可推广至任意伪时间,使得用户能够选择最适合数据集的算法。在具有较简单分化层级且起点明确的体系中,该核函数相较传统伪时间方法能提供关于终末状态与命运承诺的额外洞察。
2.3 学习基于发育潜能的向量场
伪时间推断通常需要预先给定初始状态。如果初始状态未知,可以使用CytoTRACE推断干性得分,其核心假设是:在平均意义上,未成熟细胞表达的基因数量多于成熟细胞。该假设在许多早期发育情境中十分有效,但CytoTRACE在处理大规模数据时无论在时间还是内存方面都无法扩展,且不能解析通向终末状态的具体轨迹或命运概率。基于此,研究对原始CytoTRACE方法进行了改造,开发了CytoTRACEKernel,使KNN图的边指向更高的成熟度,并能够在图谱级数据上量化细胞间的转移概率。该核函数在多个数据集上的结果与原始方法一致。此外,在包含130万细胞的小鼠器官发生图谱中,原版CytoTRACE在超过8万细胞时即失效,而新方法能在2分钟内运行完整数据集。
研究利用CytoTRACEKernel分析来源于多能细胞聚集体(类胚体)的内胚层发育。基于CytoTRACE的伪时间在各实验时间点上平滑增加,符合生物学预期,并成功识别出11个终末群体中的10个以及正确的初始状态。相比之下,Palantir与DPT在首阶段识别出一个双峰的早期细胞分布,导致其他阶段的伪时间范围被压缩。内胚层可发育为多种内脏器官,因此研究将命运概率与基因表达相关联,从而识别与谱系相关、可能参与器官发生的基因,包括MIXL1、FOXA2与SOX17等转录因子,与原始研究结果一致。
为寻找这些转录因子的潜在上游调控因子,研究将内胚层轨迹中相关性最高的基因按其在CytoTRACE伪时间中的峰值排序,并以热图展示其平滑表达趋势。结果显示,LINC00458、LINC01356、NODAL以及九个转录因子的峰值均早于FOXA2与SOX17。它们全部是已知的小鼠内胚层发育基因,而LINC00458先于LINC01356表达这一预测也已在此前研究中被观察到。
CytoTRACEKernel使得研究能够在无需预先指定伪时间初始状态的情况下,从内胚层发育的单次快照中推断细胞动态,并成功恢复终末状态、关键驱动基因及其时间激活模式。
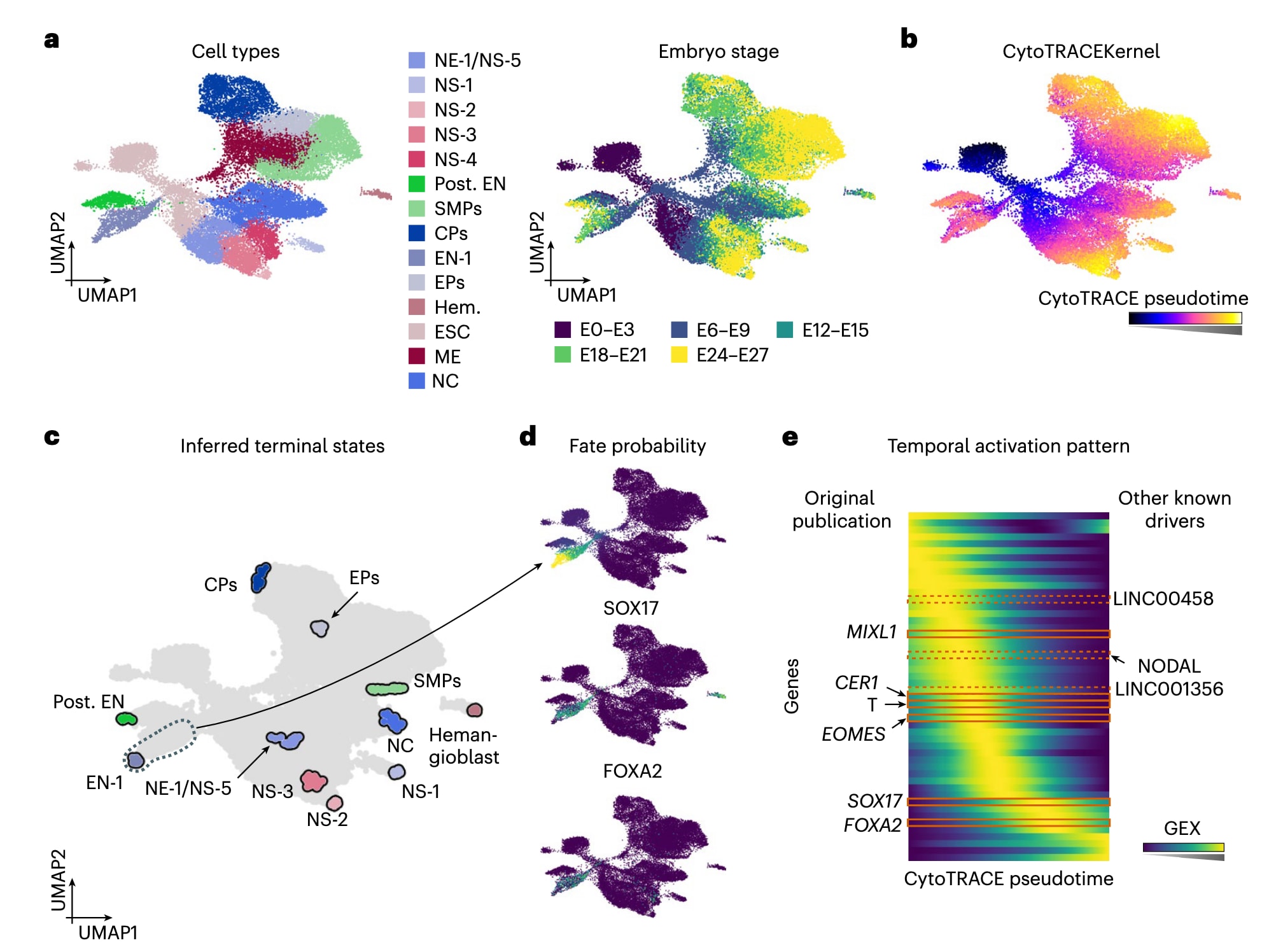
图3 | 展示了CytoTRACEKernel在恢复基因激活时间序列中的能力。 a与b部分为31,029个类胚体细胞的UMAP嵌入,分别按原始簇注释(包括心脏前体CPs、内胚层EN、心外膜前体EPs、胚胎干细胞ESC、血管母细胞Hem.、中胚层ME、神经嵴NC、神经外胚层NE、神经元亚型NS、后部内胚层Post. EN、平滑肌前体SMPs;a左图)、胚胎阶段(a右图)以及CytoTRACEKernel计算得到的伪时间(b)着色。c部分展示基于CytoTRACEKernel推断出的终末状态。d部分在UMAP上显示内胚层谱系的命运概率(上)与恢复出的驱动基因的表达量(下)。e部分展示沿CytoTRACE伪时间的前50个自动识别出的谱系相关基因的平滑表达趋势,并按照其在伪时间中的峰值排序。图中标出原始研究中报道的基因(左)以及CellRank 2额外恢复的已知驱动基因(右)。
2.4 为命运映射引入时间分辨率
单细胞时间序列在研究非稳态分化过程中愈发重要,计算上的挑战在于如何匹配不同时间点测序的细胞以重建状态变化轨迹。多数既有方法要么仅估计群体动力学,要么利用OT进行跨时间点匹配,却忽略时间点内部的状态转变,而这些局部动态信息对于判定分化方向与检测终末状态非常关键。基于此,研究者提出RealTimeKernel,将WOT计算的跨时间点转移与基于相似度的时间点内部转移结合,实现多视角建模。融入跨时间点转移后,能够在时间序列数据中无偏地识别初始与终末状态,得到更精细的命运映射。为了获得整体分化动态的初步理解,研究者在嵌入空间可视化基于RealTimeKernel的随机游走。
由于包括WOT在内的许多OT实现均采用熵正则以加速求解,会导致转移矩阵极为稠密,从而减缓下游分析速度,限制大规模数据的处理。为此提出自适应阈值稀疏化策略,使宏状态与命运概率的计算在MEF重编程数据上的速度分别提升9倍与56倍。通过比较阈值前后终末状态的命运概率,发现各谱系的相关性极高,表明稀疏化保持了信息一致性。在此基础上,RealTimeKernel取得完美的TSI分数,而VelocityKernel则明显较低。
许多应用需要连续时间信息而非离散时间点,因此研究者构建了一种新的“实时引导的拟时序”方法,在利用实验时间点的同时将其嵌入连续的表达变化空间。在MEF重编程数据上,该拟时序与实验时间的相关性显著优于传统方法,并能够沿连续轴捕获渐进的命运建立过程。
研究者将RealTimeKernel应用于咽内胚层发育分析,该组织在胚胎早期形成甲状旁腺、甲状腺与胸腺等器官。利用E9.5至E12.5的时间序列数据,自动重建初始状态并找回原研究中手动标注的11个终末状态中的10个,且在TSI表现上显著优于VelocityKernel。基于命运概率与基因表达的相关性,还成功回溯各谱系的已知关键调控因子,如Gcm2、Hhex与Foxn1。
进一步聚焦胸腺髓质上皮细胞(mTEC)的分化轨迹,研究者选取相关细胞群后,成功恢复初始状态、识别各终末状态,并在TSI上优于基于RNA velocity的推断。通过计算命运概率,发现一个具有更高mTEC分化概率的前体细胞簇,而这一簇在二维UMAP中易被忽略,凸显了高维命运推断的重要性。相关性分析识别出多种潜在调控因子,包括Fos、Grhl3、Elf5以及多个已知mTEC标记基因,并发现一些与人类不同mTEC亚群相关的基因也在高排名之列。
相比之下,WOT仅依赖跨时间点信息,无法识别该潜在mTEC前体簇;即使借助RealTimeKernel提供的前体细胞信息,WOT在驱动基因识别上依然表现较弱,原因可能在于其使用pullback分布而未考虑时间点内部动态。相较之下,CellRank 2利用跨时间点与时间点内部信息构建全局转移矩阵,从而计算连续的命运概率。传统差异表达分析在驱动基因识别上的性能同样不及相关性方法。
综上,RealTimeKernel通过整合时间内与时间间的表达变化视角,不仅能够识别新的潜在前体细胞群,也能比依赖单一数据视角的方法挖掘出更多具有生物学意义的关键调控因子。
2.5 从代谢标记中推断动力学速率与细胞命运
标准单细胞测序流程具有破坏性,无法直接追踪基因表达随时间的变化;而代谢标记技术可标记新转录的mRNA,从而获得具备分钟到小时级时间分辨率的单细胞RNA数据,极大提升对系统动态的解析能力。基于此,研究者构建了一种利用脉冲–追踪实验来学习细胞状态定向变化轨迹的方法。与既有模型类似,该方法将mRNA动态描述为一个包含转录速率与降解速率的动力系统,并利用代谢标记所携带的动态信息估计每个细胞与基因的动力学参数。具体而言,基于细胞相似性图谱,在每个细胞、基因与标记时间点上构建局部邻域,通过最小化观测与估计转录本之间的平方欧氏距离来推断转录与降解速率,据此得到高维velocity向量场,并利用VelocityKernel推断细胞间转移概率。
该方法应用于小鼠肠类器官的scEU-seq数据,成功恢复吸收细胞、肠内分泌细胞、杯状细胞与潘氏细胞四个终末状态。与经典RNA velocity相比,基于代谢标记推断的velocity场具有更高的细胞类型纯度,并在TSI评价上取得更优表现。同时,与另一种基于代谢标记推断细胞动力学的方法dynamo相比,CellRank 2不依赖稳态假设、可在全部细胞上进行参数估计、能获得细胞特异的动力学速率并提供概率化的轨迹推断,而dynamo在类器官数据中仅识别出吸收细胞这一终末状态。
在驱动基因识别方面,研究者整理了各谱系的已知关键调控因子与标记基因,并与各方法的排名进行比较。基于代谢标记的CellRank 2在四个终末状态上均取得最佳表现,显著优于不使用标记信息的CellRank 1以及使用标记信息但基于稳态假设的dynamo。推断出的细胞与基因特异的动力学参数还使得进一步探索谱系相关基因的调控策略成为可能。分析杯状细胞谱系中排名靠前的标记与调控基因可观察到两类典型策略:一类表现为转录速率升高而降解速率降低,另一类则表现为同时升高转录与降解速率,两者分别对应协同型与竞争型调控模式。同样的基因集合在吸收细胞谱系中呈现出不同方向的调控变化,进一步体现了动力学参数在解析谱系特异调控中的价值。
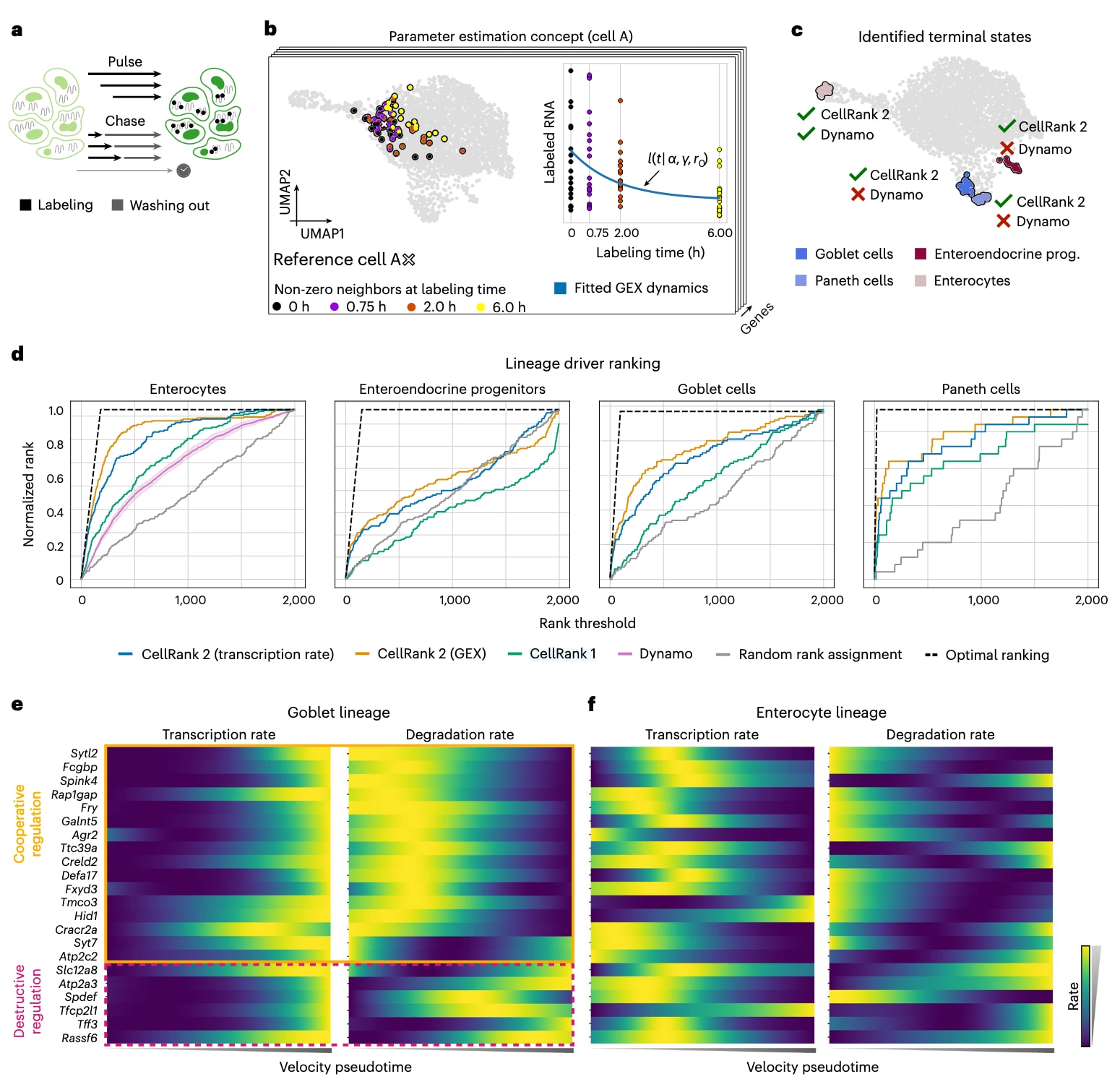
图5 | 通过代谢标记量化谱系特异性的调控策略。 **a,**在脉冲–追踪(pulse–chase)实验中对细胞进行代谢标记9,并在随后进行单细胞测序。脉冲实验通过将细胞置于含核苷类似物的培养基中不同时间,使新生 RNA 被标记;追踪实验则先让细胞在核苷类似物中长时间孵育,使细胞内 mRNA 完全掺入标记核苷,随后洗脱这些核苷并在不同时间点收集样本。**b,**对于每个细胞、每个基因以及每个标记时长,研究者寻找一个邻域,使其中包含预设数量、具有非零表达计数的细胞(示例为细胞 A)。随后利用该邻域细胞估计该细胞–基因组合的特异性转录速率 α 与降解速率 γ,以建模标记 mRNA 的动力学。**c,**UMAP 展示了使用 CellRank 2 与 dynamo 识别出的终末状态。绿色对勾表示该方法成功恢复了对应的终末状态;红色叉号表示未能识别。**d,**不同方法对各谱系驱动基因的排序结果。由于 dynamo 仅识别出肠细胞(enterocyte)为终末状态,因此只对该谱系产生基因排名。图中展示了 dynamo 的平均基因排名及其 95% 置信区间(方法)。**e,**杯状细胞(goblet)谱系中,已知高排名驱动因子的推断转录速率(左)与降解速率(右)。**f,**与 e 同理,但展示的是肠细胞(enterocyte)谱系的结果。
3 Discussion
CellRank 2构建了一个稳健、模块化且具高度可扩展性的框架,用于推断和研究单细胞水平的轨迹与命运决策。其核心思想在于将转移矩阵的推断与分析分别交由不同的kernel与估计器完成,从而在统一体系内兼容多种数据模态,克服单一数据类型方法的局限性。该工具成功用于人类造血体系的拟时序分析,并基于stemness估计解析了内胚层发育过程中的基因动态。模块化设计使各类数据模态的快速集成成为可能,且整体框架的计算可扩展性远超以往实现。
为了整合时间序列数据,研究者设计了高效的OT kernel,使模型同时考虑时间点之间与时间点内部的信息。在此基础上成功识别到髓质胸腺上皮细胞的潜在祖细胞群,而这类细胞在忽略时间点内部动态的方法中并未被捕捉。近年来,时间序列测序逐渐结合遗传谱系示踪或空间分辨技术,相关计算方法能够更准确地跨时间映射细胞。这类增强的跨时间信息可与RealTimeKernel结合应用,如在线虫与小鼠胚胎发育数据中的示例所示,也凸显了面向多模态时间序列的视角无关框架的重要性。
对于时间序列的连续分析,Mellon提出以密度估计加强OT映射的思路,有望进一步改进CellRank 2的时间整合能力。此外,在整合代谢标记数据时,CellRank 2的kernel–估计器设计显示出特别的价值。基于细胞特异性的转录与降解率推断,研究者展示了代谢标记如何弥补剪接信息推断RNA velocity时的结构性限制,从而完整识别类器官中全部谱系。进一步结合推断的动力学参数,还能够探究细胞状态变化背后的调控策略。在同类方法中,velvet无法估计转录速率并假设所有细胞共享恒定降解率;storm虽放宽该假设,但只在后处理阶段实现,且依赖确定性分析。相比之下,CellRank 2可直接估计细胞特异的动力学参数,并通过灵活的马尔可夫链提供概率化的下游分析。
随着单细胞代谢标记技术与大规模、空间或体内体系结合,分析需求的规模迅速增长,也显示出构建可扩展框架的必要性。未来的方向之一是在推断动力学参数的同时整合分化轨迹排序。研究者已提出多种可利用方向性信息的kernel:当存在至少两个或三个脉冲追踪时间即可使用代谢标记向量场推断;RealTimeKernel适用于时间点密集的时间序列;VelocityKernel适合满足RNA velocity模型假设的体系;PseudotimeKernel在能识别唯一初始状态且分化单向推进的条件下能揭示状态变化;CytoTRACEKernel则适用于初始状态未知的情形。若这些假设不满足,不同kernel会产生偏差,例如VelocityKernel在造血体系中未能正确重建已知分化层级。多种kernel可通过全局权重组合,已有研究利用其联合分析心外膜类器官发育与人类皮层褶皱形成。未来计划探索基于局部细胞位置的kernel加权,以实现情境依赖的多模态整合。
在驱动基因识别方面,目前方法依赖命运概率与基因表达的相关性,但要深入机制仍需引入扰动数据与因果推断策略。这种结合将更好地揭示分子属性与命运决策之间的因果联系。整体来看,随着单细胞数据在规模与多样性上的持续扩展,这一框架将在解析与理解细胞命运选择方面发挥关键作用。