NMI 2022 | 通过深度学习与分子模拟加速理性PROTAC设计

获取详情及资源:
0 摘要
蛋白水解靶向嵌合体(PROTAC)通过招募泛素−蛋白酶体系统来选择性降解致病相关蛋白,已成为一种重要的分子工具。然而,PROTAC的开发需要在极其庞大的化学空间中进行大量试验与筛选。为加速这一过程,研究提出一种适用于低资源情境的深度生成模型,用于实现理性PROTAC设计,并进一步结合深度强化学习,引导模型生成具有更优药代动力学特性的分子。以BRD4为示例靶标,方法共生成5,000个候选分子,并利用基于机器学习的分类器与物理模拟进行多层筛选。作为概念验证,研究从中遴选、合成并实验检测了六个BRD4降解型PROTAC,其中有三个在细胞实验和蛋白印迹中得到验证,其中一个候选物在小鼠体内表现出良好的药代特性。深度学习与分子模拟的结合有望提升PROTAC的理性设计与优化效率。
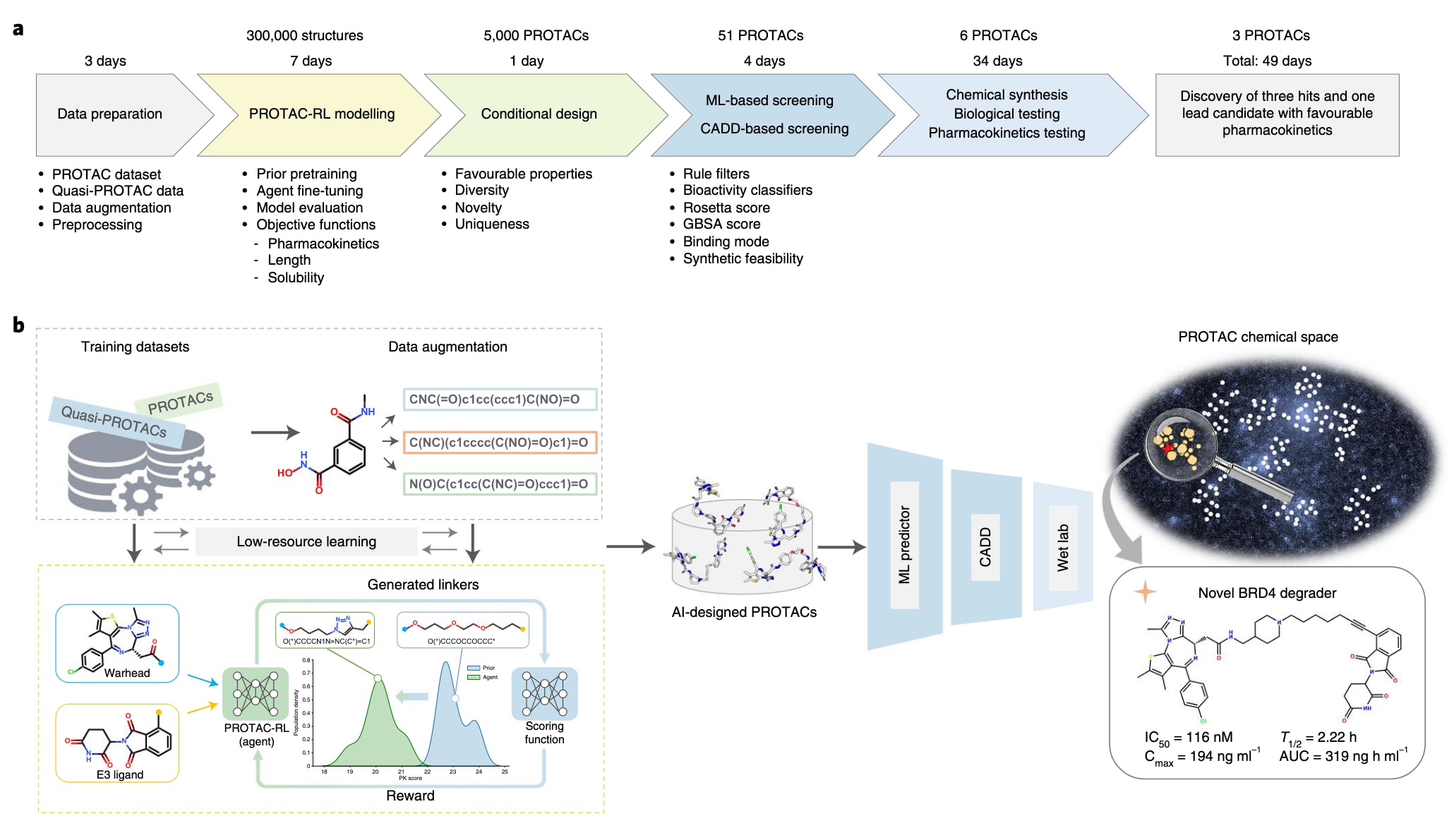
图1 | 方法概览。 a,基于人工智能的加速PROTAC设计策略的总体流程与时间线。b,利用PROTAC-RL进行先导PROTAC设计的整体工作流程。CADD表示计算机辅助药物设计。
1 引言
自从2001年首次提出PROTAC概念以来,利用泛素−蛋白酶体系统选择性降解致病相关蛋白已成为重要策略。PROTAC由三部分组成:定位靶蛋白的配体、招募E3泛素连接酶的配体以及连接两者的化学连接体。由于这种双功能结构能够同时结合靶蛋白与E3连接酶,从而形成三元复合物并促进靶蛋白的多泛素化与降解,因此只需瞬时结合即可诱导作用,这与依赖持续占据靶点的传统小分子抑制剂截然不同。此外,PROTAC不受限于传统“可成药”位点,能够利用蛋白表面的任意结合位点,实现对“非可成药”靶点的调控,这也使其设计难度远超常规小分子药物。
PROTAC的理性设计可分为三部分,其中战头与E3配体的开发与传统小分子发现相似,而连接体的实验设计却极为困难,因为在缺乏有效PROTAC的情况下,靶蛋白与E3酶本身并不发生相互作用。传统策略需要大量试验与迭代,因此效率极低。近年来研究逐渐聚焦于连接体的去 novo 设计,因为其在分子的物化性质与降解效率中起关键作用。然而,由于三元复合物结构复杂且动态性强,连接体设计依旧艰巨。与此同时,PROTAC分子通常偏大且柔性高,导致其往往无法满足口服药物的药代性质,这给后续开发带来另一个重大挑战,因此亟需新方法加速功能性PROTAC的发现。
基于深度生成模型的快速化学空间探索为分子设计带来了突破。不同类型的生成网络已成功用于小分子、肽以及抗体的设计,也被应用于连接体生成。例如,DeLinker利用图结构并直接结合三维信息,SyntaLinker则将连接体生成转化为句子补全任务。然而,这些方法依赖小分子数据训练,未能考虑小分子与PROTAC在策略与化学空间上的差异,也未纳入药代特性。此外,公开可用的PROTAC数据极少,目前最大的公开数据库也仅约2,300条,难以支撑高质量模型训练,更未在实验层面验证其生成分子的有效性。
基于此,研究构建了PROTAC-RL,一种结合增强型transformer结构与记忆辅助强化学习的全新生成模型,用于理性PROTAC设计。模型输入E3配体与战头,输出可行的连接体结构以构建完整分子。为解决数据稀缺问题,方法首先利用大量与PROTAC化学空间相近的准PROTAC小分子预训练片段连接模型,并在随机SMILES增强的真实PROTAC数据上进行微调,得到Proformer模型。随后通过基于经验奖励函数的记忆辅助强化学习,使生成分子具备更优药代属性。以BRD4为概念验证靶点,研究生成了5,000个候选分子,并通过聚类、分层机器学习分类器以及基于物理的分子模拟进行筛选。根据合成可及性,研究最终合成并测试了六个候选PROTAC,其中三个在细胞实验与蛋白印迹中表现出降解活性;另有一个候选物在Molt4细胞中展现出强抑制效果并在小鼠体内具有良好药代性质。整体流程用时仅49天,充分显示了深度学习与分子模拟组合在提升PROTAC设计与优化效率方面的巨大潜力。
2 结果
2.1 PROTAC-RL概述
对于给定的一对战头与E3配体,理性PROTAC设计的核心在于能够在庞大的化学空间中进行有效采样,生成既具化学可行性又具有优良药代动力学特性的分子。为同时实现这两个目标,方法由两个相互关联的模块组成:负责化学空间采样的“先验”网络Proformer,以及引导先验模型朝向目标性质优化的“智能体”网络PROTAC-RL。
在化学空间采样部分,方法将任务形式化为句子补全过程,输入为战头与E3配体的SMILES表示,输出为生成的PROTAC分子。基础先验模型Proformer基于transformer构建,因为这类结构在分子建模任务中展现优异性能。由于公开可用的PROTAC数量极少(不足2,300个,即PROTAC-DB),模型首先在29.4万条与PROTAC体量相近的准PROTAC小分子上进行预训练,再使用真实的PROTAC数据进行微调,并通过随机SMILES增强策略提升微调效果。
在性质优化部分,Proformer被嵌入到强化学习框架(PROTAC-RL)中,用以引导模型生成具备目标性质的分子。强化学习之所以适用,是因为分子性质函数往往复杂且无法直接求导,需通过策略引导采样实现优化。
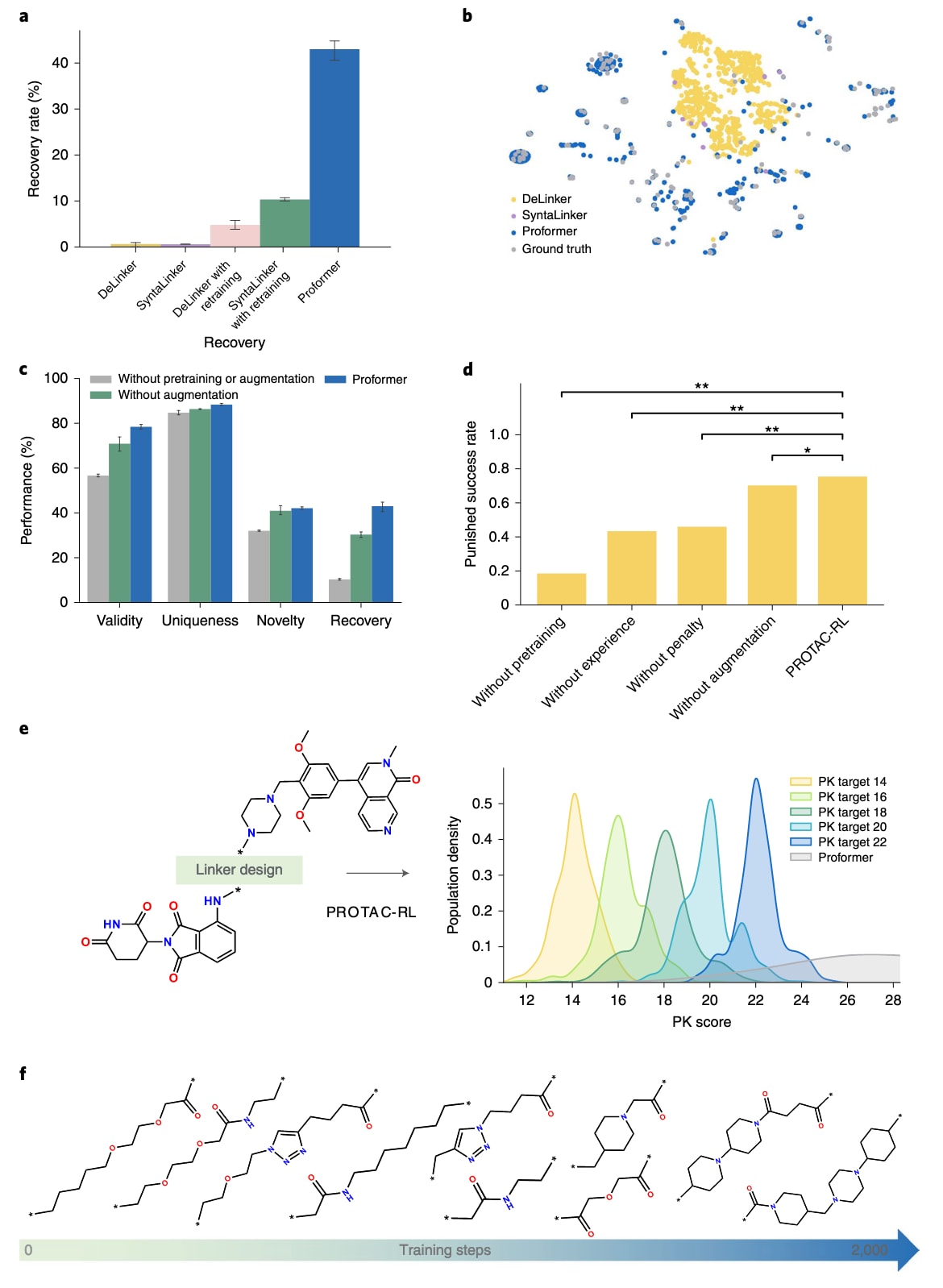
图2|PROTAC-RL的性能表现。 a,领先片段连接模型的性能比较,数据以平均值呈现,误差线表示标准差(n=3次独立运行)。b,测试集中真实PROTAC与不同模型生成的PROTAC的t-SNE可视化分布。c,不同训练策略下生成模型在有效性、独特性、新颖性与恢复率方面的性能比较,数据以平均值±标准差呈现(n=3次独立运行)。d,PROTAC-RL模型在条件生成任务中的平均惩罚成功率消融研究,每组总样本量为n=10,使用双侧t检验(从上至下的精确P值分别为
2.2 Proformer实现了通用的PROTAC生成
为了评估模型性能,研究借鉴以往片段连接工作,将PROTAC数据集按8:1:1划分为训练集、验证集与测试集。对于测试集中每一对战头与E3配体,模型生成前十个候选分子,并统计真实连接体在这些生成结果中被成功复现的比例,即恢复率,用于衡量模型表现。
研究将Proformer与当前领先的片段连接方法进行比较,包括基于图结构的Delinker与基于语言模型的SyntaLinker,以及它们在PROTAC训练集上重新训练的版本。结果显示,Proformer的恢复率达到
在使用PROTAC训练集重新训练Delinker与SyntaLinker后,它们的恢复率分别提升至4.8%与10.4%,但仍远低于Proformer的43.0%。这种差距部分来自模型结构与训练策略的优势,因为移除SMILES增强会导致恢复率下降12.7%,进一步移除预训练步骤会再降低19.9%。总体来看,Proformer的优越表现源于在有效性、独特性与新颖性三方面取得的平衡,相较其他方法更能准确与多样地生成合理的PROTAC结构。
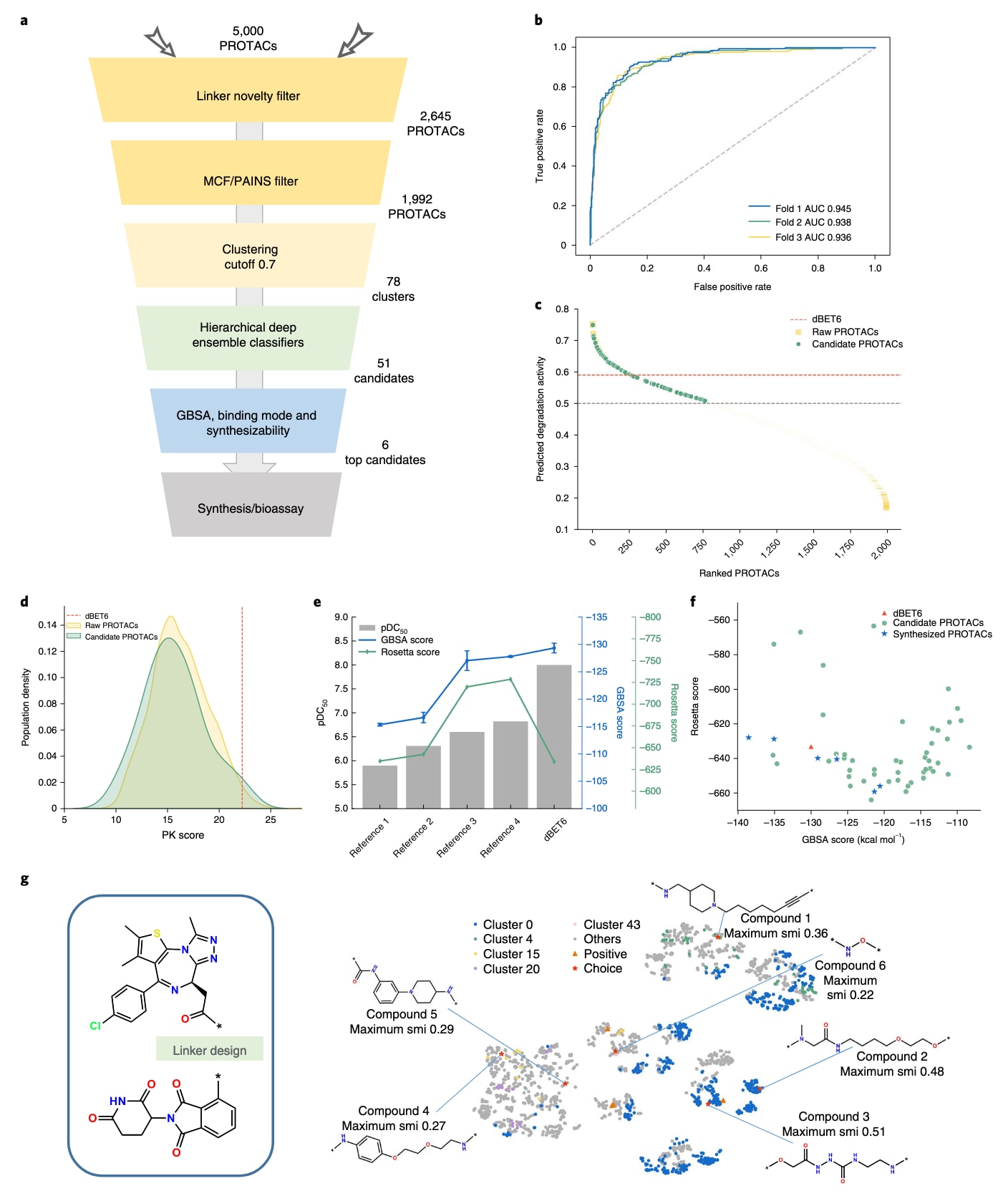
图3|计算筛选流程。 a,生成后体内筛选的整体流程。b,采用三折交叉验证评估集成活性预测器性能的ROC曲线。c,原始生成的BRD4 PROTAC与最终候选分子的降解活性预测得分排序。d,生成分子与候选分子的PK得分分布。e,对已知活性与非活性BRD4 PROTAC进行ProsettaC(Rosetta得分)与分子动力学模拟(GBSA能量)的回顾性分析,显示三次独立运行的均值与标准差。f,候选分子、已合成分子与参考分子的Rosetta得分与GBSA得分散点图。g,所有生成分子(灰色)、已知活性BRD4 PROTAC(橙色)、代表性聚类(蓝、绿、黄、浅黄、紫)以及已合成候选(红色)的t-SNE分布。mCF指药物化学过滤;pDC50指Log半最大降解浓度;smi指相似性。
2.3 强化学习实现了条件性的PROTAC生成
另一个阻碍PROTAC开发的关键问题是其普遍较差的药代动力学性质。为生成满足期望PK得分(方法中定义,数值越低越好)的分子,研究采用强化学习策略,并加入记忆经验机制与重复惩罚得分以缓解RL过程中常见的模式崩塌问题。研究使用经验性PK目标函数,并将平均PK得分从25.6降低至14.0,这是对PROTAC而言较为合理的范围。由于RL过程并不依赖特定的PROTAC结构,研究从测试集中随机选择十对战头与E3配体,通过2,000步训练,每对生成100个候选。若生成分子的PK得分落在目标值±2.0范围内,则视为成功。
结果显示,PROTAC-RL生成的分子平均PK得分为14.5,平均惩罚成功率达75.5%。如此高的成功率源自四个关键模块(预训练、增强、记忆经验与惩罚机制)协同作用,而移除任意模块都会显著降低性能。例如,去除预训练会将成功率降至15.0%,原因在于仅依赖小规模训练集的模型难以获取足够结构多样性,凸显预训练在低资源场景中的必要性。若去除惩罚或记忆模块,惩罚成功率也将分别下降至37.2%与39.5%,显示这些模块对于提升全局搜索能力、避免陷入局部最优至关重要。
以BI-7273(靶向BRD7)和CRBN配体沙利度胺为例,研究尝试生成PK得分分别约为22、20、18、16和14的分子,并未将PK得分极低化,因为过低的PK往往对应连接体过短,不利于稳定三元复合物的形成。多个阈值的设置有助于保持生成结果的结构多样性。结果如图2e所示,生成分子分别有97.4%、94.2%、88.8%、95.2%和95.4%分布在目标PK范围内±2.0区域;相比之下,先验模型生成的PK<18分子仅占不到0.5%。对于其余九组配对亦呈现类似趋势。
图2f展示了RL过程中结构随奖励增强的演化趋势,生成分子逐渐具有更复杂的骨架、更丰富的环系与取代基。此外,研究进一步以辛醇–水分配系数logP与连接体长度作为奖励函数进行测试,结果再次证明PROTAC-RL能够引导先验模型生成具备指定性质的分子,表现出良好的泛化能力。
2.4 针对BRD4的计算机虚拟筛选
为进一步展示方法的有效性,研究选取BRD4作为案例研究对象。BRD4是癌症治疗的重要靶点,但目前尚无进入临床阶段的BRD4 PROTAC分子。尽管已有研究利用JQ1作为战头、以CRBN为E3连接酶开发了BRD4降解剂dBET6,但其渗透性与药代性质不佳(PK得分22.24)。该研究旨在通过RL生成PK性质更优的BRD4 PROTAC,以PK目标值20、18、16、14和12启动RL过程,并将连接体长度限定在2–16个原子范围内,因为这是BRD4 PROTAC的最佳长度区间。
在RL训练中,研究在第1,000步至2,000步之间每隔250步保存一次模型,并使用四个模型各生成250条连接体,最终共获得5,000个候选PROTAC,其中有效SMILES为4,860条、唯一结构为2,894条。由于缺乏标准化的PROTAC筛选流程,研究设计了一套综合筛选策略以逐步缩小候选范围。首先,为确保分子新颖性,剔除与训练集中BRD4 PROTAC的ECFP指纹Tanimoto相似性高于0.6的分子(剩余2,645条),随后利用PAINS过滤掉具有不利结构的分子(剩余1,992条)。这些候选按Tanimoto相似性通过Butina算法聚成78个簇,其平均簇内相似性为0.184,表明结构间差异较大。
随后,研究使用基于四种异构机器学习模型集成训练的降解活性预测器,对所有分子进行DC50与降解百分比评分,其三次ROC-AUC分别为0.945、0.938与0.936。由于训练样本有限,预测器的泛化能力有限,因此仅用于剔除低评分分子,并通过分层策略在保持分子多样性的同时筛选出高评分结构。最终获得51个候选分子,其PK得分分布显著优于原始生成集合。
对于这51个结构,研究利用PRosettaC流程结合分子动力学模拟构建三元复合物模型。观察到已知BRD4 PROTAC在Rosetta评分与MM/GBSA能量上的一致性并不完全对应。对于51个候选分子,其中34个的Rosetta得分优于dBET6,另有3个的MM/GBSA得分更佳,两者合并共有36个表现优异的结构。根据合成可行性,研究选取其中6个进行实验验证。所有生成分子与筛选出的活性候选的分布图显示,这六个最终候选结构多样性良好,与已知活性PROTAC的相似性较低。
2.5 湿实验室验证
经过计算筛选后,共确定六个先导候选分子,并在第39天前成功完成合成。首先在HEK293T细胞系上通过蛋白印迹实验开展初筛,以评估BRD4的降解能力。结果显示,化合物1–3能够降低BRD4水平,其中化合物1在100 nM即可下调BRD4,而化合物2与3则在300 nM时诱导下降;化合物4–6未表现出明显降解活性。
随后选取化合物1–3在Molt4细胞系上测试其体外抗增殖作用。结果显示三者均具有抑制活性,IC50分别为116 nM、5.1 μM与21 μM。除了活性评价外,人类hERG通道抑制实验亦是药物心脏毒性的关键指标。对化合物1的检测显示其hERG抑制值为27.4 μM,属于较低水平,表明其具有进一步开发潜力。
化合物1还表现出良好的理化性质,水溶性LogS为1.42,分配系数LogD为3.27。鉴于PROTAC-RL旨在优化分子的药代特性,研究进一步在啮齿类动物中测试化合物1的体内表现。以2 mg·kg−1剂量腹腔注射给予小鼠,三次给药均获得约2.22 h的半衰期。初次给药后血药峰浓度Cmax为194 ng·mL−1,并在给药后0.5 h达到峰值,明显优于参考化合物dBET6(Cmax仅176 ng·mL−1,半衰期0.52 h)。这些结果表明化合物1兼具强效BRD4降解活性与优良药代性质,其整体特性总结见补充表6。
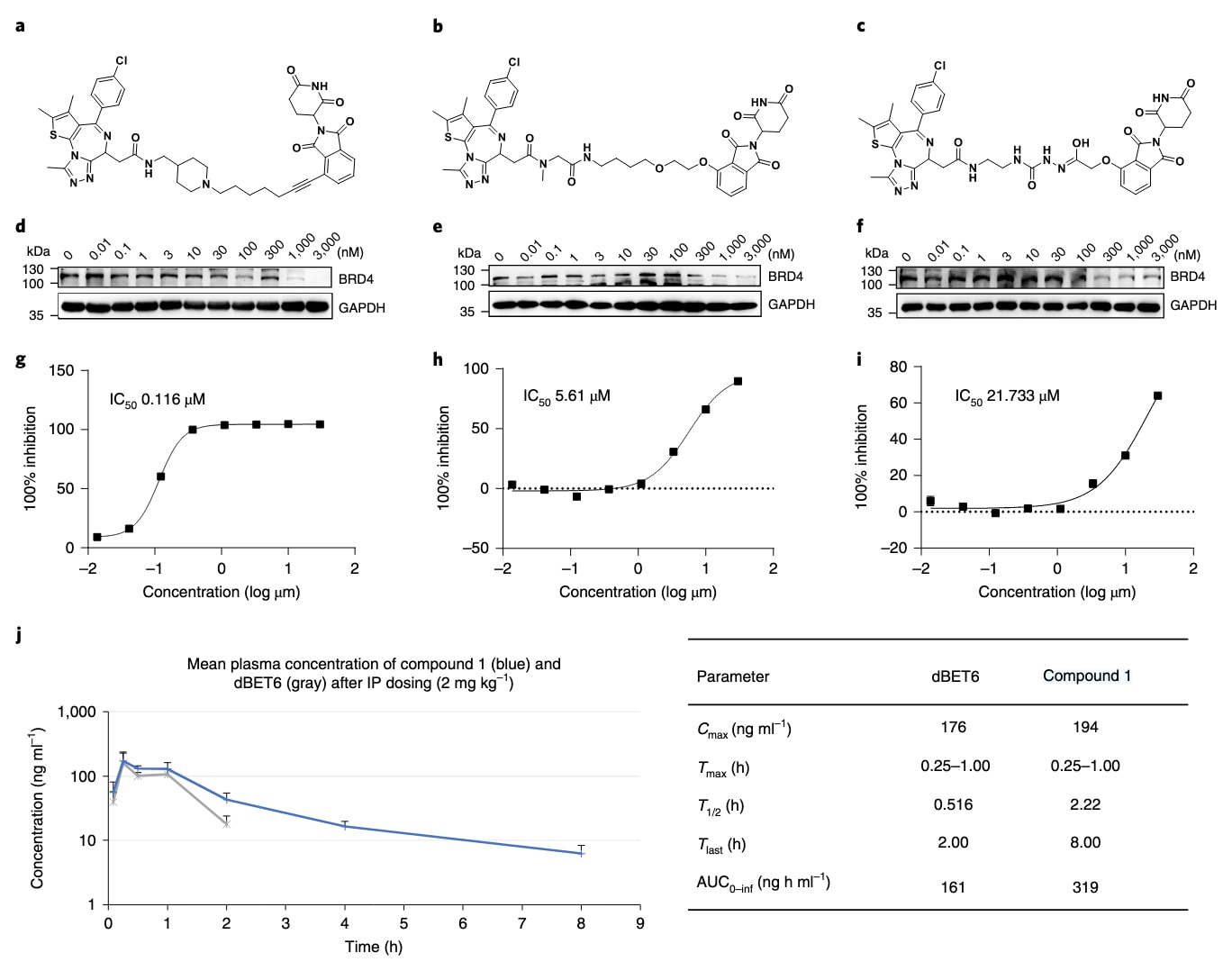
图4|生物活性与药代特性表征。 a–c,化合物1(a)、2(b)与3(c)的结构。d–f,HEK293T细胞经三小时给药后BRD4与GAPDH的蛋白印迹结果,分别对应化合物1(d)、2(e)与3(f)。g–i,Molt4细胞经72小时处理后使用CellTiter-Glo检测的细胞活力,分别对应化合物1(g)、2(h)与3(i),数据以平均值±标准误(n=3重复)呈现。j,CD1小鼠在腹腔注射2 mg·kg−1剂量后进行的药代研究,制剂为capitol/water体系,数据以平均值±标准误(n=3重复)呈现。Cmax指血药峰浓度;Tmax指达峰时间;T1/2指半衰期;Tlast指最后可测时间点;AUC指从零到无限的曲线下面积。
2.6 结构与机制分析
为了理解降解作用的机制基础,研究进一步比较了dBET6与化合物1所形成的三元复合物。结果如图5a–c所示,化合物1能够维持与dBET6相同的关键相互作用,包括BET战头中三唑结构分别与BRD4的CYS470与ASN474形成的两条重要氢键,以及CRBN配体中戊二酰亚胺部分与CRBN的HIS326与SER327主链之间的氢键。这表明化合物1能够保持与参考化合物类似的结合方式。
值得注意的是,化合物4虽然在预测中表现为更好的降解活性(基于GBSA与pRosettaC评分)且结合模式也与参考化合物相似,但在实验中未呈现明显降解效果。为探究潜在原因,研究对化合物1与化合物4进行了引导分子动力学模拟,以评估各自三元复合物的稳定性。有趣的是,由化合物1形成的复合物显著比化合物4更稳定(图5d)。这种稳定性的差异很可能解释了化合物4在湿实验中缺乏生物活性的原因。
3 讨论
该研究提出一套全自动计算框架,将条件生成模型、机器学习以及基于物理的模拟相结合,用于实现PROTAC的理性去 novo 设计,并对生成分子进行了实验验证,证明其具有良好的抗癌活性与药代特性。所发现的分子能够有效降解BRD4,而BRD4在多种人类癌症中过度表达。湿实验室的结果验证了该策略的高效性,仅合成并测试少量候选分子便取得约50%的成功率,并在49天内完成整个流程,凸显了结合人工智能驱动的计算策略与实验的巨大价值。
由于PROTAC-RL的设计具有通用性,该方法适用于多类条件生成任务,并能同时处理多个优化目标。因此未来可进一步加入更多限制条件,例如细胞通透性或口服生物利用度,以提升设计分子的药物可开发性。当然,当前框架下仍存在若干潜在局限。首先,作为强化学习方法,奖励函数的设计对最终输出极为关键。然而由于缺乏足够的实验数据,能够使用的稳健预测器仍然有限。该研究采用了部分经验性评分函数作为替代,也可考虑以对接打分函数引导模型生成潜在候选。其次,当前的三元复合物建模仍依赖基于物理的模拟。结果显示,对接得分最好的结构并不一定具有活性,而引导分子动力学模拟能够提供额外帮助。随着超级计算能力的发展,这类模拟的成本有望显著降低,同时也可以尝试新的深度学习结合界面预测的方法。
第三,连接点对降解能力与选择性有根本影响。目前PROTAC-RL沿用既有策略,根据已有活性数据预设连接点。若有晶体结构,可依据高分辨率的共晶结构中暴露于溶剂的区域确定连接位置;若缺乏结构数据,则可结合构效关系、可解释人工智能方法以及分子动力学模拟来推断合适的连接点。最后,蛋白结构预测与蛋白–蛋白相互作用预测的进步有望进一步提升三元复合物建模的准确性。
总体而言,该研究的结果表明,现代深度学习技术正迎来用于PROTAC发现的最佳时机。这类努力不仅能够提升新分子实体的发现速度,还可降低药物开发中对资源与成本的需求。相信该研究提出的策略将为未来的PROTAC设计工作提供重要启示。