NC 2025 | ChemEnzyRetroPlanner:用于自动化有机–酶催化混合合成规划的虚拟平台
今天介绍的这项工作来自 Nature Communications。ChemEnzyRetroPlanner是一套面向复杂分子合成的开源有机-酶促混合逆合成规划平台,核心目标是在同一套自动化规划流程中同时利用有机反应的策略广度与酶催化的高立体选择性、温和条件和潜在绿色优势。平台以RetroRollout*为关键搜索框架,在And-OrTree与路径深度引导等机制下提升多步路线搜索的效率与求解率,并支持将有机单步逆合成模型与酶促单步模型协同调用,从而更容易生成真正的混合路线。除多步规划外,系统还集成反应合理性过滤、反应条件预测、酶促反应识别、酶类别推荐以及基于EasIFA的活性位点注释与验证,形成从“能不能做”到“用什么酶做、位点是否匹配”的闭环。平台提供Web服务界面与可编程API,并构建了可离线部署的LLM驱动智能体,能够自动完成安全检查、工具选择与策略切换等决策;在USPT、PaRoutes和天然产物等基准上表现优于多种对照方法,在多种药物与生物活性分子的案例中可复现文献路线,同时给出具备可解释验证信息的酶促步骤建议,为药物研发中更合理、成本更优且更可持续的合成路线设计提供了一体化工具链。

获取详情及资源:
- 📄 论文: https://doi.org/10.1038/s41467-025-65898-3
- 💻 代码: https://github.com/wangxr0526/ChemEnzyRetroPlanner
0 摘要
有机合成与酶催化的结合,为复杂分子的高效、可持续构建提供了重要途径。有机合成擅长覆盖广泛的反应类型,而酶催化能够在温和条件下提供更高的立体选择性,从而在成本效益与环境影响方面带来优势。尽管如此,现有酶催化合成规划算法在形成稳健的有机-酶催化混合策略时仍存在明显瓶颈,主要体现在混合规划方法不易构建,以及对基于模板的酶推荐过度依赖,进而限制了在多样场景下的适应性。ChemEnzyRetroPlanner被提出为一个开源的合合成规划平台,通过AI驱动的决策机制协同有机与酶催化策略。平台集成了多项计算模块,包括混合逆合成规划、反应条件预测、可行性评估、酶促反应识别、酶推,以及对酶活性位点的计算机模拟验证。其核心创新之一是RetroRollout*搜索算法,在多个数据集上针对有机化合物与天然产物的合成路线规划表现优于现有工具。该平台同时提供直观的图形界面与可扩展的编程API,并结合chain-of-thought策略与Llama3.1模型,以在不同任务情境下自动激活混合合成策略。整体结果表明,这一全自动、开源系统有望提升分子合成规划的效率与可持续性。
1 引言
有机合成与酶促合成在能力上互补,将二者结合可为复杂化合物的合成规划提供更强的策略空间。传统有机合成覆盖反应类型广、适用性强;酶作为高效生物催化剂,在关键步骤的立体化学控制方面表现突出,有助于减少保护-脱保护操作。同时,酶促反应通常在温和条件下进行,反应介质多为水或较温和的有机溶剂,与常依赖贵金属的有机催化反应相比,往往更具环境可持续性与成本优势。近年来,定向进化的进展显著拓展了酶催化的化学空间,使其能够作用于非天然底物并适配更复杂的合成需求,从而为构建有机-生物催化混合策略创造了新机会,在传统有机合成与生物催化之间搭建起面向现代复杂分子设计与制造的桥梁。
多数计算机辅助合成规划算法沿袭化学家的逆合成分析思路,从目标分子出发递归寻找更可能的反应前体,直至得到合适的简单构建模块。数据驱动的有机合成规划系统通常由若干关键环节构成,包括用于生成搜索动作的单步预测模型、反应合理性评估器、多步搜索策略、可定制的分子构建模块库、反应条件推荐模块,以及路径排序或评分函数。传统单步逆合成模型多在USPTO或Reaxys等数据集上训练,其中酶促反应样本相对有限,导致这类模型往往难以准确预测酶促反应,也使得如何将酶促规划有效融入既有框架成为突出问题。
为此,近年出现了全酶促合成规划算法,其单步预测模型往往依赖KEGG、MetaNetX、Rhea等专门的酶促反应数据库进行训练,从而具备预测酶促反应的能力;但这类模型的覆盖化学空间相对较窄,在合成策略广度上通常不及基于传统有机化学大规模数据训练的模型。围绕混合路线规划,已有研究提出多种有机与酶促反应的整合思路:例如,利用混合整数线性规划在由文献报道的有机与酶促反应构成的混合网络中寻找从起始原料到目标产物的最优路线,适于对已知部分路线做可控优化,但难以产生全新的合成策略;也有工作采用协同预测框架,并行使用两个基于模板的单步逆合成模型,分别对应有机与酶促反应,共同设计混合路径;另有方法先由纯有机规划生成完整路线,再用评分模型定位效率最低的步骤并以酶促规划工具替换,以此引入酶催化改造。还有研究基于Transformer训练两套TripleTransformerLoop架构,分别处理有机反应的单步逆合成、条件推荐与正向验证,以及酶促反应的单步逆合成、酶建议与正向验证,并在路线规划中结合路线惩罚分数与启发式最佳优先树搜索,实现混合路线生成。不同于在搜索过程中直接混用模型的策略,也有方案将基于模板的酶促反应识别模块嵌入既有多步逆合成框架,在完成合成规划后再识别可由酶催化的步骤。
在平台层面,面向有机-酶促混合合成规划的工具也在快速发展。例如,RetroBioCat同时提供交互式与全自动规划策略,以基于模板的预测算法设计合成路径,并依赖专家定义的酶促反应模板在路径中识别潜在酶促步骤;与之不同,BIONAVI采用基于Retro*的搜索算法,并融合基于Transformer的有机-酶促混合单步逆合成预测模型以提升路径规划成功率。尽管该领域已有进展,当前酶促反应识别与酶推荐仍主要依赖模板匹配与基于相似度的反应排序,严格的模式匹配可能显著限制对其他潜在混合策略的探索;同时,关于反应-酶匹配的进一步验证机制仍相对不足,例如对酶活性与关键活性位点的识别与校验覆盖不够,容易引入误差,也凸显对更精确可靠验证策略的需求。另一个共性问题是自动化程度不足,在任务定义、工具选择等环节仍需要专家投入大量精力;相较成熟的商业合成规划平台,混合有机-酶促规划平台的自动化水平尤为有限。
随着ChatGPT、Llama3等通用大语言模型的发展,LLM驱动的专用Agent逐渐成为趋势。既有研究表明,通过提示工程,LLM可自主完成多类研究相关任务,降低领域门槛并提升流程效率。在合成化学方向,此类工具增强型LLM也开始出现:ChemCrow展示了LLM结合chain-of-thought推理策略后,能够编排多种合成工具以应对复杂化学问题;随后,Coscientist与LLMRDF进一步通过集成网页浏览、文献分析、代码解释器与自动化合成平台,推动端到端的自主实验设计、执行与优化;在材料与高分子方向,也有工作将LLM与知识图谱结合,提出面向聚合物合成的自动化路线规划与优化框架。尽管此类方法的能力会受其可调用工具的边界影响,仍体现出LLM能够自主配置工具组合以最大化工具效用的潜力。
在有机-酶促混合规划场景中,酶促反应数据稀缺会限制酶促单步模型对大化学空间搜索的支撑能力,而传统有机单步逆合成模型又难以有效预测酶促步骤,因此需要探索多种混合规划策略,包括单步模型的组合方式,以及反应条件与酶推荐策略的协同。相关任务往往涉及复杂的配置与决策流程,将其交由LLM承担有望提升规划效率与预测准确性,也使得发展具备更强自动决策能力的混合有机-酶促合成规划Agent成为值得关注的方向。
在这一背景下,提出了混合有机-酶促合成规划平台ChemEnzyRetroPlanner,整体框架如图1所示。平台提供7个关键计算模块,包括多步合成规划器、单步逆合成预测器、反应条件预测器、反应速率预测器、酶促反应识别器、酶推荐器,以及酶活性位点标注器EasIFA。各模块在合成规划过程中的协同逻辑在补充信息图S10中给出,并且每个模块都可通过编程API独立调用,以适配定制化场景下的自动合成规划需求。在此基础上,平台结合chain-of-thought方法使用Llama3.1:70b联动多工具,构建出可执行一组有机-酶促混合合成规划任务的Agent,并基于Web服务开发了更易用的图形化界面。该研究还提出RetroRollout搜索框架,以And-OrTree与路径深度评分函数为引导,在Retro基础上引入在线模拟步骤与兄弟节点跳转搜索策略,以提升规划速度并提高目标分子路线求解成功率;同时整合来自不同数据源的多种单步策略,使系统能够面向不同规划情境选择合适的混合有机-酶促策略;围绕生物催化合成构建工具链,覆盖路径中的酶促反应识别、酶推荐与酶活性位点的计算机模拟验证;并通过所开发的编程API构建了完全开源的混合有机-酶促合成规划Agent,在继承ChemCrow的化学信息学工具、安全工具与通用工具的同时,为反应类工具提供纯开源替代方案,并支持酶促反应工具的调用。
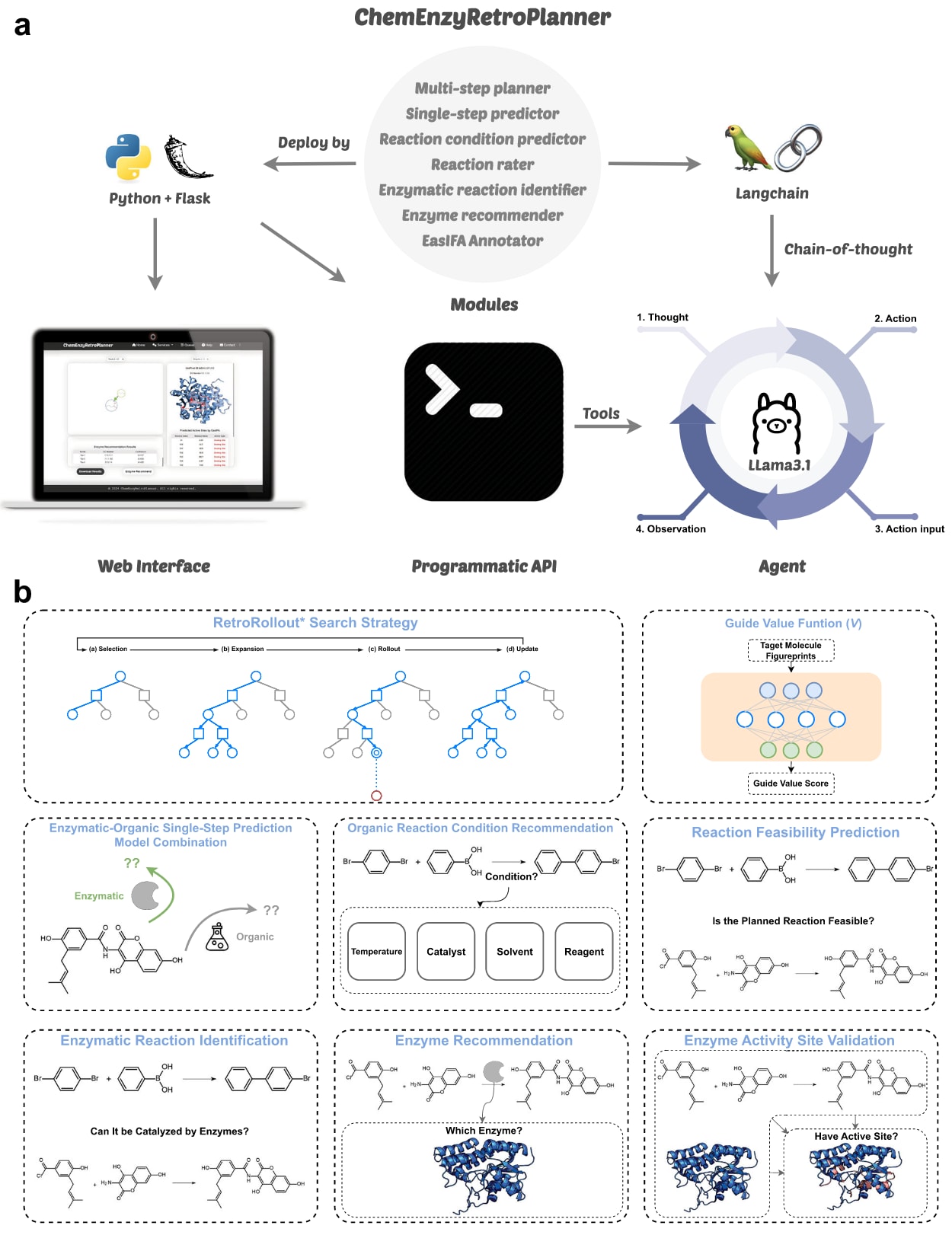
图1 | 展示了有机-酶催化混合合成规划平台ChemEnzyRetroPlanner的总体概览。 a展示了ChemEnzyRetroPlanner的技术框架以及平台支持的三种交互模式,包括图形界面、编程API与智能体。b概述了平台所包含的主要算法模块。API指应用程序编程接口。
2 结果
2.1 多步合成规划基准评测
平台的多步规划能力通过覆盖三类典型多步合成场景的测试集进行评估,其中两类来源于有机合成专利数据,另一类基于天然产物。所用数据包括USPTO-multistep-190测试集,该数据最初在Retro*算法评测中被整理并在后续研究中广泛采用;还包括Genheden等人构建的PaRoutes标准评测数据中的PaRoutes-Set-n1与Set-n5的随机子集,这两套数据同时提供多步合成路径数据、评测阶段使用的单步反应训练数据以及构建模块分子数据集;此外还使用了Zeng等人评测BioNavi平台时采用的天然产物测试集及其配套的可购构建模块数据集。
在USPTO-multistep-190测试集上的评测,比较了搜索框架、跳跃搜索策略、引导函数类型、可购构建模块数据集质量以及单步逆合成模型性能,对目标分子合成路径搜索表现的影响。基线算法选用原始Retro*,测试分子为从USPTO中提取的190个具有代表性的复杂化合物。评测结果见表1,其中不使用引导函数的搜索框架记为“SearchFrameName-0”。当搜索迭代限制为100轮,单步逆合成策略网络采用GraphFP,并使用修改后的Zinc可购构建模块分子数据集时,RetroRollout在是否使用引导函数的两种设置下,均能够为更多分子求解出合成方案,优于Retro。GraphFP是基于模板的单步逆合成预测模型,在USPTO-all-remapped数据集上训练,模型结构、训练流程以及可购构建模块数据集的细节在补充信息中给出。与Retro*-0相比,RetroRollout*-0额外解决了11个分子,路径求解率提升5.79%;与Retro相比,RetroRollout多解决12个分子,求解率提升6.32%。
表1 | 展示了ChemEnzyRetroPlanner中,RetroRollout在USPTO-multistep-190测试集上的表现,并与Retro进行对比。
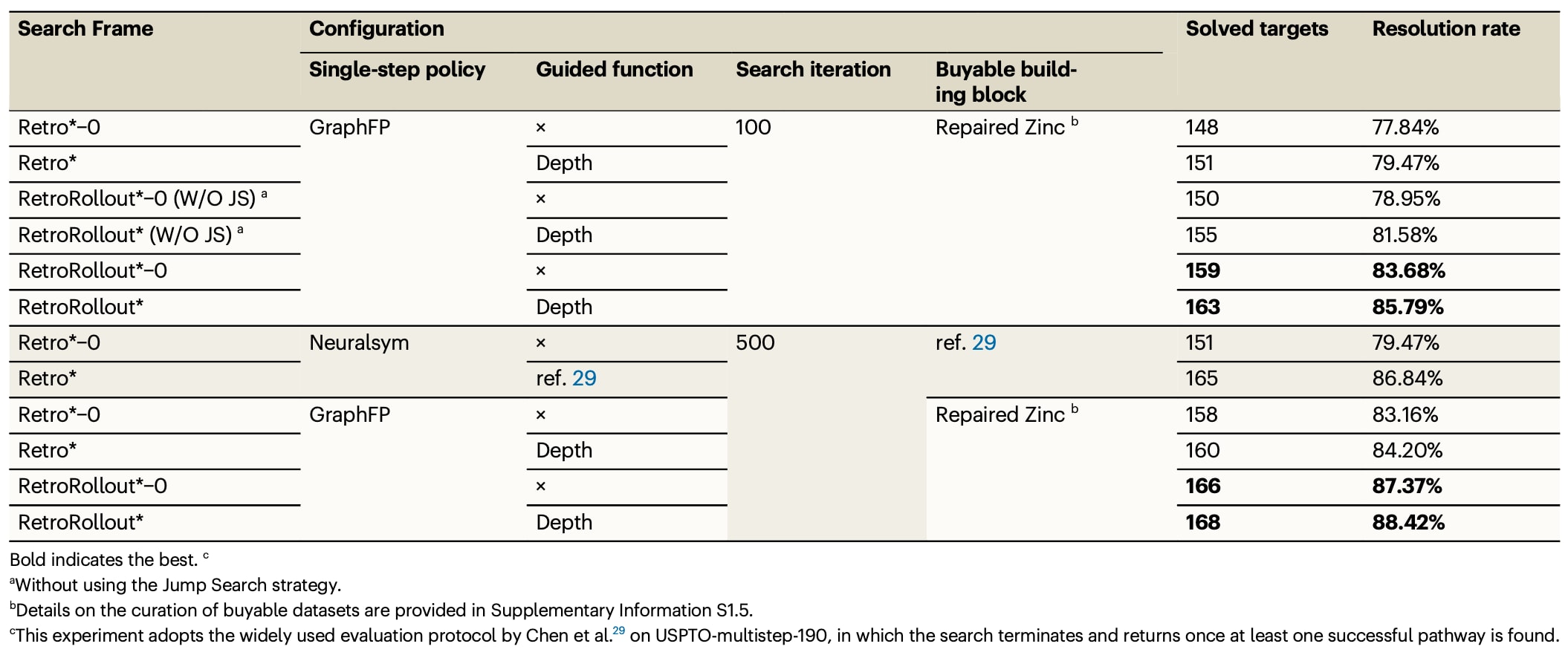
引导函数的作用也被单独对比:在深度相关的引导函数加入后,可求解目标分子数量进一步增加,Retro相较Retro-0多解决3个分子,RetroRollout相较RetroRollout-0多解决4个分子。当最大搜索轮数提升到500轮时,RetroRollout与RetroRollout-0相对各自在100轮设置下分别又多解决7个与5个分子,其中500轮搜索的RetroRollout能够解析88.42%的目标分子合成路径。进一步对比了Chen等人提出的原始Retro配置,其单步策略网络使用Neuralsym,并沿用原始引导函数模型与构建模块数据集;在相同迭代轮数下,RetroRollout仍能解决更多分子,且对引导函数的依赖更弱。值得注意的是,即便不使用引导函数,RetroRollout-0相对使用反应成本引导函数的Retro*仍多解决0.53%的分子。
跳跃搜索策略的影响通过消融实验评估,去除跳跃搜索的两个变体分别记为RetroRollout*-0(W/OJS)与RetroRollout*(W/OJS),对应无引导函数与采用深度引导函数的两种框架设置。结果显示,与包含跳跃搜索的RetroRollout*-0与RetroRollout相比,去除跳跃搜索后分别少解决4.7%与4.2%的目标分子,说明跳跃搜索能够同时提升搜索效率与成功率。同时,在相同单步预测模型与引导函数设置下,即便不使用跳跃搜索策略,所提出的搜索框架仍比原始Retro方法多解决1.1%与2.1%的目标分子,表明框架本身具有优势。
此外,在相同数据集上采用连续搜索策略进一步比较Retro与RetroRollout的路径表现。以100轮搜索为预算,RetroRollout不仅解决更多目标分子,还为每个目标分子平均额外提供57.8条成功路径,其中58%的路径长度短于参考的真实路径,而Retro为51%。在仅100轮迭代的预算下,RetroRollout的目标分子求解率甚至高于Retro在350轮迭代下的表现,优势为3.16%,同时平均搜索时间减少7.54s。尽管在该设置下RetroRollout每个目标分子产生的成功路线数量(170.2)少于Retro(409.5),但仍能有效给出结构复杂且更难求解目标的有效合成方案,体现出整体规划能力的稳健性。这也暗示RetroRollout的搜索行为更倾向于进行更深层的探索,从而更早发现复杂路径,并保持较好的计算效率。综合来看,相较Retro,RetroRollout*在路线质量、搜索效率与应对挑战性目标的能力之间实现了更优的平衡。
PaRoutes随机抽取测试集上的评测,采用标准化的多步逆合成评测流程,系统评估多步算法框架的综合表现。分别使用PaRoutesSet-n1与Set-n5从USPTO反应数据中抽取的路径子集训练GraphFP单步逆合成模型,模型训练细节在补充信息中给出。两套数据中路径的叶子节点分子被用作可购构建模块数据集,并分别针对Set-n1与Set-n5训练深度引导函数。Set-n1的分布更接近USPTO反应数据的总体分布,而Set-n5包含更多长路径样本。评测指标不仅包括可求解目标分子数量,还包括路径搜索平均耗时、完整合成路线命中的top-k准确率,以及两项反映路线质量的指标:规划路径更短的比例与叶子节点分子平均重叠度。结果见表2,路线评分与排序采用Badowski等人报道的方法。
表2 | 展示了ChemEnzyRetroPlanner中,RetroRollout在PaRoutes测试集上的表现,并与Retro进行对比。
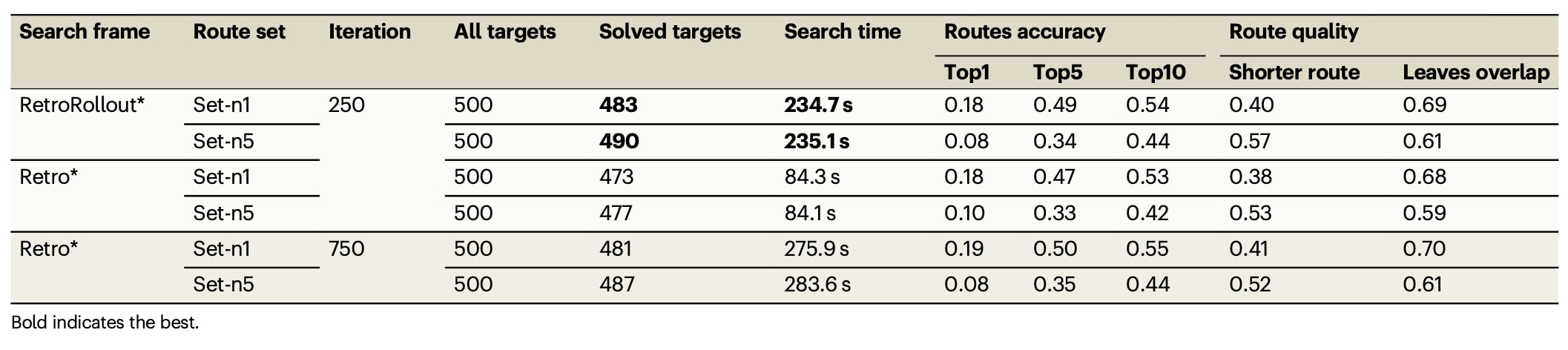
当搜索迭代限制为250轮时,在Set-n1中RetroRollout解决了500个目标分子中的483个,而Retro解决473个。两种框架在top-1准确率上接近,但RetroRollout在top-5与top-10准确率更高,且规划路径平均更短,叶子节点与标准路径的重叠度更高。在Set-n5中,RetroRollout解决490个分子的路径,Retro解决477个;在top-1准确率上Retro略高于RetroRollout*,但RetroRollout在top-10准确率上领先。尽管两者在Set-n5的top-k表现各有优势,RetroRollout规划路线的质量整体更高,表现为更短的路线与更高的叶子节点重叠度。为进一步客观比较,将Retro的搜索迭代提高到750轮,此时Retro对每个分子的平均搜索时间显著高于RetroRollout在250轮下的耗时,每个目标分子平均多耗约40到50s,但可求解分子数量仍低于RetroRollout。尽管在该设置下Retro的top-k准确率与路线质量略高于RetroRollout,差异并不显著。总体而言,在两种PaRoutes标准评测设置下,RetroRollout能够以更短时间解决更多分子合成路径问题,同时保持与Retro相当或略高的路径规划质量。
天然产物数据集上的评测,使用Zeng等人用于评估BioNavi平台的天然产物数据集来检验ChemEnzyRetroPlanner在天然产物场景下的路径求解能力,并与已报道平台进行对比。实验设置与相关工作保持一致,并使用相同的可购构建模块数据集。平台在路径搜索中评估了多种单步策略模型,这些模型在文中以其训练数据集命名,包括来自ASKCOS的4种基于模板的单步逆合成预测算法:Pistachio ringbreaker、BKMS metabolic、Reaxys以及Reaxys biocatalysis,其中BKMS metabolic与Reaxys biocatalysis由酶促反应数据训练得到。除上述模型外,还测试了在USPTO-all-remapped训练的基准模型GraphFP(与USPTO-multistep-190评测中一致),以及Zheng等人报道的基于Transformer、在酶促与有机反应混合数据集上训练的单步逆合成模型。该部分结果见表3。
表3 | 展示了ChemEnzyRetroPlanner中,RetroRollout*在天然产物数据集上的表现,并与其他方法进行对比。
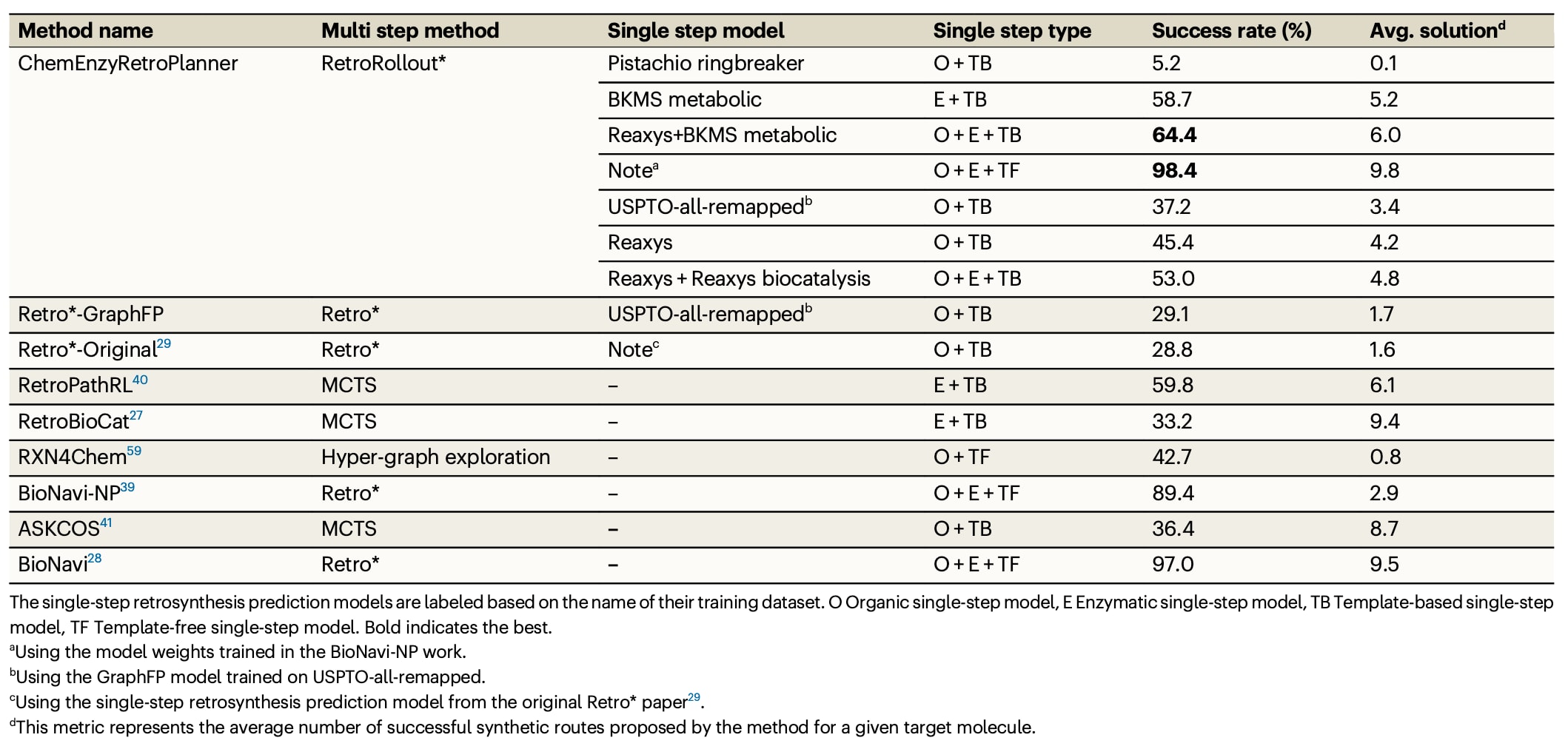
结果表明,当ChemEnzyRetroPlanner与RetroRollout结合,并搭配主流开源单步逆合成预测模型时,目标分子的路径求解成功率得到显著提升。采用两种不同的基于模板单步模型组合策略,分别由Reaxys与BKMS metabolic数据集训练,其表现优于RetroPathRL、RetroBioCat、ASKCOS以及原始Retro等基准方法,路径求解成功率达到64.4%,明显高于模板基方法中表现最佳的RetroPathRL(59.8%)。此外,当RetroRollout*与一种与BioNavi-NP相同的无模板方法结合时,同样实现更高的路径求解成功率,目标分子的合成路径求解率达到98.4%,并略高于在更广数据集上训练的BioNavi方法。
2.2 酶促反应识别模型与酶推荐器的基准评测
该部分系统评估了ChemEnzyRetroPlanner平台内置的酶促反应识别模型与酶推荐器的表现,并与当前具有代表性的基线方法进行对比。针对酶促反应识别,选取了应用较广的基线方案:基于反应模板匹配的方法。该方法使用RetroBioCat发布的135个酶促反应模板,通过判断给定反应步骤是否与任一预定义模板匹配来确定其是否为酶促反应。评测使用由混合数据集构成的测试集,其中USPTO-1000-TPL作为负样本,ECReact作为正样本,数据集划分细节在补充信息S1.3中给出,评测结果汇总于表4。结果显示,平台内置的基于RXNFP的酶促反应识别模型在“酶促/非酶促”二分类任务上表现稳定且准确,除召回率为0.9895外,其余指标均超过0.9900,同时推理速度约为模板匹配基线的5倍。相比之下,模板匹配方法的分类性能明显偏低,主要原因在于其高度依赖人工整理反应规则的区分能力;如表4所示,该方法的预测能力难以满足平台对高精度的需求。通过将酶促反应识别表述为二分类问题,并利用带注意力机制的可学习反应指纹,该模型在鲁棒性与可扩展性方面表现突出,为合成规划中的大规模酶促反应筛选提供了更可靠的基础。
表4 | 展示了ChemEnzyRetroPlanner内置的酶促反应识别模型的基准测试结果,并与基于反应模板匹配的识别方法进行对比。

针对酶推荐器,核心任务是将酶促反应分类到对应的EC编号。对比对象选择了两类主流方法:CLAIRE与Selenzyme2023。CLAIRE基于对比学习,以RXNFP与DRFP指纹的组合对酶促反应进行编码,并使用tripletmarginloss使同一EC编号样本的嵌入距离更近、不同EC编号样本的距离更远;其原始实现支持到EC编号第三级(EC-L3)的预测。由于酶预测任务需要更细粒度的EC编号推荐,该方法被调整为支持完整四级EC编号(EC-L4)预测,同时保留在EC-L3层级进行性能评估的能力;为保证对比公平,CLAIRE在与平台酶推荐器相同的数据划分上重新训练。Selenzyme2023则是基于相似度的酶推荐工具,通过在内部数据库中检索最相似的酶促反应并返回对应酶作为推荐;为评估其真实推理表现,该工具在本地部署并在EC-L3与EC-L4两个层级上同时评估预测准确性与计算效率。三种方法的对比结果汇总于表5。
表5 | 展示了ChemEnzyRetroPlanner内置的酶推荐模型的基准测试结果,并与其他主流酶推荐方法进行对比。
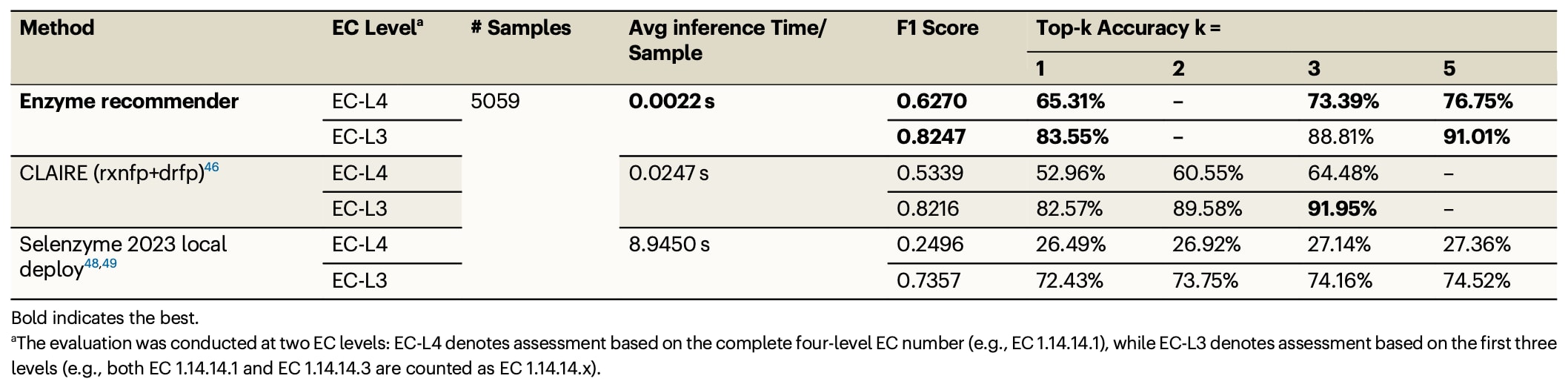
在EC-L3预测任务上,ChemEnzyRetroPlanner内置酶推荐器与CLAIRE整体相当,Top-1准确率分别为83.55%与82.57%,Top-3准确率分别为88.81%与91.95%。在更具挑战的EC-L4预测上,平台内置酶推荐器的Top-1准确率达到65.31%,显著高于CLAIRE的52.96%,差距主要来自于在更细粒度EC编号下正样本不足时,CLAIRE的对比学习更难获得稳定有效的表示。Selenzyme2023在EC-L3层级的Top-1准确率为72.43%,但在EC-L4层级性能大幅下降,Top-1准确率仅为26.49%。在推理速度方面,平台内置酶推荐器约为CLAIRE的10倍,约为Selenzyme2023的4000倍,体现出很高的计算效率,更适合嵌入需要对大量反应节点进行快速、频繁预测的合成规划工作流中。
2.3 反应合理性评估器的基准评测
该部分首先基于USPTO反应数据构建了用于评估反应合理性评估器基线能力的基准数据集,称为template-shuffling数据集。负样本通过模板打乱(template-shuffling)策略生成,数据生成流程示意如图S5所示。最终数据集包含1,842,509条反应样本,其中正样本完全来自USPTO数据,占比70%,负样本占比30%。数据集按8:1:1随机划分为训练集、验证集与测试集,用于筛选最优的反应表示方式与模型结构。评测比较了三类反应表示: Morgan指纹、RXNFP与DRFP。结果显示,采用Morgan指纹并结合双分支多层感知机结构的模型(结构见图S6,记为RXNFilter-benchmark)在整体表现上最佳。在该基准数据集的测试集上,RXNFilter-benchmark取得AUC为0.9583,precision为0.9155,recall为0.9160,f1score为0.9145,整体略优于基于RXNFP与DRFP的模型。
在确定最佳模型结构与反应表示后,进一步在更大规模的反应合理性预测数据集上进行训练,该数据集称为FaissTemplate sampling数据集,由Faiss相似度采样与反应模板选择模型联合构建。该数据集包含2,745,104条样本,按9:0.5:0.5随机划分为训练集、验证集与测试集,并保持正负样本1:1平衡。在该数据集的内部测试集上,部署版本模型RXNFilter-deployed取得AUC为0.9093,precision为0.9331,recall为0.8330,f1score为0.8329,体现出较强且相对均衡的分类能力。
为进一步比较模型区分不同类型反应样本的能力,从FaissTemplate sampling数据集的测试集中抽取44,280条样本构建了一个与基准数据集不重叠的公共测试集。该测试集包含三类样本:真实反应、由模板选择神经网络以高置信度生成的“可信正样本反应”,以及在模板模式上成立但在化学上不合理的“不可可信负样本反应”(对应模板选择神经网络的低置信度)。三类样本比例为4:1:5。该公共测试集上对比了RXNFilter-benchmark、RXNFilter-deployed以及ASKCOSv2系统内置的fastfilter,结果汇总于表S13。RXNFilter-deployed在该测试集上表现最为均衡,AUC为0.9000,accuracy为0.8209,precision为0.8211,recall为0.8212,f1score为0.8210。相比之下,仅在template-shuffling数据上训练的RXNFilter-benchmark出现明显更高的假阳性率:尽管recall达到0.9441,accuracy却降至0.6420,precision降至0.5885。ASKCOSv2fastfilter也呈现类似特征,accuracy为0.6487,precision为0.6008,recall为0.8865,表明其对不可行反应的识别能力有限。
总体而言,RXNFilter-deployed能够提供更准确且更稳健的分类效果,因此更适合作为实际合成规划工作流中的过滤模块,用于识别并剔除化学上不合理的反应。
2.4 ChemEnzyRetroPlanner的Web服务器界面
在输入与参数设置方面,平台支持直接粘贴SMILES,也支持点击“Draw”按钮在JSME容器中手绘分子结构。示例中以分子CCCC(=O)O作为输入,如图2a所示。用户可点击“Options”按钮为混合合成规划选择运行参数,包括“Keep search after solved one route”“Use reaction plausibility evaluator”“Use guiding function”“Predict reaction conditions”“Identify enzymatic reactions”“Recommend enzymes”。平台提供4套预定义的可购构建模块数据集供选择,并允许同时勾选多个数据集。与此同时,平台还提供7个可选的单步逆合成预测模型,其中3个基于常规有机反应数据集训练,分别为GraphFP(USPTO-all-remapped)、PistachioRingbreaker与Reaxys;另有3个基于酶促反应数据集训练,分别为Transformer(USPTO-NPL+BioChem)、BKMSMetabolic与ReaxysBiocatalysis。这些模型以训练数据集命名,并采用MLP与基于反应模板的单步算法实现,模型细节见补充信息S1.1。在单步逆合成模型选择框中,允许同时选择多个模型进行协同搜索,将酶促单步逆合成模型(例如BKMSMetabolic)与有机反应单步逆合成模型(例如Reaxys)结合,有助于规划有机-酶促混合合成路径。此外,搜索算法的迭代次数也可按需设定。完成参数配置后,点击“Submit”即可启动对输入目标分子的合成路线搜索。算法运行过程与输出信息会显示在“Log”框中,并返回一个用于查看结果的key。搜索完成后,“ViewResults”按钮会被激活,点击后将跳转到结果界面,通过上传对应key来查看计算结果。
在结果展示方面,当提供相应的key文件后,界面会跳转至结果页,如图2b上半部分所示。结果页主要包含三部分:用于在多条合成路线间切换的下拉菜单、可交互的合成路线展示图,以及单步反应信息展示框。对每条展示的合成路线,可点击反应节点查看该步的详细信息,其中酶促反应以绿色标注,常规有机反应以黄色标注。对应信息会在单步反应展示框中依次呈现,包括单步反应方程、平台推荐的反应条件、酶促反应识别结果以及酶推荐结果。当某一步单步反应被识别为酶促反应后,“EnzymeRecommend”按钮会被激活;点击后平台将调用UniProt数据库API,在推荐的EC编号下检索相关酶,并使用EasIFA算法预测活性位点。此时会弹出酶结构展示栏,支持对酶结构进行交互式查看,并在结构下方的表格中展示更详细的活性位点信息。
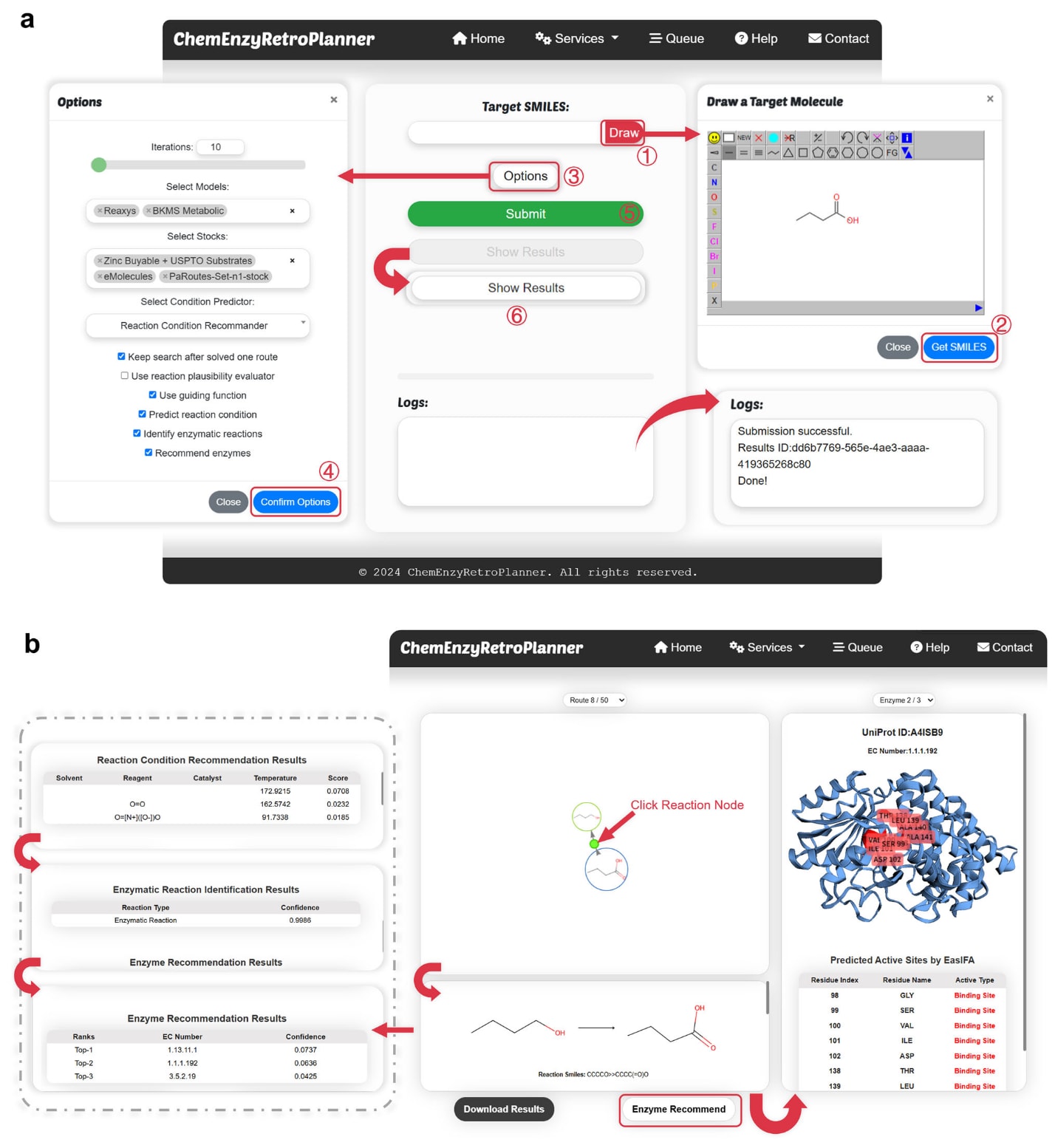
图2 | 展示了有机-酶催化混合合成规划平台ChemEnzyRetroPlanner的平台界面。 a为参数选择界面与日志输出界面;b为平台的合成路径展示界面与酶活性位点展示界面。
2.5 合成规划案例研究
为进一步评估ChemEnzyRetroPlanner的实用性,该平台被用于6种具有药物相关性或生物活性的化合物,开展逆合成路线规划。算法采用基于模板且可解释的单步逆合成预测器,整合Reaxys与BKMSmetabolic反应知识库。起始原料库选用RepairedZinc与BioNavi中使用的构建模块,并额外加入约束,将每个化合物的最大碳原子数限制为8。搜索迭代次数设为10次,所得所有候选路线均与文献报道的合成路线相匹配。其中,Esmolol、Desmethylxanthohumol以及3-(Benzenesulfonyl)-6-methyl-4-(4-methylpiperidin-1-yl)quinoline的预测路线体现了有机-酶促混合合成策略(补充信息图S12),而Celiprolol、S-(-)-warfarin与AZD7545对应的则是完全有机合成路径(补充信息图S13)。
在Esmolol案例中,合成从可商业购买的起始原料开始,包括p-iodophenol、methylacrylate、allylbromide与isopropylamine(在图S12中以虚线框标注)。策略首先通过酯化与亲核取代构建关键中间体,其包含芳香环骨架与烯丙基侧链;随后在单加氧酶催化下对末端双键进行区域选择性环氧化,生成环氧中间体,该酶对应UniProtID为A0A1D6GQ67,EC编号为1.14.14.159。最后,环氧化物与isopropylamine发生开环反应,得到Esmolol的β受体阻滞剂骨架。
在Desmethylxanthohumol案例中,使用了3种可商业购买的构建模块: trihydroxyarylketone、phenylpropeneether以及dimethylallyldiphosphate(DMAPP)(图中虚线框)。文献支持的aldol缩合首先组装出chromanone核心中间体,随后chromanone与DMAPP发生亲核取代反应生成带有异戊烯基化取代的天然产物衍生物Desmethylxanthohumol。该关键步骤被平台判定为可能由prenyltransferase催化,对应UniProtID为A0A0B5A051,EC编号为2.5.1.136,并与已知的酶底物特异性相一致。
第三个示例以3-(Benzenesulfonyl)-6-methyl-4-(4-methylpiperidin-1-yl)quinoline为目标。合成路线从多种可商业购买化合物出发,包括benzenesulfonitrile、methanol、triethylorthoformate、p-toluidine与p-methylpiperidine(虚线框)。其中,benzenesulfonitrile首先在nitrilase催化下水解生成相应酸,该酶对应UniProtID为Q42966,EC编号为3.5.5.1,随后与methanol酯化得到methylbenzenesulfonylacetate。并行路线中,p-toluidine与一种亲电性酮中间体发生缩合与环化形成quinolinone核心;随后使用phosphorylchloride(POCl3)进行脱水氯化,得到chloroquinolinone中间体,最后与p-methylpiperidine进行亲核取代引入关键哌啶侧链,完成合成。
在降压药Celiprolol的逆合成规划中,平台给出了一条可行路线,起始原料为可商业购买的1-(2-hydroxy-5-nitrophenyl)ethanone、tert-butylamine与diethylcarbamoylchloride(图S13虚线框)。合成首先按照文献报道对1-(2-hydroxy-5-nitrophenyl)ethanone的酚羟基进行环氧化,引入环氧基团;随后中间体与tert-butylamine发生亲核开环反应,生成含tert-butylamide结构的中间体。接着硝基被还原为相应的苯胺衍生物,并在酰胺化步骤中与N-ethyl-N-chloroacetamide反应,安装含脲结构的侧链,从而完成Celiprolol的合成。
第五个案例聚焦抗凝药S-(-)-warfarin。合成起始原料为可商业购买的o-hydroxyacetophenone、diethylcarbonate、benzaldehyde与acetone。路线一开始由o-hydroxyacetophenone与diethylcarbonate发生环化-缩合反应生成4-hydroxybenzopyran-2-one;并行地,benzaldehyde与acetone进行aldol缩合得到1-phenylbut-1-en-3-one。随后这两个中间体通过Michael加成偶联,有效生成最终产物S-(-)-warfarin。
第六个案例中,平台成功复现了文献报道的AZD7545完整合成路线。该路线以3种可商业购买构建模块为起点,包括2-chloroaniline、dimethylamine与(R)-3,3,3-trifluoro-2-hydroxy-2-methylpropionicacid,并通过4步反应序列得到AZD7545,与文献合成过程一致。
总体来看,这6个代表性示例不仅展示了ChemEnzyRetroPlanner在复杂生物活性化合物逆合成规划中的广泛适用性,也体现出其在设计有机-酶促混合路线与常规纯有机合成路线方面的双重能力,并在准确性与可操作性上表现出较高水平。
2.6 智能体案例研究
ChemEnzyRetroPlanner包含一个基于编程API的智能体应用。该智能体除可在完全离线部署条件下执行ChemCrow所支持的合成规划任务外,还能够处理反应步骤中的酶促步骤规划与验证任务。图3a展示了一个工作流示例,智能体可自主评估单步反应的可行性,判断其是否为酶促反应,并进一步给出酶推荐。具体而言,智能体首先调用ReactionRater工具评估4-(hydroxymethyl)-2-furancarboxaldehydephosphate(4-HFC-P)的合成反应是否可行;在确认反应可行后,再使用EnzymaticRXNIdentifier判断该反应步骤是否可能由酶催化,即是否属于酶促反应。该工具以0.9975的置信度识别该单步反应为酶促反应。随后智能体调用EnzymeRecommender,给出最可能催化该反应的5类酶,其中最可能的酶对应EC编号为4.2.3.153,且该推理结果被认为是准确的。

图3 | 展示了ChemEnzyRetroPlanner智能体的酶推荐案例研究。 a展示了反应可行性判断、酶促反应识别与酶推荐的推理示例,所用反应案例为4-(hydroxymethyl)-2-furancarboxaldehydephosphate(4-HFC-P)的合成。b展示了针对4-HFC-P合成的具体酶推荐结果,该推荐基于a中预测得到的最可能EC编号4.2.3.153,并包含对活性位点的验证。
图3b进一步展示了EasIFA模块在酶实体推荐与活性位点预测验证中的作用,用于检查在给定酶促反应下推荐酶是否存在相应活性位点。结果显示,EasIFA推荐的酶为UniProtID为Q58499的(5-formylfuran-3-yl)methylphosphatesynthase,该酶包含两个活性位点LYS27与LYS85,并与UniProt记录的反应位点信息一致。此外,补充信息图S14还给出了该智能体在多步合成规划任务中的推理结果:与ChemCrow类似,该智能体能够自主查询化合物的SMILES信息,执行安全检查,并最终调用ChemEnzyRetroPlanner合成规划工具的编程API生成合成路线,同时对关键合成步骤进行归纳总结。ChemEnzyRetroPlanner及其智能体平台支持完全离线部署,具备构建全自主有机-酶促混合合成规划自动化平台的潜力。

图4 | 展示了ChemEnzyRetroPlanner智能体对天然产物phenoxyradicalVII合成规划的分析结果。 智能体首先调用CotrolChemCheck工具判断输入分子是否属于受管制化学品,随后调用RetroPlannerRetrosynthesis工具进行逆合成分析。当纯有机合成规划配置未能产生结果时,智能体会自动采用混合合成策略,最终成功得到合成规划结果,并与实际合成案例相对应。
在更复杂的合成规划任务中,ChemEnzyRetroPlanner智能体体现出较强的自动决策能力,能够自主决定并优化单步逆合成预测模型的混合配置策略,从而更高效地为处于不同化学空间的分子规划合成路径。例如,图4展示了智能体对天然产物phenoxyradicalVII的合成路径分析,该分子是大豆中由isoliquiritigenin生物合成hispidol过程中的一个中间体。智能体首先评估该化合物是否属于受控化学品,在确认其与受控化学品相似度较低后,调用RetroPlannerRetrosynthesis工具进行路径规划。初始阶段采用纯有机合成规划策略但未能为该天然产物生成路径;随后智能体自主决定切换为混合合成规划策略,将有机合成与酶促合成方法结合,成功生成131条候选合成路径,其中最优路径与phenoxyradicalVII的真实生物合成路径精确一致。与此同时,智能体还对该最优路径每一步所需的基础反应条件进行了系统总结,并在采用酶促合成策略时,给出了应优先考虑的酶类型建议。补充信息图S15进一步给出了智能体对另一种天然产物合成路径的分析,两种天然产物的合成路径示意图见图S16。这些案例不仅体现出混合合成规划策略在解决合成路径难题方面相较纯有机策略的潜在优势,也展示了该智能体在复杂配置决策中的稳健自动化能力。
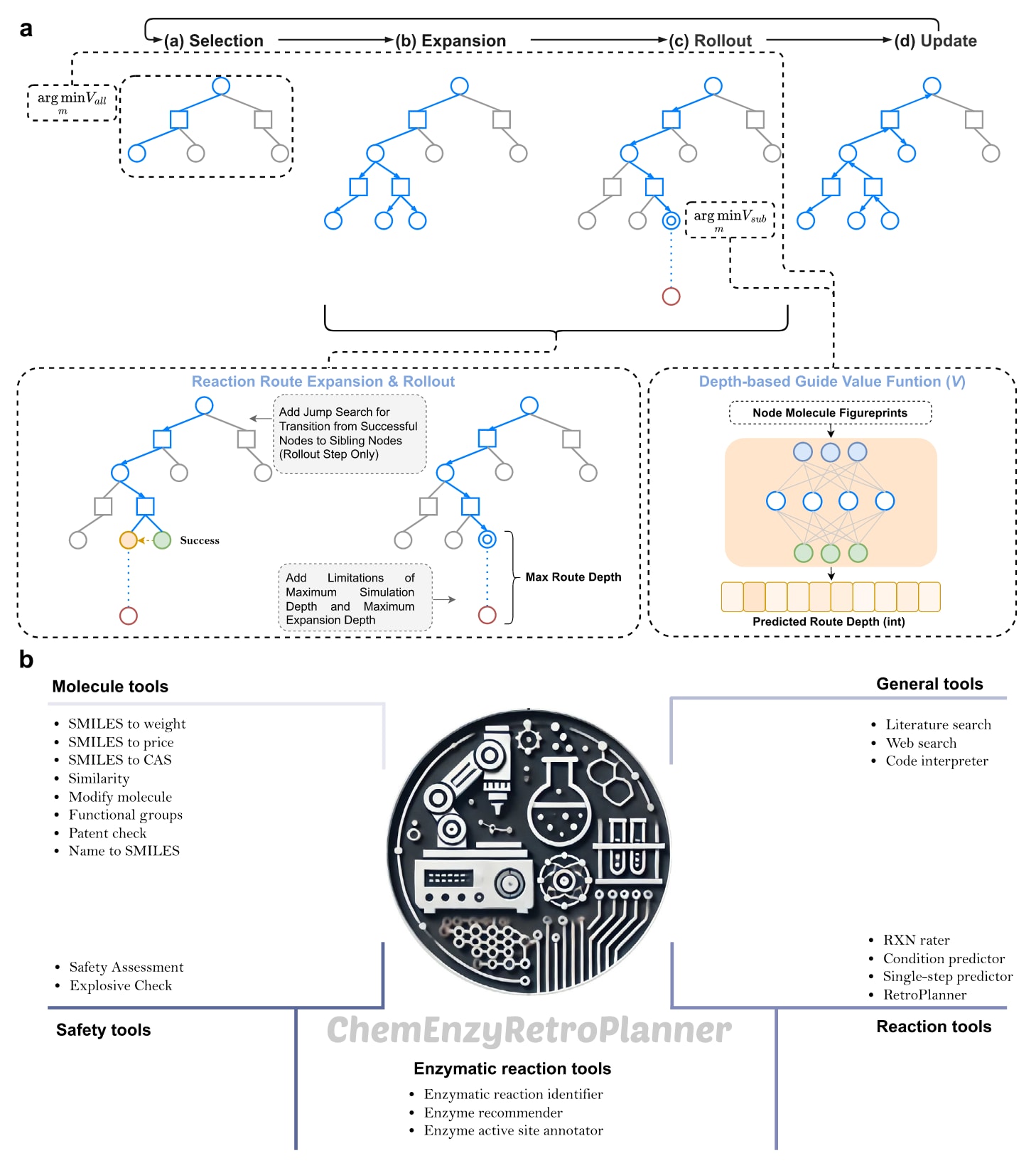
图5 | 展示了ChemEnzyRetroPlanner中搜索框架与智能体工具集增强的总体概览。 a展示了RetroRollout*在算法层面的改进示意,关键步骤包括Selection、Expansion、Rollout与Update,并给出了在Rollout阶段进行跳跃搜索(jumpsearch)的过程示意;算法伪代码见补充信息S1.8。这里,m表示一个分子节点,Vall表示全局成本列表,Vsub表示与分子节点m对应的所有子节点的累计子树成本。b展示了ChemEnzyRetroPlanner智能体实现的工具集,包括分子工具、反应工具、安全评估工具、通用工具与酶促反应工具,共同支持灵活且可扩展的智能体行为。RXN表示reaction。
3 总结
该研究提出了有机-酶促混合合成规划平台ChemEnzyRetroPlanner,在整合一套较完整的有机合成规划工具的同时,也纳入了面向酶促合成规划的计算与验证工具。平台支持有机单步逆合成预测模型与酶促反应单步逆合成预测模型之间的联合搜索,并在多个与有机合成及天然产物相关的测试集上相较同类算法平台展现出明显优势。为提升实际使用体验,平台还引入自动化配置流程,并基于Web服务器实现了更易用的图形界面。同时,平台提供工具级编程API与完全开源的智能体应用,在自主实验室部署与可扩展性方面具有较强潜力。
总体而言,ChemEnzyRetroPlanner能够规划将有机反应与酶促反应相结合的混合合成路线,并在路径规划过程中支持反应条件预测、酶类别推荐以及酶活性位点标注等功能。该开源平台旨在帮助药物研究人员识别更合理、更具成本效益且更符合环境可持续性的合成路线,同时为酶促反应的应用提供关键的活性位点验证信息,从而给出更具依据的合成解决方案。
由于混杂变量是不可观察的,该研究团队尝试实现基于代理变量的混杂调整方法(参考文献44)。然而,当将其集成到深度学习管道中时,遇到了显著的计算挑战,特别是在矩阵求逆过程中分布的可靠估计和数值不稳定性方面。值得注意的是,目前的生物学数据集限制了代理变量仅能使用序列衍生的特征,这可能不足以捕捉到完整的混杂生物学机制的范围。未来引入多模态数据(如结构或功能注释)可能通过提供正交信息源来增强代理变量的质量,从而更好地逼近潜在的混杂因素。尽管因果推断理论为不可观察的混杂因素提供了有原则的解决方案,但适应深度学习框架仍然不是一项简单的任务。该研究团队在讨论中明确承认这些局限性,并将在未来的工作中优先弥合这一方法论差距,特别是关注多模态代理变量的优化。
尽管该研究团队对