NMI 2024 | DiG: 利用深度学习预测分子体系的平衡分布
今天介绍的是一项发表在 Nature Machine Intelligence 上的研究,该工作提出了一种名为DiG的深度学习框架,用于预测分子体系的平衡分布。与以往侧重单一最可能结构的分子建模方法不同,该研究关注分子在平衡态下的整体构象分布,因为许多关键的宏观性质与功能行为正是由这种分布所决定。研究受到热力学退火与扩散过程的启发,利用基于Graphormer的深度生成模型,在分子描述符条件下高效生成多样化构象,并同时估计状态密度。DiG在蛋白质构象采样、蛋白质–配体结合、催化剂表面吸附以及性质引导的材料设计等多个任务中展示了良好的泛化能力和显著的计算效率优势。该工作推动了分子建模从“预测一个结构”迈向“刻画一个分布”,为理解分子体系的统计行为和加速分子与材料设计提供了新的思路。

获取详情及资源:
0 摘要
深度学习的进展极大提升了分子结构预测的准确性。然而,许多与实际应用密切相关的宏观观测量并不是由单一分子结构决定的,而是来源于结构的平衡分布。获取这类分布的传统方法,例如分子动力学模拟,计算成本高昂且在很多情况下难以实施。在此提出一种名为Distributional Graphormer(DiG)的深度学习框架,旨在预测分子体系的平衡分布。DiG受到热力学退火过程的启发,利用深度神经网络在给定分子体系描述符(如化学图或蛋白质序列)的条件下,将一个简单分布逐步变换为平衡分布。该框架能够高效生成多样化构象,并对状态密度进行估计,其速度相比传统方法提升了数个数量级。进一步展示了DiG在多种分子任务中的应用,包括蛋白质构象采样、配体结构采样、催化剂–吸附物采样以及性质引导的结构生成。DiG在分子体系统计理解的方法学上取得了显著进展,为分子科学研究开辟了新的可能性。
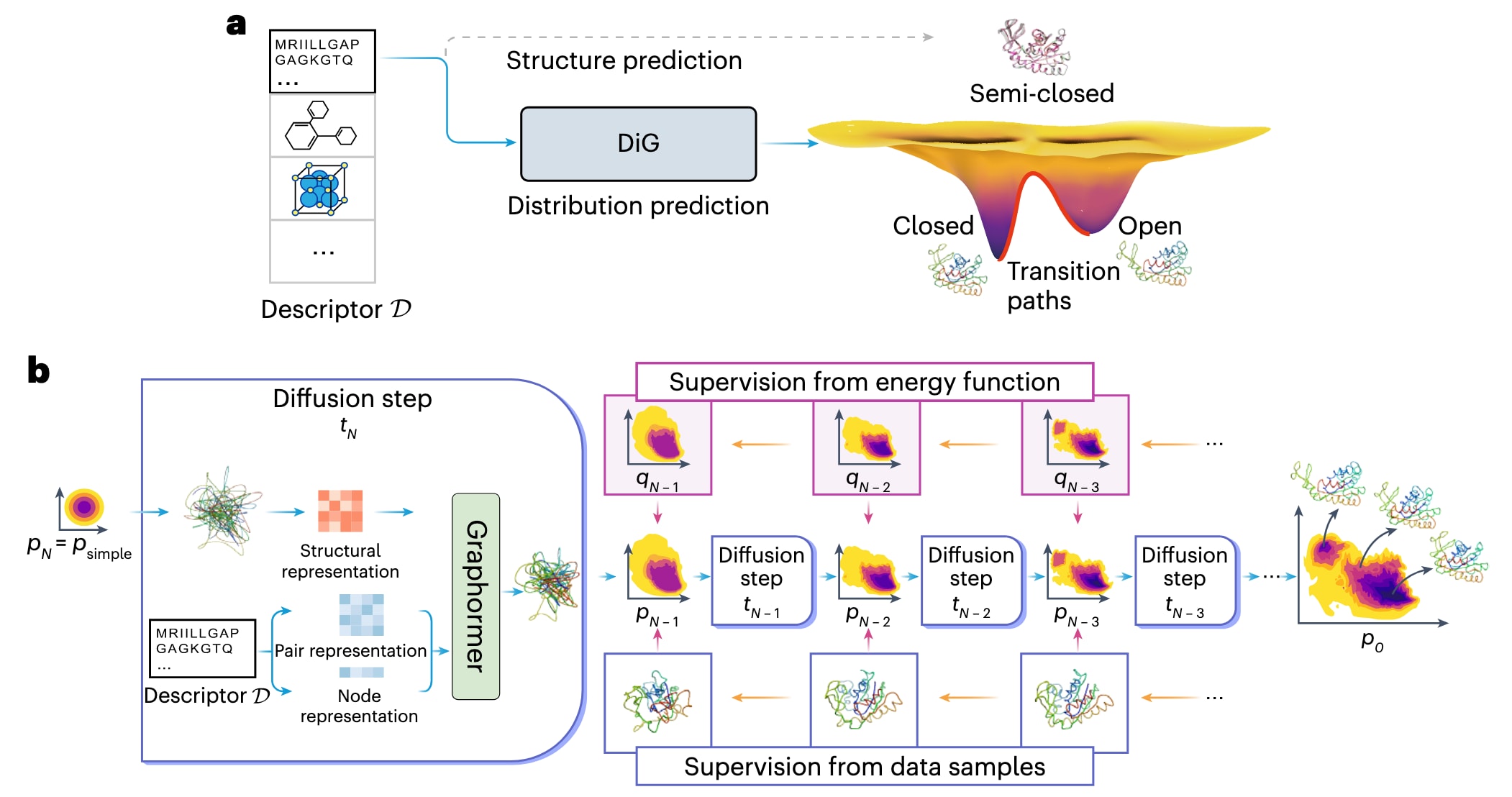
图1|利用DiG框架预测构象分布。 a,DiG以目标分子体系的基础描述符DD作为输入,例如氨基酸序列,生成一个结构概率分布,该分布旨在近似体系的平衡分布,并采样不同的亚稳态或中间态。相比之下,诸如AlphaFold等静态结构预测方法的目标是预测分子的单一高概率结构。b,用于预测分子结构分布的DiG框架。该框架以深度学习模型Graphormer作为核心模块,用于预测一个扩散过程(→),将简单分布逐步转化为目标分布。模型的训练目标是使每一个中间扩散时间步i所得到的分布
1 引言
深度学习方法在高效预测分子结构方面表现突出。例如,AlphaFold能够以原子级精度预测蛋白质结构,从而推动了结构生物学中的新应用;基于神经网络的对接方法可以预测配体的结合构象,支持药物发现中的虚拟筛选;深度学习模型还被用于预测催化剂表面吸附物的结构。这些进展展示了深度学习在分子结构与分子状态建模方面的巨大潜力。然而,仅预测最可能出现的单一结构只能揭示分子体系在平衡态下的部分信息。分子通常具有较高的柔性,而平衡分布对于准确计算宏观性质至关重要。例如,生物大分子的功能可以通过结构概率来推断,以识别亚稳态;而熵和自由能等热力学性质则可以利用统计力学方法,从结构空间中的概率密度计算得到。
图1a展示了传统结构预测与分子体系分布预测之间的差异。腺苷酸激酶具有两种不同的功能构象,即开放态和闭合态,且均已通过实验确定,但常规预测结构通常对应于高度可能的亚稳态或某种中间态。如该示意所示,亟需一种方法来采样具有多种功能状态的蛋白质的平衡分布。不同于单一结构预测,平衡分布的研究仍主要依赖经典且计算代价高昂的模拟方法,而深度学习在这一方向的发展仍然不足。通常,平衡分布通过分子动力学模拟进行采样,但其计算成本高,在许多体系中难以实现。增强采样模拟和马尔可夫状态模型能够加速稀有事件的采样,但依赖于体系特定的集体变量,难以推广。另一类方法是粗粒化分子动力学,其中已引入深度学习策略,这些方法在单一分子体系上取得了良好效果,但尚未证明其通用性。玻尔兹曼生成器通过从简单参考态构建概率流来生成平衡分布,同样面临难以推广到不同分子的挑战。尽管在小肽体系中,生成更长时间步长的流模型已显示出一定的泛化能力,但尚未扩展到大型蛋白质体系。
在该研究中,提出了DiG这一深度学习方法,用于近似预测分子体系的平衡分布,并高效采样多样且与功能相关的结构。结果表明,DiG能够在不同分子体系之间实现泛化,并提出与实验观测相一致的多样化结构。DiG的思想来源于模拟退火,该过程通过逐步演化将均匀分布转化为复杂分布。具体而言,DiG模拟了一种扩散过程,将一个简单分布逐渐变换为目标分布,从而近似给定分子体系的平衡分布。如图1b右侧箭头所示,由于初始简单分布被设计为可独立采样且具有解析形式的密度函数,DiG不仅能够实现平衡分布的独立采样,还可以通过跟踪扩散过程得到该分布的密度函数。该扩散过程还可以引入偏置,用于面向性质的反向设计,并支持在高概率区域内实现结构之间的连续插值。
这一扩散过程由基于Graphormer架构的深度学习模型实现,并以目标分子的描述符作为条件,例如化学图或蛋白质序列。DiG可以利用实验数据或分子动力学模拟得到的结构数据进行训练。针对数据稀缺的情形,进一步提出了一种物理信息引导的扩散预训练方法,利用体系的能量函数,如力场,对模型进行训练。无论是在数据驱动模式还是能量监督模式下,模型在每一个扩散步骤中都能获得独立的训练信号,从而实现高效训练,避免了长链反向传播带来的计算负担。
在蛋白质结构分布、结合口袋中配体构象分布以及催化剂表面分子吸附分布这三项预测任务上对DiG进行了评估。结果显示,DiG能够生成真实且多样的分子结构。对于该研究所研究的蛋白质体系,DiG可以高效生成与主要功能状态相似的构象。此外,通过引入偏置分布以偏好具有目标性质的结构,DiG还能够促进分子结构的反向设计,从而拓展在数据不足情形下的分子设计能力。上述结果表明,DiG推动了分子领域中深度学习方法从单一结构预测迈向结构分布预测,为高效计算分子的热力学性质奠定了基础。
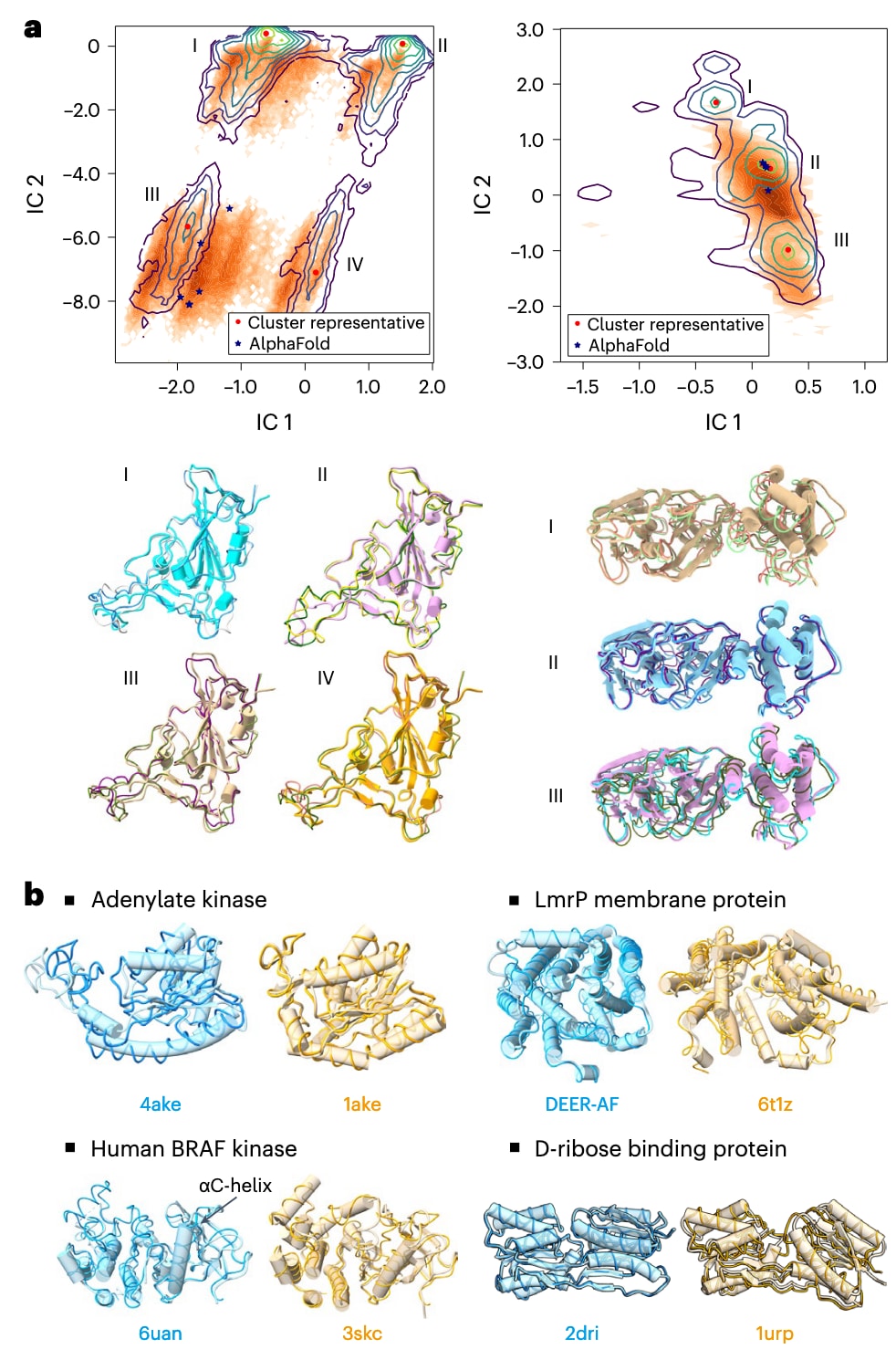
图2|蛋白质构象的分布与采样结果。 a,DiG生成的结构能够重现毫秒尺度分子动力学模拟中观察到的多样化构象。分子动力学模拟得到的结构被投影到由两个时间滞后独立成分分析坐标张成的低维空间中,即独立成分1和2,并通过等高线形式展示其概率密度。左图中,针对受体结合结构域蛋白,分子动力学模拟在该二维空间中揭示了四个高占据区域。DiG生成的结构被映射到同一二维空间中,以橙色点表示,其分布由颜色深浅反映。在分布图下方,将DiG生成的结构(细带状表示)与代表性结构进行叠加对比,同时给出了AlphaFold预测结构在该空间中的位置(以星号表示)。右图展示了SARS-CoV-2主蛋白酶的对应结果,并与分子动力学模拟和AlphaFold预测结果进行比较。等高线图显示出三个构象簇,DiG生成的结构主要覆盖簇II和簇III,而簇I中的结构相对不足。b,DiG在生成蛋白质多构象方面的表现。将DiG生成的结构(细带状表示)与实验解析结构进行对比,其中实验结构以其PDB编号标注,DEER-AF除外,该结构为AlphaFold预测模型,以圆柱状示意表示。对于腺苷酸激酶、LmrP膜蛋白、人类BRAF激酶以及D-核糖结合蛋白这四种蛋白质,DiG均能够较好地重现两种功能状态的结构,不同状态以青色和棕色加以区分。
2 结果
在该节中展示了DiG在多种分子问题中的应用潜力,包括蛋白质构象研究、蛋白质–配体相互作用以及分子在催化剂表面的吸附行为。此外,还通过将DiG应用于具有目标电子带隙的碳同素异形体生成,考察了其在分子结构反向设计方面的能力。
2.1 蛋白质构象采样
在生理条件下,大多数蛋白质分子表现出多个功能状态,并通过动力学过程相互联系。对这些构象进行采样对于理解蛋白质性质及其与其他分子的相互作用至关重要。近期研究表明,AlphaFold可以通过操控多序列比对等输入信息,为部分蛋白质生成替代构象。然而,该方法依赖于改变多序列比对的深度,难以推广到所有蛋白质,尤其是同源序列数量较少的情况。因此,亟需发展先进的人工智能模型,能够在构象空间中一致地反映能量景观,并采样多样化的蛋白质结构。在此展示了DiG生成多样且具有功能相关性的蛋白质结构的能力,这一特性对于高效采样平衡分布至关重要。
由于蛋白质构象的平衡分布在实验或计算上都难以直接获得,可用于训练和评测的高质量数据较为匮乏。为此,研究收集了公共数据库中的实验结构和模拟结构用于模型训练,并通过构建分子动力学模拟数据集以及提出物理信息引导的扩散预训练方法来缓解数据不足的问题。DiG的性能从两个层面进行了评估:一是将其预测的构象分布与长时间尺度的全原子分子动力学模拟结果进行对比,二是在具有多种已知构象的蛋白质体系上进行验证。
如图2a所示,针对来自SARS-CoV-2病毒的两种蛋白质,即刺突蛋白的受体结合结构域和主蛋白酶,给出了由分子动力学模拟得到的构象分布。这两种蛋白质是病毒的重要组成部分,也是新冠治疗中的关键药物靶点。毫秒时间尺度的分子动力学模拟能够广泛采样构象空间,因此其结果可视为平衡分布的近似。以蛋白质序列作为DiG的输入描述符生成结构后发现,尽管这些模拟数据并未用于模型训练,DiG生成的结构仍与模拟得到的构象分布高度一致。在受体结合结构域的二维投影空间中,分子动力学模拟形成了四个主要区域,而DiG能够覆盖全部这些区域,并较好重现对应的代表性结构。对于主蛋白酶,模拟中观察到的三种代表性构象同样能够被DiG预测出来。需要注意的是,其中一个构象簇的覆盖效果仍有限,显示出模型仍有改进空间。从构象覆盖度来看,在二维流形中,仅使用一万个由DiG生成的结构,即可覆盖约70%的模拟构象空间。
由于全原子分子动力学模拟的计算成本极高,毫秒尺度的蛋白质模拟通常仅能在专用硬件或结合马尔可夫状态模型的大规模分布式计算中实现。为进一步评估DiG在生成多样化结构方面的能力,研究转而考察了具有多种实验解析结构的蛋白质体系。如图2b所示,DiG能够为四种蛋白质生成多种构象,并与实验结构良好对应。以腺苷酸激酶为例,DiG生成的结构分别接近其开放态和闭合态。对于药物转运蛋白LmrP,DiG生成的构象覆盖了两种已知状态,其中一种来源于实验解析,另一种则是由AlphaFold预测并得到双电子–电子共振实验支持的构象。在人类BRAF激酶中,两种状态的整体差异相对较小,主要体现在A环区域及其邻近的αC螺旋上,而DiG生成的结构能够准确捕捉这些局部差异。对于D-核糖结合蛋白,两结构域之间的相对排布是主要差异来源,DiG能够生成对应于直立构象以及扭转或倾斜构象的结构,并在对其中一个结构域进行比对时,为另一结构域提供合理的中间态。
此外,DiG还能够通过潜在空间插值生成合理的构象转变路径,展示了其刻画构象动力学过程的潜力。总体而言,DiG不仅限于蛋白质的静态结构预测,还能够生成对应不同功能状态的多样化结构,为蛋白质平衡构象分布的高效采样提供了新的途径。
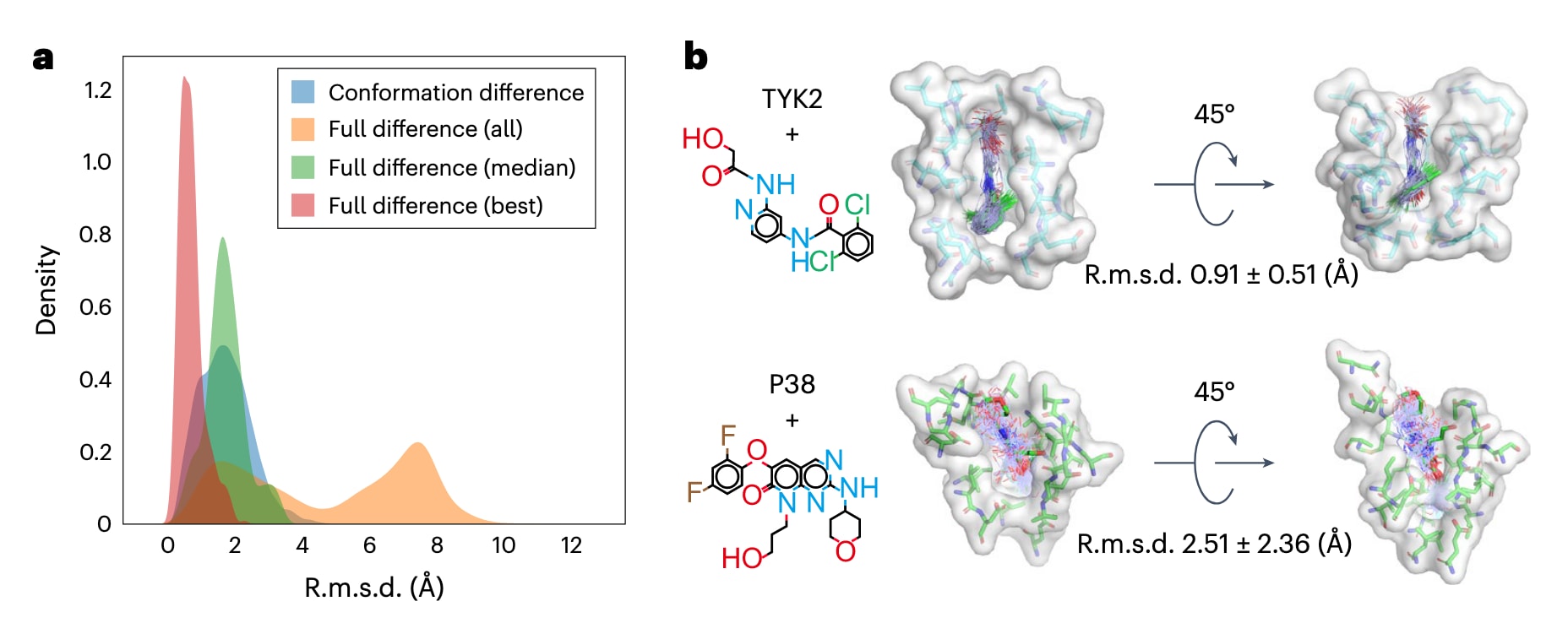
图3|DiG在蛋白质口袋周围配体结构采样中的结果。 a,DiG在配体与蛋白质结合口袋中的构象预测结果。与晶体结构相比,DiG生成的配体结构及其结合姿态具有较高的准确性,这一点体现在均方根偏差统计上,其中红色直方图表示最佳匹配情况下的r.m.s.d.分布,绿色直方图表示r.m.s.d.中位数的统计结果。当对每个体系考虑全部50个预测结构时,可以观察到明显的多样性,其特征由黄色直方图所示的r.m.s.d.分布反映,并进行了归一化处理。所有r.m.s.d.数值均基于配体在复合物结构中的坐标进行计算。b,代表性体系展示了配体结构的多样性,且这种预测到的多样性与结合口袋的性质密切相关。对于TYK2蛋白这类具有深且狭窄结合口袋的体系(上方面板中以表面表示),DiG为配体预测得到的结合姿态高度相似(同样以上方面板中的原子键表示)。相比之下,在P38蛋白中,结合口袋相对平坦且较浅,预测得到的配体结合姿态更加多样,并表现出较大的构象柔性(下方面板,表示方式与TYK2示例相同)。在复合物结构旁标注了平均r.m.s.d.值及其对应的标准差。
2.2 结合位点周围的配体结构采样
蛋白质构象采样的一个直接拓展是对可成药口袋中的配体结构进行预测。为建模蛋白质与配体之间的相互作用,通过对1,500个复合物进行分子动力学模拟来训练DiG模型。随后,在409个未包含于训练集中的蛋白质–配体体系上对模型性能进行了评估。DiG的输入信息包括蛋白质口袋的相关描述,如原子类型与空间位置,以及配体的描述符,即SMILES字符串。为处理口袋周围原子数量不同以及SMILES字符串长度不一致的问题,对输入的节点与成对表示进行了零填充。模型的预测结果为配体与蛋白质口袋的原子坐标分布。
对于蛋白质口袋而言,预测得到的原子位置相较输入值的变化在r.m.s.d.意义下可达1.0 Å,反映了在生成配体结构过程中口袋的柔性。对于配体结构,偏差主要来源于两个方面:一是生成结构与实验解析结构之间的构象差异,二是由于配体平移和旋转导致的结合姿态差异。在所有测试体系中,构象差异总体较小,平均r.m.s.d.为1.74 Å,表明生成的配体结构在构象上与晶体结构高度相似。若将结合姿态的偏差一并考虑,则观察到更大的差异,但DiG仍能够为每个体系预测出与实验结构非常接近的构象。在每个体系生成的50个结构中,几乎所有409个测试体系的最佳匹配结构与实验数据之间的r.m.s.d.均小于2.0 Å。
配体生成结构的准确性与结合口袋的特征密切相关。例如,在TYK2激酶蛋白的案例中,图3b上方面板所示配体相对于晶体结构的平均偏差仅为0.91 Å。相比之下,在P38靶点中,配体表现出更加多样的结合姿态,这可能源于其结合口袋相对较浅,使得最稳定的结合姿态相对于其他姿态并不占据绝对优势。分子动力学模拟揭示了与DiG生成结果一致的趋势,即配体在TYK2中的结合更为紧密,而在P38中则相对较弱。总体而言,DiG生成的配体结构与实验中观测到的结合姿态高度一致。
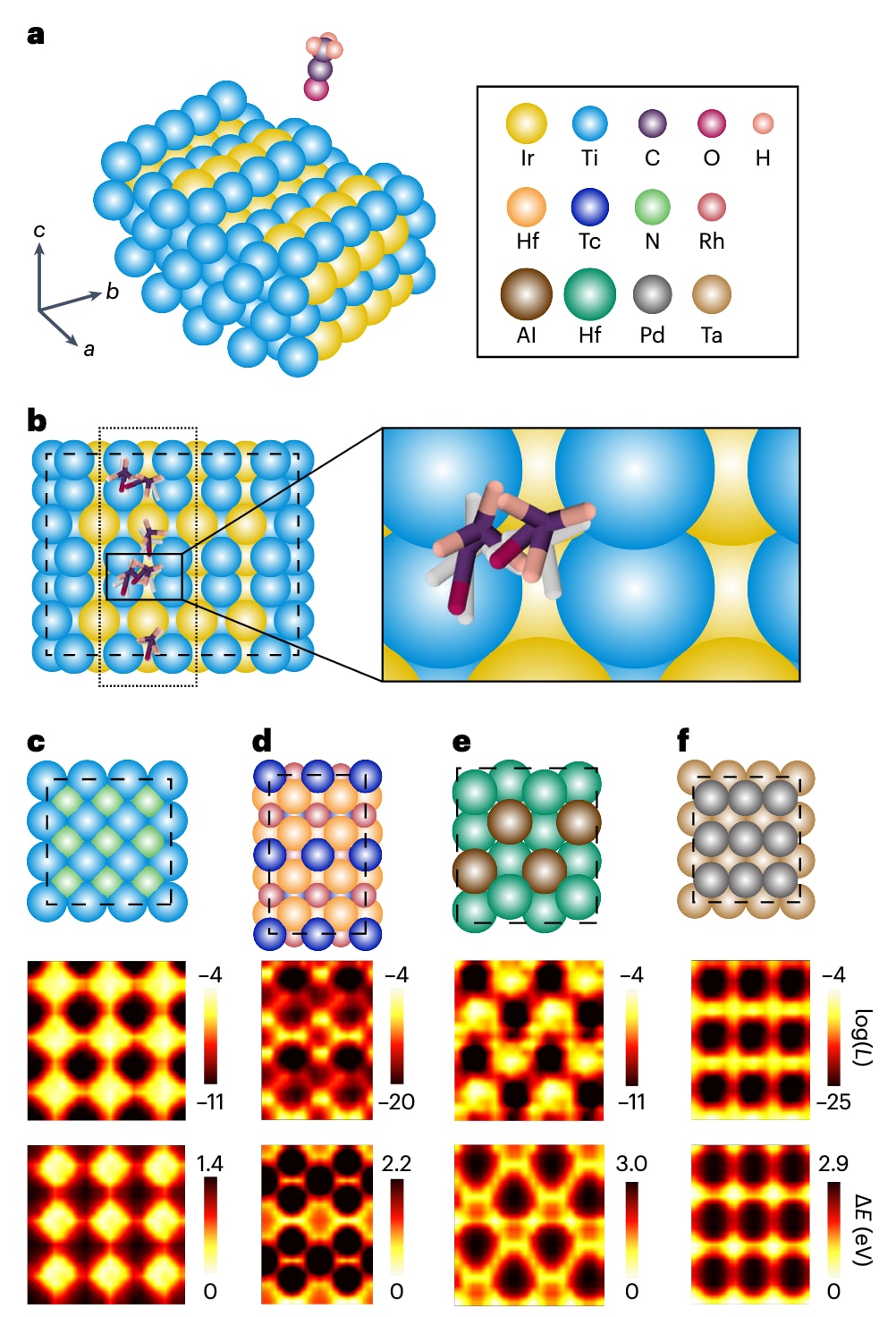
图4|DiG在催化剂–吸附物采样问题中的结果。 a,问题设定:预测吸附物在催化剂表面上的吸附构型分布。b,DiG预测得到的吸附位点及其对应构型(彩色显示)与密度泛函理论计算结果(白色显示)的对比。DiG能够识别出所有吸附位点,且预测得到的吸附物结构与密度泛函理论计算结果高度接近,有关吸附位点与构型的详细信息见补充资料。c–f,单个N或O原子在TiN、RhTcHf、AlHf和TaPd催化剂表面上的吸附预测结果,并与密度泛函理论计算结果进行对比。上方面板展示催化剂表面结构,中间面板给出了吸附物在对应催化剂表面上的概率分布,以对数尺度表示,下方面板展示了通过密度泛函理论方法计算得到的吸附物与催化剂之间的相互作用能。预测得到的吸附位点及其概率分布与密度泛函理论所揭示的能量景观具有高度一致性。
2.3 催化剂–吸附物采样
识别活性吸附位点是非均相催化中的核心问题之一。由于表面与分子之间相互作用高度复杂,该任务通常高度依赖密度泛函理论等量子化学方法,并结合分子动力学模拟或网格搜索等采样技术,从而带来巨大的计算开销,在许多情况下甚至难以承受。为评估DiG在该任务中的能力,研究基于Open Catalyst Project中提供的催化剂–吸附物体系分子动力学轨迹对模型进行训练,并进一步在训练集中未包含的随机吸附物与表面组合上进行测试。DiG以衬底中原子的类型与初始位置、衬底的晶格矢量以及分子吸附物的初始结构作为联合输入。同时,引入交叉注意力子层以处理周期性边界条件,相关细节见补充信息。
在给定催化剂衬底和分子吸附物后,DiG能够预测吸附位点及稳定的吸附构型,并为每一种构型给出相应的概率估计。图4a,b展示了酰基在阶梯状TiIr合金表面上的吸附构型,DiG预测出了多个可能的吸附位点。为验证这些预测构型的合理性并评估其覆盖程度,进一步采用基于密度泛函理论的网格搜索进行对比分析。结果表明,DiG能够成功预测出网格搜索所发现的所有稳定吸附位点,且对应吸附构型之间具有良好一致性,r.m.s.d.约为0.5–0.8 Å。需要指出的是,图4b所示的衬底与吸附物组合并未包含在训练数据集中,因此该结果体现了DiG在催化吸附预测中的跨体系泛化能力。正文中仅展示了俯视图,补充图4还给出了吸附构型的正视图。
除准确预测吸附位点外,DiG还能为每一种吸附构型提供概率估计。该能力在单原子吸附体系中得到了进一步展示,包括H、N和O原子在十种随机选择的金属表面上的吸附。对于每一种吸附物与催化剂组合,DiG预测了吸附位点及其概率分布。为验证预测结果,对相同体系开展了基于密度泛函理论的网格搜索计算,以获得吸附位点及对应能量。以网格搜索识别的吸附位点作为参考,DiG在这十种金属催化剂表面上的单原子吸附体系中实现了81%的位点覆盖率。图4c–f进一步展示了四个体系的详细结果,即单个N或O原子在TiN、RhTcHf、AlHf和TaPd金属表面上的吸附情况。上方面板给出了具体体系,中间面板展示了投影到与催化剂表面平行平面上的吸附概率分布,下方面板则为基于密度泛函理论计算得到的吸附能。可以看到,预测得到的概率分布与吸附能之间具有高度一致性。
值得注意的是,与密度泛函理论方法相比,DiG在计算效率上具有显著优势。对于一个催化剂–吸附物体系,在单个现代图形处理单元上,DiG采样所有吸附位点仅需约1 min,而使用VASP进行一次密度泛函理论结构弛豫至少需要2 h,且该成本还会随着网格分辨率的提高而增加一个大于100的倍数。如此快速且准确的吸附位点预测及其分布特征,在揭示催化反应机理以及指导新型催化剂设计方面具有重要价值。
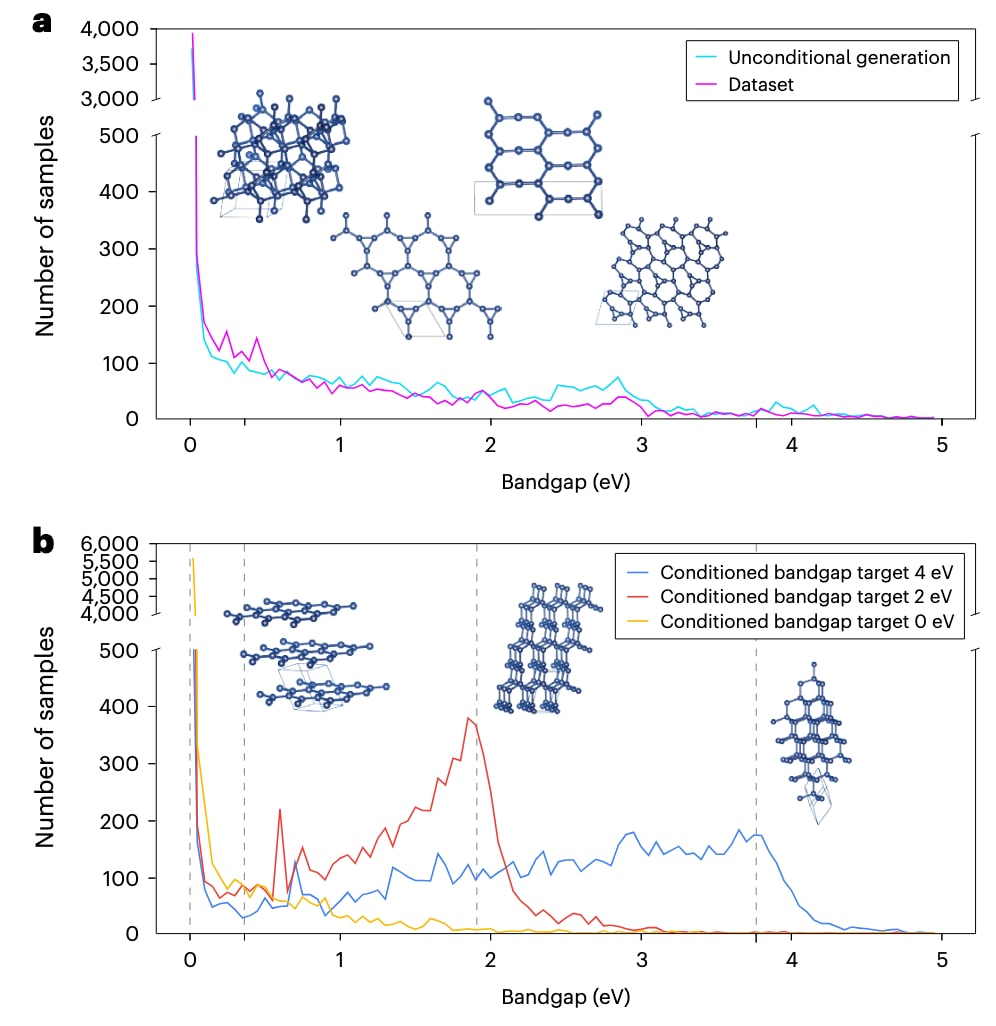
图5|具有特定带隙的碳结构的性质引导生成结果。 a,在未对带隙作任何限定的情况下,由训练后的DiG生成结构对应的电子带隙分布。生成结构在带隙取值上未表现出明显偏好,其分布与训练数据集中的带隙分布高度相似。b,针对三种目标带隙(0、2和4 eV)生成的结构结果。生成结构的带隙分布在期望值附近出现峰值。具体而言,当目标带隙为0 eV时,DiG生成的结构呈现出类石墨特征;而当目标带隙为4 eV时,生成的结构则主要类似于金刚石。图中的竖直虚线表示生成结构中接近0、2和4 eV的带隙位置。插图展示了若干代表性结构。
2.4 性质引导的结构生成
尽管DiG在默认情况下按照训练数据中学习到的分布生成结构,其输出分布也可以被有意地引导和偏置,从而使生成的结构满足特定需求。在此利用这一特性,将DiG用于反向设计,具体方法见“利用DiG进行性质引导的结构生成”一节。作为概念验证,研究以具有目标电子带隙的碳同素异形体搜索为例展开。类似任务在新型光伏材料和半导体材料的设计中具有重要意义。
为训练该模型,通过基于密度泛函理论计算得到的能量分布开展结构搜索,构建了一个由碳材料组成的数据集。对应能量极小值的结构构成了用于训练DiG的数据集,并进一步用于生成新的碳结构。电子带隙的性质预测采用了基于M3GNet架构的神经网络模型,该模型作为性质评估器,被引入到碳结构的性质引导生成过程中。
图5展示了由DiG生成的碳结构所对应的带隙分布。在原始训练数据集中,大多数结构的带隙集中在约0 eV附近。当将目标带隙作为附加条件输入DiG后,模型能够生成具有期望带隙的碳结构。在带隙预测模型的引导下,生成分布向目标值发生偏移,并在目标带隙附近呈现出明显峰值。图5中给出了若干代表性结构。在以4 eV为目标带隙的条件生成中,DiG生成了与金刚石相似的稳定碳结构,其特征是较大的带隙;而在以0 eV为目标带隙的情况下,生成的结构则更接近石墨,表现出较小的带隙。图5a还展示了若干无条件生成得到的结构示例。
为评估DiG生成碳结构的质量,使用PyMatgen软件包中的StructureMatcher工具,计算生成结构与数据集中经弛豫结构相匹配的比例。对于无条件生成,匹配率达到99.87%,在所有采样结构中,基于分数坐标计算得到的归一化r.m.s.d.平均值为0.16。对于条件生成,匹配率提高至99.99%,但平均归一化r.m.s.d.增大至0.22。这表明,在提高生成目标带隙结构概率的同时,条件生成在一定程度上会影响结构质量,相关讨论见补充信息。
这一概念验证研究表明,DiG不仅能够在高维构型空间中刻画具有复杂特征的概率分布,还可以在与性质量化模型,例如机器学习预测器,相结合的情况下,用于材料的反向设计。由于性质预测模型与DiG的扩散模型彼此解耦,该方法可以方便地推广到针对其他目标性质的材料反向设计任务中。
3 讨论
预测分子状态的平衡分布是分子科学中的一项重大挑战,对于理解结构–功能关系、计算宏观性质以及分子与材料设计具有广泛影响。现有方法通常需要大量单分子的实验测量或模拟采样,才能刻画平衡分布。该研究提出了DiG这一深度生成框架,旨在预测平衡概率分布,从而在不同分子体系中高效采样多样化构象并估计状态密度。DiG受到退火过程的启发,通过一系列深度神经网络将状态分布从简单形式逐步转化为目标分布,并在合适数据条件下近似学习体系的平衡分布。
DiG被应用于多种分子任务,包括蛋白质构象采样、蛋白质–配体结合结构生成、催化剂表面的分子吸附以及性质引导的结构生成。结果表明,DiG能够生成化学上合理且具有高度多样性的结构,在部分任务中,其分布在低维投影空间内与分子动力学模拟结果高度相似。借助先进的深度学习架构,DiG可以从分子描述符中学习构象表示,例如蛋白质的序列或化合物的化学式。同时,扩散模型在刻画复杂多峰分布方面的能力,使DiG能够捕捉高维空间中的平衡分布。
基于上述特性,该框架为分子科学研究开启了丰富的应用前景。DiG能够为分子体系提供统计层面的理解,支持自由能和热力学稳定性等宏观性质的计算,而这些信息对于研究分子体系中的物理和化学现象至关重要。此外,DiG能够从平衡分布中生成相互独立且同分布的构象,相较于传统采样或模拟方法,例如马尔可夫链蒙特卡洛或分子动力学模拟,具有显著优势,后者往往需要依赖罕见事件来跨越能垒。在所测试的两个蛋白质案例中,DiG覆盖了与毫秒尺度分子动力学模拟相当的构象空间。基于OpenMM的性能基准,若在NVIDIA A100上模拟刺突蛋白受体结合结构域1.8 ms的时间尺度,约需7–10个GPU年的计算资源,而在不进行推理加速的情况下,使用单个A100 GPU通过DiG生成5万个结构仅需约10天。类似甚至更高的加速比,也已在催化剂表面吸附分布预测任务中得到体现。
在给定状态下实现平衡分布的定量预测仍依赖于数据的可获得性,但DiG在广阔且多样的构象空间中进行探索的能力,有助于发现新的功能性分子结构,包括蛋白质结构、配体构象以及吸附构型。DiG为连接分子体系的微观描述与宏观观测提供了新的途径,有望对生命科学、药物设计、催化研究以及材料科学等多个分子科学领域产生深远影响。