NMI 2025 | GerNA-Bind: RNA–配体结合特异性解析
今天介绍的这项工作来自Nature Machine Intelligence。靶向RNA的小分子药物被认为是调控生物通路和治疗复杂疾病的一种重要策略,但RNA结构的高度柔性、多构象特征以及高质量复合物结构数据的匮乏,长期限制了RNA–配体相互作用的系统研究。传统基于物理建模的方法虽然能够提供原子尺度的相互作用解释,但对高分辨率结构数据依赖较强,难以扩展到大规模筛选场景;而基于序列或简单理化特征的方法虽然高效,却难以刻画RNA的三维空间特性和动态行为。近年来,深度学习方法在RNA–配体相互作用预测中展现出潜力,但在低同源性体系、不确定性评估以及结合位点解析等方面仍存在明显不足。通用型结构预测模型虽然可以生成RNA或RNA–配体复合物构象,却往往需要额外的特异性评分和筛选步骤,才能服务于RNA靶向药物设计。在这一背景下,GerNA-Bind被提出作为一种面向RNA–配体结合特异性预测的几何深度学习框架。该方法通过同时整合RNA与小分子的多层级表示,包括一维序列、二维结构图以及三维空间构象,系统建模RNA–配体之间的空间几何关系与相互作用模式。模型不仅能够预测是否发生结合,还能够显式给出碱基–原子层面的相互作用矩阵,并通过基于证据的学习策略对预测不确定性进行量化。该文围绕GerNA-Bind的模型设计、性能评估与生物学应用展开。研究首先系统验证了该方法在多个公开数据集上的预测能力,展示其在低同源性RNA–配体对上的显著优势;随后分析了模型在不确定性估计和结合位点预测方面的表现;最后通过针对MALAT1三股螺旋结构的大规模虚拟筛选与实验验证,展示了GerNA-Bind在RNA靶向药物发现中的实际应用潜力。整体而言,该工作为将几何深度学习引入RNA结构生物学与药物设计提供了一个具有代表性的范式。

获取详情及资源:
- 📄 论文: https://www.nature.com/articles/s42256-025-01154-z
- 💻 代码: https://github.com/GENTEL-lab/GerNABind
0 摘要
RNA分子在多种生物学过程中发挥着关键的调控作用,同时也是多种疾病中极具潜力的治疗靶点。然而,由于RNA结构高度复杂且高分辨率结构数据十分有限,发现能够选择性结合特定RNA构象的小分子仍然面临巨大挑战。针对这一问题,该研究提出了一种名为GerNA-Bind的几何深度学习框架,通过整合多状态RNA–配体的结构表示及其相互作用,实现对RNA–配体结合特异性的预测。GerNA-Bind在多个基准数据集上取得了当前最优的预测性能,在低同源性RNA–配体对的相互作用预测中尤为突出。在结合位点预测任务中,其精度相比AlphaFold3提升了20.8%。此外,该方法能够给出信息充分且校准良好的预测结果,并内置不确定性量化机制,为后续决策提供支持。在大规模虚拟筛选应用中,GerNA-Bind成功识别出18种结构多样的小分子,能够靶向致癌相关的MALAT1 RNA,且实验验证表明其结合亲和力达到亚微摩尔水平。其中一种领先化合物能够选择性结合MALAT1的三股螺旋结构,降低其转录本水平,并抑制癌细胞迁移。这些结果表明,GerNA-Bind在RNA靶向药物发现中具有重要潜力,不仅具备高预测精度,同时还能提供具有生物学意义的结构洞见。
1 引言
利用小分子靶向核糖核酸RNA被视为调控生物通路和开发治疗手段的一种变革性策略,通过选择性作用于RNA结构,该策略在应对复杂疾病方面展现出重要潜力。深入理解RNA–配体相互作用的内在机制,是推动RNA靶向药物发现和理性设计高效治疗分子的关键前提。近年来,相关研究主要依赖基于物理原理的方法来解析RNA–配体的结合机制,重点考察RNA与配体之间的静电相互作用、π–π堆叠作用以及氢键等分子作用力。这类方法能够提供原子尺度的细致认知,但其预测能力受到高分辨率结构数据稀缺的显著限制,而这些数据往往依赖X射线晶体学或冷冻电镜等资源消耗巨大的实验技术。为缓解结构数据不足带来的瓶颈,另一类方法尝试通过序列相似性和理化特征描述符来预测RNA–配体结合。这些方法实现简单、计算效率高,但由于难以刻画RNA固有的结构柔性和构象变化,在动态RNA体系中的结合特异性预测能力有限。尽管深度学习模型为RNA–配体相互作用预测引入了新的思路,但在低同源性RNA–配体对上仍面临挑战,同时由于模型不确定性较高,并不完全适用于高通量虚拟筛选场景。与此同时,诸如AlphaFold3和RoseTTAFold All-Atom等大规模生成式或预测模型开始展现出在RNA或RNA–配体复合物建模方面的潜力。然而,这类通用模型通常需要在下游步骤中引入额外的过滤和特异性评分,才能保证预测结果具有生物学相关性,尤其是在RNA特异性结合问题中更为明显。这一现状凸显了对能够有效处理多样RNA结构及其相互作用的稳健工具的迫切需求。
近期,基于序列的生成模型推动了无需结构输入的可扩展生物分子设计,但这类方法往往忽略了分子识别过程中至关重要的空间几何约束。随着RNA结构预测精度的不断提升,将结构先验信息引入模型,为提高预测准确性和泛化能力提供了一条互补途径。针对上述挑战,GerNA-Bind被提出作为一种用于RNA–配体结合预测的几何深度学习框架。该模型融合多种结构表示与相互作用分析,同时利用实验解析结构和高通量筛选数据构建功能嵌入。这些嵌入能够同时刻画基于图的拓扑连接关系以及三维空间相互作用特征,从而提升预测精度,并通过不确定性量化评估结果的可信度。在RNA训练数据有限且结构多样性显著的背景下,不确定性估计在引导可靠预测中发挥着关键作用,尤其有助于优先筛选高置信度的RNA–配体候选对用于后续实验验证。
GerNA-Bind通过三个方面展示了几何深度学习在RNA–配体相互作用研究中的潜在变革意义。首先,该方法在多个RNA–配体基准数据集上取得了当前最优的预测性能,在低同源性RNA–配体对中表现尤为突出。其次,借助三角几何模块,GerNA-Bind能够解析不同RNA家族中的结合模式,为RNA–配体相互作用提供结构层面的洞见。最后,在虚拟筛选应用中,该方法识别出18种结构多样、对致癌相关长链非编码RNA MALAT1具有亚微摩尔亲和力的小分子。其中一种代表性化合物能够选择性结合MALAT1的三股螺旋结构,抑制其转录本水平并降低癌细胞迁移能力。通过将几何深度学习与RNA结构生物学相结合,GerNA-Bind构建了一个加速RNA靶向治疗分子开发的稳健框架。该工具的开放发布有望推动计算生物物理学与转化RNA医学领域的进一步创新。
2 结果
2.1 GerNA-Bind概述
图1展示了GerNA-Bind的整体架构。该模型以RNA和小分子配体作为输入,其中RNA主链结构既可以来源于实验验证结果,也可以通过RNA折叠模型RhoFold进行计算预测,小分子则以其三维分子构象形式引入模型。GerNA-Bind在预测精度、鲁棒性和可解释性方面表现突出,适用于RNA–配体结合特异性预测、RNA靶向虚拟筛选、结合位点识别以及结合机制分析等多种任务场景(图1b)。
GerNA-Bind通过整合多状态RNA–配体表示及其相互作用来实现高效建模,具体包括一维RNA序列、二维RNA二级结构、三维RNA构象,以及小分子的二维分子图和三维分子构象(图1c)。该模型在设计上力求简洁而高效,通过多状态输入实现对RNA–配体结合特异性的精确预测。在具体实现中,模型采用等变图Transformer对RNA的三维构象进行编码,使用图神经网络刻画RNA的二维二级结构,并通过序列编码器提取一维RNA序列特征。对于小分子配体,则分别利用图Transformer和图神经网络对其三维构象和二维分子图进行编码。在模型核心部分,GerNA-Bind在亲和力预测模块中引入双重注意力机制,同时建模RNA–配体复合物中的二维图连接关系和三维空间相互作用(图1d)。该模块通过记忆缓存机制融合碱基–原子层面的成对特征、RNA表示以及分子表示,从而预测结合亲和力,并进一步用于估计结合特异性。此外,模型引入基于证据参数化的Dirichlet分布,对每一个RNA–配体复合物的结合概率及其不确定性进行联合估计,使模型不仅能够给出结合判断,还能够提供相应的置信度信息,为后续决策提供支持。
为进一步提升预测性能,GerNA-Bind整合了几何约束模块,对RNA碱基与类药小分子原子之间的成对相互作用矩阵进行优化。该模块不仅增强了成对相互作用预测的准确性,还通过显式输出碱基–原子相互作用矩阵提升了模型的可解释性。基于该相互作用矩阵的细化建模,模型能够更准确地评估RNA–配体的结合特异性。关于模型的更详细描述可参见Methods和补充说明。
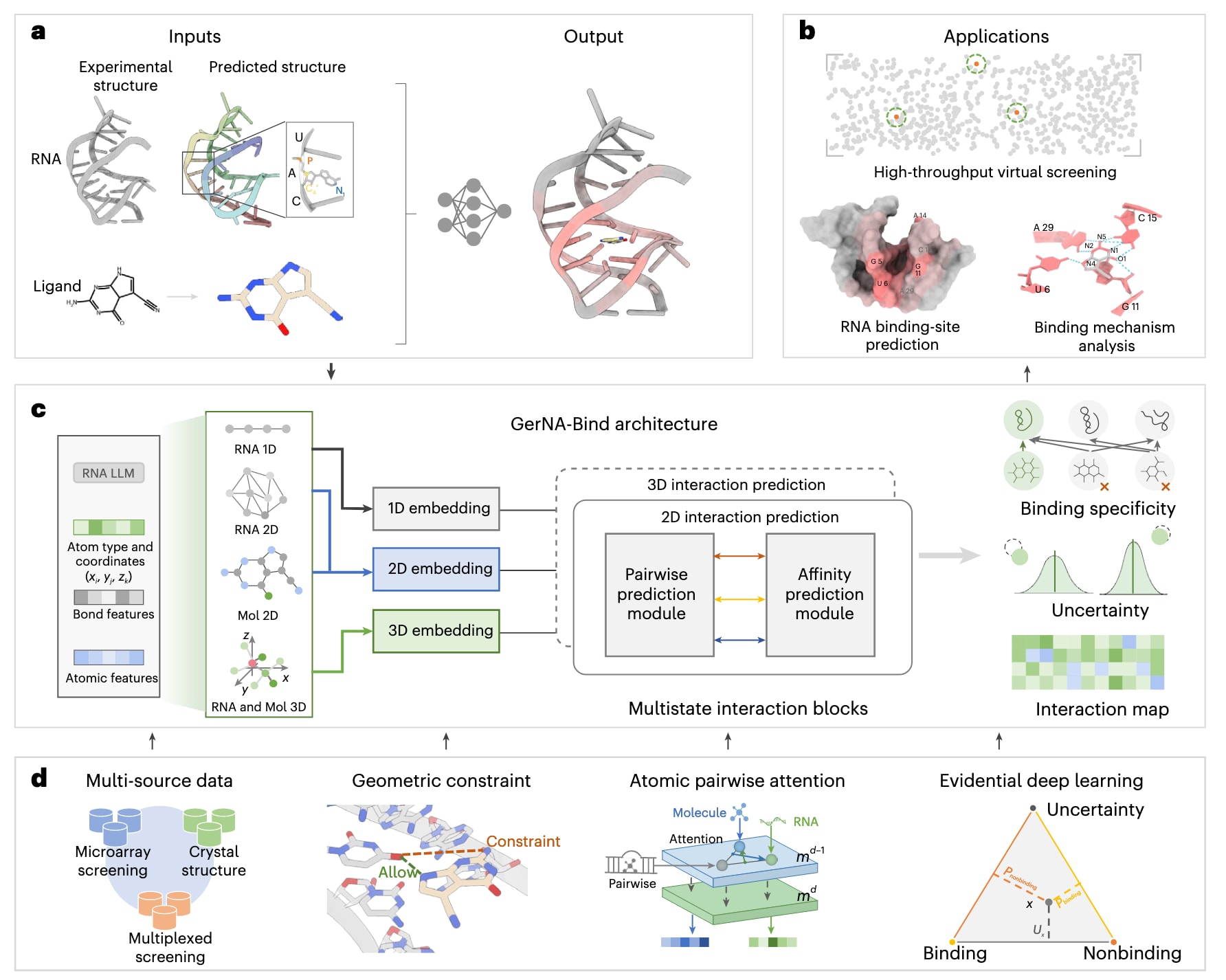
图1|GerNA-Bind概述。 a,GerNA-Bind以RNA和小分子配体作为输入。RNA以主链碱基和原子的三维坐标表示,图中以PDB条目3GCA为示例;小分子配体则以RDKit计算得到的三维分子构象表示。b,GerNA-Bind可应用于多种任务场景,包括RNA靶向虚拟筛选、RNA结合位点预测以及结合机制分析。c,GerNA-Bind是一种用于RNA–配体结合特异性预测的多模态神经网络,整合了一维RNA序列、二维RNA二级结构、三维RNA构象、二维分子图以及三维分子构象。模型通过成对预测模块和亲和力预测模块,同时捕捉二维图结构连接关系与三维空间相互作用,并输出不确定性评分,用于评估每一次预测结果的置信度。d,GerNA-Bind在多来源数据上进行训练,包括微阵列筛选、多重筛选实验以及晶体结构数据。模型利用几何约束优化RNA–配体的成对相互作用,并在亲和力预测模块中引入原子级成对注意力机制以识别RNA结合位点。其中,md和md−1分别表示原子成对注意力网络中的第d层和第d−1层。模型结合基于证据的深度学习方法,在输出结合特异性预测的同时给出不确定性评分,用于评估预测可信度。x表示预测点,$$P_{nonbinding}$$和$$P_{binding}$$分别表示不结合和结合的概率,U表示不确定性,Ux表示预测点x的不确定性。LLM表示大语言模型。
2.2 RNA–配体结合特异性的精确建模
该研究首先利用两个公开的、经实验验证的RNA–配体相互作用数据集Robin和Biosensor评估GerNA-Bind在RNA–配体结合特异性预测方面的能力。这两个数据集同时包含大量发生相互作用和未发生相互作用的RNA–配体对,为模型在具有生物学意义且数据充足的条件下学习区分结合与不结合状态提供了良好基础。为严格评估模型性能,研究采用了四种不同的数据划分策略,用于模拟训练集与测试集在RNA或配体层面不重叠的域内与域外预测场景。有关数据集构建及划分方式的详细说明见Methods部分。
在性能对比中,GerNA-Bind与多种现有方法进行了系统比较,包括当前具有代表性的RNA–配体相互作用预测模型RSAPred,以及三种由蛋白–配体相互作用模型改造而来的方法DeepDTIs,DeepConv-DTI和GraphDTA。模型性能通过多项指标进行评估,包括受试者工作特征曲线下面积AUROC、准确率、F1值以及精确率–召回率曲线下面积AUPRC,从而全面比较不同方法在结合特异性预测任务中的表现。
结果如图2a所示,GerNA-Bind在所有评测场景中均显著优于对比方法。在Robin数据集中,在同源性和分子指纹联合划分条件下,GerNA-Bind的AUROC相较于表现最优的GraphDTA提升了6.7%,相较于RSAPred提升了12.4%。在Biosensor数据集中也观察到类似趋势,GerNA-Bind分别较GraphDTA和RSAPred提升了9.1%和12.6%。除在最具挑战性的划分策略下取得领先表现外,GerNA-Bind在其余三种评估划分中同样表现出显著优势,表明该方法在RNA–配体结合特异性预测中具备良好的鲁棒性和泛化能力。AUPRC、准确率和F1值等指标与AUROC结果保持一致,进一步验证了GerNA-Bind的整体性能优势。
GerNA-Bind取得上述性能提升的关键原因在于其能够系统处理多状态RNA–配体表示及其相互作用,这使其区别于主要依赖单一模态信息的现有方法。如图2b所示,GerNA-Bind整合了RNA和小分子的多层级信息,将序列、图结构和三维构象特征联合建模,从而显著提升预测精度。为评估不同RNA结构模态的贡献,研究在多种数据划分条件下进行了消融实验,结果汇总于图2b及相关补充表格中。与仅使用单一模态输入的模型相比,GerNA-Bind在Biosensor数据集上的AUROC提升达到14.4%,在Robin数据集上提升达到8.6%。即使在最严格的数据划分条件下,模型仍分别在Biosensor和Robin数据集中实现了9.1%和5.5%的性能提升。
此外,针对网络模块的消融分析结果表明(图2c),无论采用何种数据划分策略,GerNA-Bind始终能够取得最优性能,显示出其结构设计的稳定性。不同分子图网络对特征提取效果的影响也被系统分析,进一步揭示了模型结构选择对预测性能的作用。总体而言,这些结果表明,通过融合序列、图结构和三维构象等多状态RNA–配体表示,GerNA-Bind构建了一个在RNA–配体结合特异性预测任务中高度有效且具有普适性的计算框架。
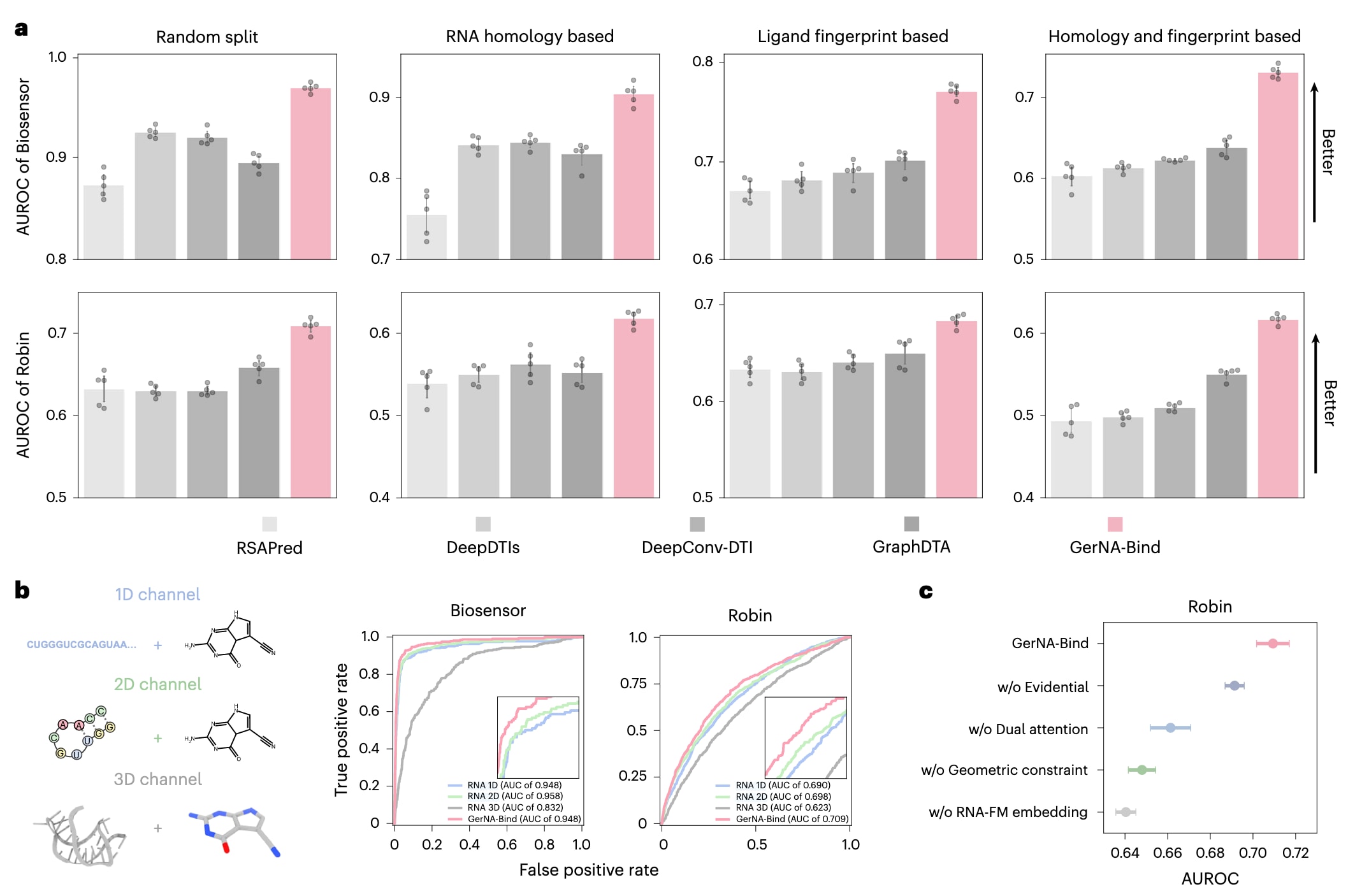
**图2|GerNA-Bind在RNA–配体结合特异性预测中的表现。**a,GerNA-Bind与多种基线模型在RNA–配体结合特异性预测任务中的性能对比,包括RSAPred、DeepDTIs、DeepConv-DTI和GraphDTA。模型性能采用AUROC指标进行评估,结果显示GerNA-Bind在Robin和Biosensor数据集的四种不同数据划分策略下均取得了当前最优的预测结果。b,GerNA-Bind基于多模态框架建模RNA–配体相互作用,分别刻画RNA序列与配体分子图、RNA二级结构图与配体分子图,以及RNA三维结构与配体三维构象之间的相互作用。消融实验系统评估了多状态表示在模型中的贡献。与仅使用单一模态输入的一维RNA序列、二维RNA结构图或三维RNA构象的模型相比,同时整合三种RNA模态显著提升了GerNA-Bind对RNA–配体结合特异性的预测能力。c,对GerNA-Bind中关键模块进行去除后的消融分析结果,性能同样以AUROC衡量。结果表明,缺失任一核心模块都会导致模型性能下降,进一步验证了各组成模块在整体框架中的重要作用。图a和图c中的柱状图均表示五次独立实验结果的均值±标准差。
2.3 RNA–配体相互作用预测中的不确定性量化
GerNA-Bind将不确定性估计作为衡量预测可信度的重要指标,用于辅助决策过程,从而提升虚拟筛选中识别潜在药物候选分子的效率。该能力基于一种证据驱动的深度学习框架实现,模型在对每一个RNA–配体结合事件进行预测的同时,会分配相应的不确定性评分。关于不确定性估计模块的具体实现细节在Methods部分中进行了详细说明。
为评估结合预测的可靠性以及不确定性信息在决策中的实际价值,研究首先在Robin数据集(图3)上定量分析了不确定性估计与预测性能之间的关系。具体而言,所有预测结果按照不确定性从低到高排序,并划分为不同百分位区间,在每个区间内计算累计AUROC。如图3a所示,GerNA-Bind的不确定性评分与预测性能之间呈现出显著相关性,且整体表现优于集成学习和随机失活等常见不确定性估计方法。在随机数据划分条件下(图3b)的分析结果进一步表明,GerNA-Bind在不确定性估计与预测性能之间实现了0.96的Spearman相关系数,明显高于集成学习方法的0.79和随机失活方法的0.82。这些结果表明,GerNA-Bind在预测结果及其不确定性评估方面均具有较高的稳健性,能够为RNA靶向药物发现中的虚拟筛选和实验验证提供有效指导。
除相关性分析外,研究还利用Robin数据集评估了GerNA-Bind在不同RNA结构背景下预测RNA结合口袋与小分子亚结构匹配关系的能力。该分析聚焦于已定义的RNA结合口袋与配体药效团结构之间的匹配情况。首先,研究从Robin数据集中24,572种类药分子中筛选出27种在超过1,000个分子中出现的常见亚结构。随后,利用Inforna方法识别出19种具有生物学意义的RNA二级结构模体,最终构建了513种RNA模体与分子亚结构组合,用于系统评估结合预测性能。尽管这些RNA模体的筛选基于小分子结合潜力而非转录组定位,其中部分模体在结构上与已知功能元件具有相似性,例如前体microRNA发卡结构或非翻译区的茎环结构,提示其潜在的生物学相关性。
子集测试结果如图3c所示,颜色越深代表AUROC越高,同时也对应更优的预测性能,这一趋势在具有明确生物学意义的RNA模体中尤为明显。值得注意的是,富含腺嘌呤和尿嘧啶的区域更倾向于与含有氨基或羟基官能团的小分子发生相互作用,提示氢键作用可能在稳定RNA–配体结合并增强亲和力方面发挥重要作用。这些结果表明,GerNA-Bind能够识别具有特定小分子结合潜力的RNA结构模体,为理解RNA–配体相互作用提供了有价值的线索。
此外,研究还系统评估了GerNA-Bind在不同RNA结构类型中的预测性能,包括三股螺旋、G-四链体、三向连接结构、发卡结构以及假结结构。如图3d所示,GerNA-Bind在NEAT2 RNA上的表现最佳,AUROC达到0.791,并在其他RNA结构类型中保持了相对稳定的预测性能, AUROC值大多维持在0.6左右。整体来看,这些结果进一步验证了GerNA-Bind在多种RNA结构背景下进行RNA–配体结合预测时的稳定性和鲁棒性,凸显了其在药物发现实践中的应用价值。
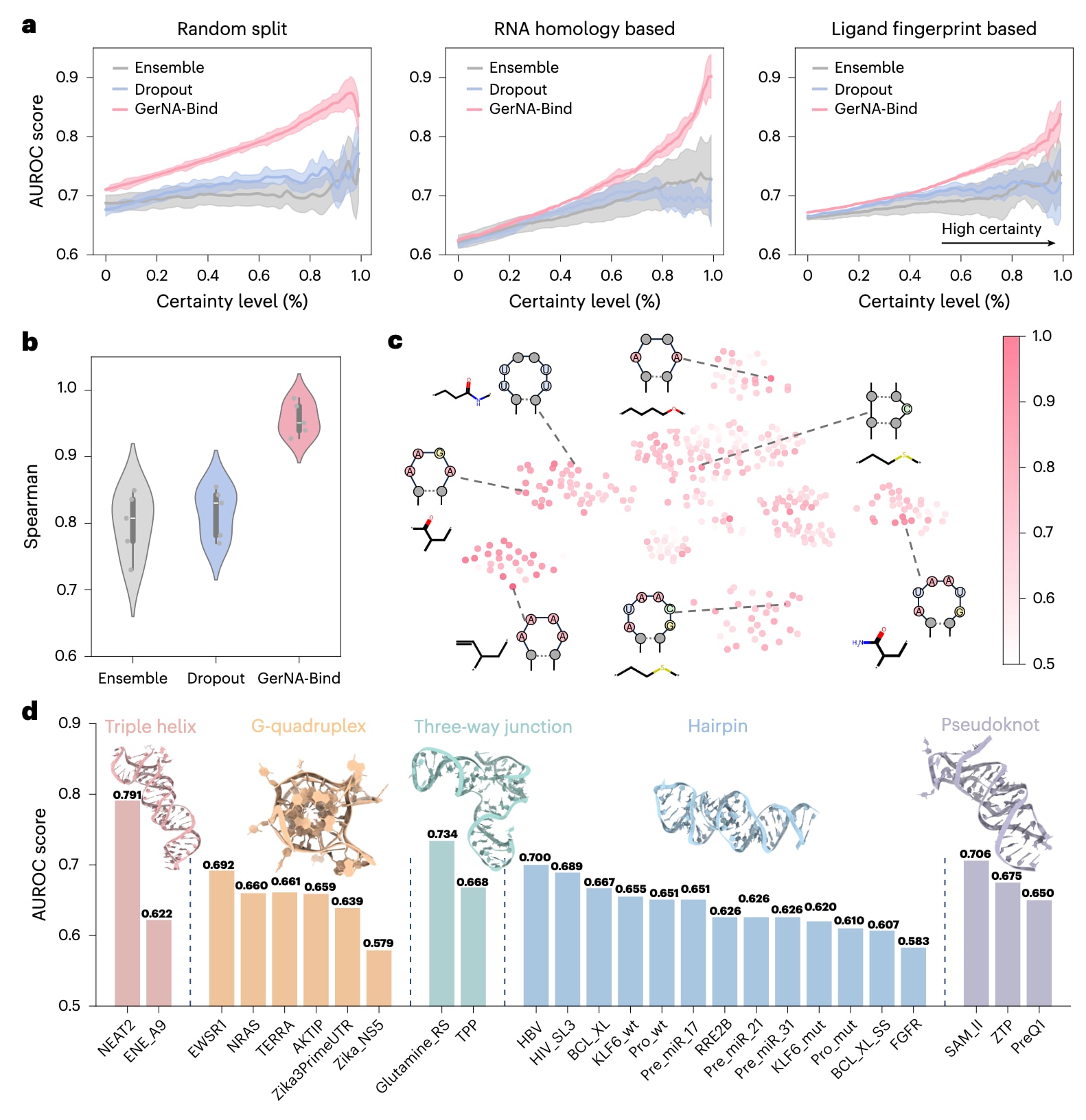
**图3|GerNA-Bind在RNA–配体相互作用预测中的不确定性量化。**a,不确定性估计与RNA–配体结合预测性能之间的相关性分析。在随机划分、同源性划分和分子指纹划分三种设置下,GerNA-Bind与集成学习方法和随机失活方法进行了对比。横轴表示按不确定性排序后的结果,其中100%分位对应最高置信度预测。b,在随机数据划分条件下进行的Spearman相关性分析,结果显示GerNA-Bind的模型置信度与结合预测性能之间存在显著的正相关关系。箱线图展示了五次随机训练与测试划分得到的统计结果,其中中位数由箱体中央线表示,四分位距对应箱体范围,须线延伸至1.5倍四分位距内的数据点。c,Robin数据集中不同子测试集上的分类性能表现。每一个子测试集由特定RNA二级结构模体与类药小分子亚结构组合定义。颜色越深表示AUROC值越高,并与更优的预测性能相关联。d,GerNA-Bind在多种RNA结构类型上的预测性能比较,包括三股螺旋、G-四链体、三向连接结构、发卡结构和假结结构。图中展示了26个RNA靶点在结合特异性预测任务中的AUROC得分。图a中的实线表示五次独立实验得到的平均AUROC,阴影区域表示对应的标准差。
2.4 GerNA-Bind在RNA结合位点识别中的优势
GerNA-Bind不仅能够预测RNA–配体的结合特异性,还能够对二者之间的相互作用进行直接建模。借助几何约束模块,模型学习得到一个用于刻画RNA–配体相互作用的成对接触矩阵(图1d)。如图所示,GerNA-Bind采用分阶段训练策略。模型首先在Robin数据集上进行为期30个epoch的预训练,该数据集包含大量随机配对的RNA–配体样本,并标注了结合与不结合标签,但缺乏实验解析的结构信息。这一阶段使模型能够学习通用的结合特征和区分模式。随后,模型在Hariboss数据集上进行微调,该数据集提供了359个经实验验证的RNA–配体晶体结构,具有高质量的结构注释。在微调过程中,发布时间不晚于2020年的318个复合物用于训练,而2021年之后发布的19个和22个复合物分别用于验证和测试。
在该设置下,RNA–配体接触矩阵中的每一个元素表示RNA中某一碱基与类药小分子中某一原子之间的最小原子距离。训练阶段中,这些距离通过三档阈值策略转化为软相互作用标签:距离小于3.5Å赋值为1.0,小于5Å赋值为0.6,小于10Å赋值为0.2。在评估阶段,接触标签则基于5Å阈值进行二值化处理。为获得碱基层面的相互作用评分,对每一个RNA碱基选取其与所有配体原子之间预测接触值的最大值。基于这一处理方式,可以计算AUROC和AUPRC等分类指标,从而量化模型区分相互作用区域与非相互作用区域的能力。此外,成对接触矩阵还被用于估计不同RNA靶点与类药分子之间的结合亲和力变化,进一步提升整体预测性能。
该模型在RNA结合位点预测任务中与多种主流结构预测框架进行了对比评估,包括RNASite、Chai-1和AlphaFold3,评估指标涵盖召回率、精确率、F1值、AUPRC以及Matthew相关系数(图4a)。如图所示,GerNA-Bind在所有指标上均显著优于对比方法,相较于AlphaFold3,其精确率提升了20.8%,F1值提升了13%,AUPRC提升了10.6%,Matthew相关系数提升了15.6%,显示出在RNA结合位点预测方面的卓越精度。为进一步评估模型的鲁棒性,研究还考察了在不进行Hariboss数据集微调以及移除几何约束模块情况下的结合位点预测性能(图4a)。结果表明,在缺乏任务特异性监督时,模型仍具备一定的结合位点预测能力,但整体性能受限,主要原因在于可用于训练的真实复合物结构数据较少。在引入Hariboss数据集微调并结合几何约束模块后,模型在结合位点预测任务中的AUROC由0.557提升至0.808。这表明几何约束模块与结构数据微调的协同作用对于提升RNA结合位点预测性能至关重要。通过在微调阶段引入RNA–配体距离映射并强化空间几何约束,模型能够有效捕捉RNA–配体相互作用中的关键几何特征。为进一步验证模型的泛化能力,研究构建了一个新的独立测试集,筛选标准包括复合物发布时间晚于2023年1月,且与Hariboss训练集的序列相似性严格低于0.8。结果显示,GerNA-Bind在该独立测试集上仍保持了良好的预测性能,验证了其在结合位点识别任务中的稳健性。相关PDB编号信息在补充说明中给出。此外,研究还对测试集中的22种RNA结构进行了分类分析,结果表明GerNA-Bind在五类RNA结构中有四类取得了最高的AUROC得分(图4b),进一步凸显了其在不同RNA结构背景下的广泛适用性和预测精度。结合位点预测结果通过对预测相互作用矩阵中每个RNA碱基的最大值进行可视化,表示该碱基成为结合位点的可能性,并使用ChimeraX进行渲染。来自不同结构簇的五个RNA–配体复合物示例进一步表明,该模型能够在多样化RNA构象中准确识别结合靶点(图4c)。值得注意的是,预测得到的结合位点能够与小分子形成有效的氢键相互作用,这是RNA–配体相互作用中最主要的作用类型之一(图4d)。由于现有结构注释中堆叠相互作用的代表性较低,且仅依赖距离几何标准难以准确刻画该类作用,当前建模框架未显式引入堆叠相互作用,其在RNA–配体结合中的潜在贡献可能因此被低估。总体而言,上述结果确立了GerNA-Bind作为一种在RNA–配体结合位点精确识别方面具有领先优势的前沿工具。
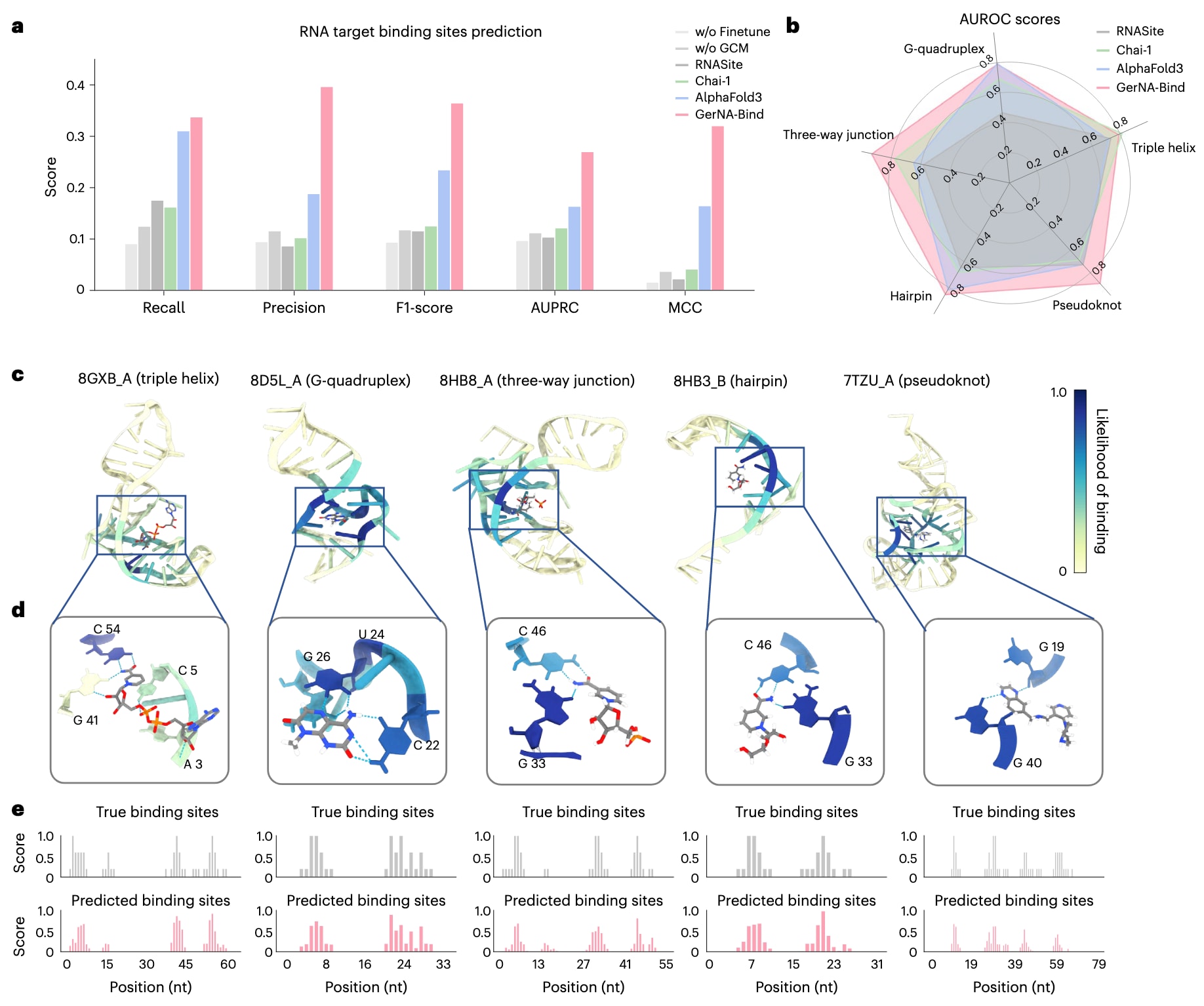
**图4|GerNA-Bind在RNA结合位点识别任务中的验证结果。**a,GerNA-Bind在RNA靶点结合位点预测中的整体性能评估结果。与RNASite、Chai-1和AlphaFold3等模型相比,GerNA-Bind表现出显著优势,预测性能明显更高。GCM表示几何约束模块。b,不同RNA结构类型下的结合位点预测AUROC得分,涵盖三股螺旋、G-四链体、三向连接结构、发卡结构和假结结构。结果表明,GerNA-Bind能够在多种RNA结构背景下稳定地预测结合位点。c,对应上述五类RNA结构的具体示例展示。图中颜色越深表示模型预测该碱基成为结合位点的可能性越高,直观呈现了结合位点在RNA结构中的空间分布。d,通过对上述实例中局部结构的分析可以看出,模型预测的RNA结合位点能够与小分子形成氢键相互作用,这是RNA–配体相互作用中最为常见且关键的作用形式之一。e,真实结合位点与模型预测结合位点分布的直方图对比结果,进一步验证了GerNA-Bind在识别RNA相互作用热点区域方面的准确性。
2.5 利用GerNA-Bind对MALAT1三股螺旋结构进行虚拟筛选
RNA靶向药物发现中的一个关键难题在于获得既具备良好疗效又具有高度选择性的小分子。基于这一背景,研究认为GerNA-Bind能够在无需依赖实验结构数据的前提下,以远高于传统物理方法的效率评估所设计活性位点的准确性与预组织状态,从而为小分子与RNA相互作用的分析提供新的手段。
为验证该方法的实际效果,研究围绕一个具有明确治疗相关性的RNA靶点开展了大规模虚拟筛选,即致癌相关的转移相关肺腺癌转录本1号长链非编码RNA MALAT1。MALAT1在多种癌症的转移和肿瘤生长过程中发挥重要作用,通过不同策略降低其水平可产生显著的抗增殖效应,因此被视为潜在的抗癌治疗靶点。MALAT1靶向药物开发中的主要挑战在于发现能够选择性结合其三股螺旋结构的小分子。该结构通过保护转录本免于降解而促进MALAT1的累积,且MALAT1高表达与多种癌症密切相关。此前仅有一项研究报道过一种具有二苯并呋喃结构的小分子可作用于该结构,可见相关化学空间仍有待系统探索。
筛选流程首先基于RNA靶向小分子数据集中识别出的常见分子亚结构,构建了一种亚结构匹配算法。该数据集整合了Robin、Biosensor和Hariboss数据集中具有活性的RNA结合小分子。利用这一策略,研究从包含8,357,246种化合物的商业化合物库Topscience refine set中剔除了绝大多数不具备相关亚结构的分子,最终构建了一个包含21,659种化合物的RNA靶向聚焦化合物库。随后,采用GerNA-Bind的预测集成模型筛选可能结合三股螺旋结构的分子,筛选标准为预测得分大于0.8且不确定性小于0.15,同时筛选预测为不与茎环结构结合的分子,其标准为预测得分小于0.2且不确定性小于0.15。为避免潜在的数据泄漏,研究进一步基于Tanimoto相似性阈值小于0.6剔除了与已知活性分子高度相似的化合物,并排除了与训练集中RNA序列同源性超过80%的样本。经过上述筛选,候选分子数量分别缩减至140种和49种。随后,研究基于分子相似性对化合物进行聚类,并从各簇中选取预测排名最高且相互作用模式和几何构型合理的分子,最终获得28种具有潜力的候选化合物(图5a)。
为评估这28种候选分子与MALAT1 RNA的结合能力,研究采用了噻唑橙TO置换实验。在进行TO置换实验之前,通过圆二色谱确认实验体系中MALAT1三股螺旋结构的完整性。已报道的MALAT1配体Compound 16作为阳性对照。在TO置换实验中,TO与RNA结合后产生荧光信号,当小分子与RNA竞争结合位点时,荧光强度随之降低,置换比例越高表明小分子对RNA的结合亲和力越强。实验结果显示,在28种候选分子中,有64%的化合物能够置换超过50%的TO,表明其对MALAT1 RNA具有较强结合能力。其中,此前未报道的Mol14、Mol10和Mol6的置换率分别达到80.9%、69.8%和69.3%,均高于阳性对照的68.4%。基于上述结果,结合能力最强的Mol14作为代表性化合物被选用于后续深入研究。
为评估这些命中分子的结构新颖性,研究将其与既往报道的30种MALAT1结合分子进行了对比分析。结果表明,新发现的化合物与已知配体之间的Tanimoto相似性多处于0.17至0.36之间,其中Mol14仅为0.17。这一结果表明,GerNA-Bind不仅能够识别已知的RNA结合结构特征,如芳香性和杂环骨架,还能够推广发现新的化学类型,例如噻嗪类化合物。
随后,研究选取多种与疾病相关的RNA,包括miR-17、miR-20a、miR-18a、HOTAIR、Pre-miR-21和QSOX-1,用于评估Mol14对MALAT1的选择性。结合亲和力通过竞争性置换值DC50进行表征,该值表示实现50% TO置换所需的小分子浓度。结果显示(图5c),Mol14对MALAT1具有高度选择性,其DC50为58nM,而对其余六种RNA的结合能力显著较弱,DC50均大于10μM。
鉴于Mol14作为MALAT1选择性配体的突出表现,研究进一步分析了其与MALAT1 RNA的结合模式。通过突变构成三股螺旋的碱基以形成不具备三股螺旋结构的茎环RNA,并比较Mol14在两种结构中的结合能力,结果发现Mol14与茎环结构的结合极弱,表明其主要作用位点位于三股螺旋区域(图5d)。进一步测试发现,Mol14几乎不与富含U•A–U的经典RNA三股螺旋结构结合,说明其识别并非RNA三股螺旋的普遍特征。MALAT1中48和49位点突出的鸟嘌呤以及周围的三股螺旋构型共同形成了一个独特的结合口袋(图5e)。结构分析表明(图5f),Mol14能够与U7、U47和A66形成氢键,这些碱基直接参与MALAT1三股螺旋结构的稳定,且GerNA-Bind准确预测了U7和A66作为关键结合位点。这些结果不仅解释了当上述位点突变破坏三股螺旋结构时结合亲和力显著下降的原因,也从结构层面揭示了Mol14对MALAT1三股螺旋区域高度特异性的结合机制。
上述结果清楚地表明Mol14能够选择性结合MALAT1。鉴于靶向MALAT1的小分子可能通过破坏RNA结构导致其在细胞内水平下降,研究进一步通过实时定量PCR检测Mol14对A549肺腺癌细胞中MALAT1水平的影响。实验结果显示,Mol14能够以剂量依赖的方式降低MALAT1的mRNA水平。同时,细胞迁移实验表明,在Mol14处理后细胞迁移速率显著下降。由于MALAT1在肿瘤细胞迁移中具有重要作用,这一现象进一步说明Mol14通过影响RNA水平发挥功能效应。与阳性对照相比,Mol14在降低MALAT1表达和抑制细胞迁移方面表现出更强作用,与TO置换实验结果保持一致。总体而言,这些研究结果验证了Mol14对MALAT1的高效且特异性结合,并进一步凸显了GerNA-Bind在RNA靶向药物发现中的应用潜力。
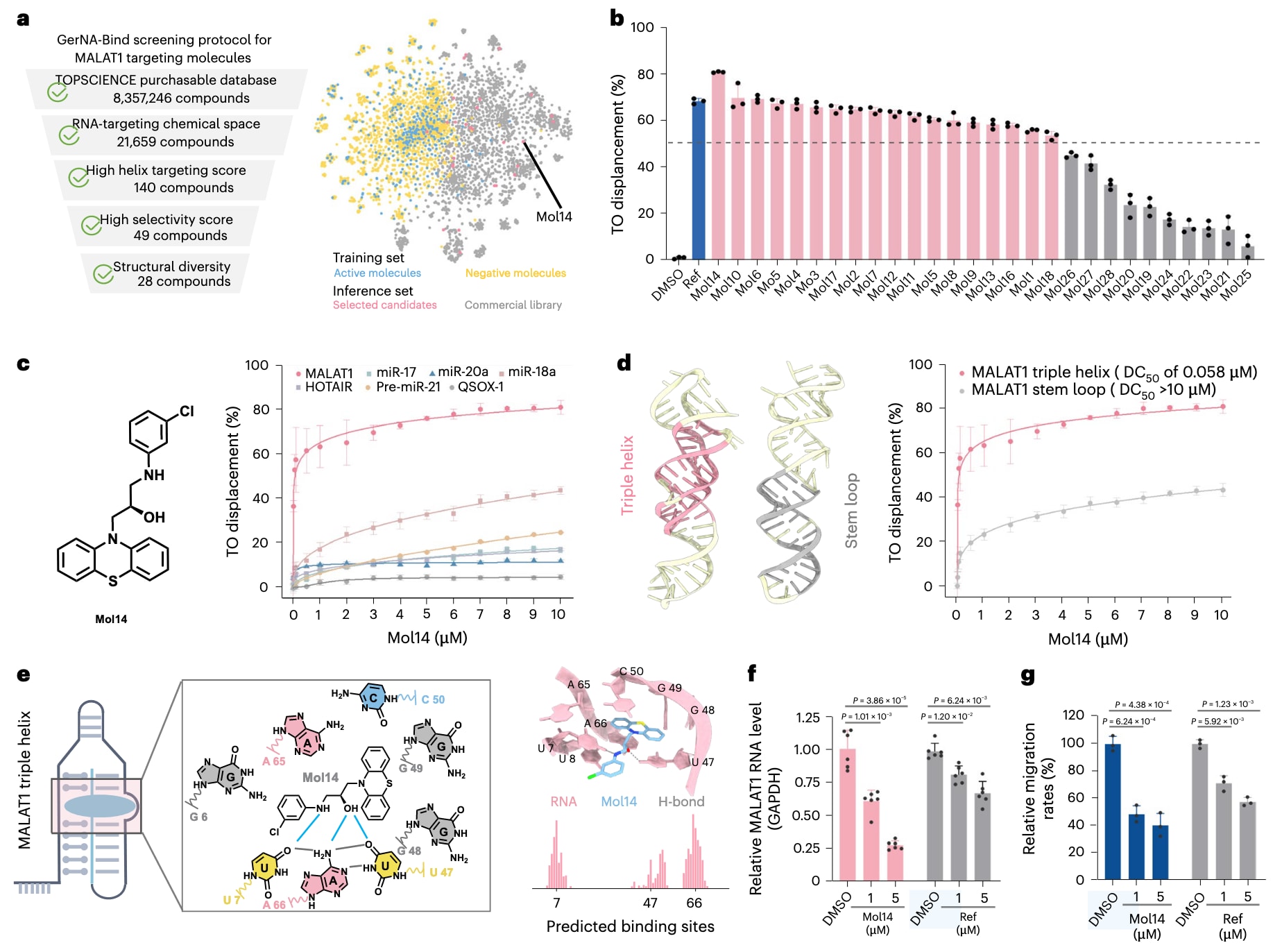
**图5|GerNA-Bind辅助RNA靶向药物发现的湿实验验证。**a,左侧展示了GerNA-Bind针对MALAT1靶点筛选候选小分子的完整流程,右侧为所筛选药物分子在化学空间中的分布示意图。b,在10μM浓度条件下,利用噻唑橙TO置换实验评估GerNA-Bind筛选得到的28种候选分子与MALAT1的结合能力。结果显示多种化合物能够有效置换TO,表明其对MALAT1具有较强的结合亲和力。c,在逐渐增加Mol14浓度的条件下,不同RNA体系中TO置换的剂量–反应曲线。该结果用于评估Mol14对不同RNA的结合选择性。d,分别以MALAT1三股螺旋结构和茎环结构为靶点,在不同Mol14浓度下测得的TO置换剂量–反应曲线,用于比较Mol14对不同RNA构象的结合偏好。e,MALAT1与小分子Mol14之间相互作用的结构分析。左侧为RNA三股螺旋束的整体结构,中间为结合口袋内RNA二级结构的局部放大图。右上角为AlphaFold3预测的复合物结构,其中RNA以红色表示,Mol14以蓝色表示,灰色虚线表示氢键相互作用。右下角为GerNA-Bind模型预测的RNA结合位点分布。f,在A549细胞中分别使用Mol14或阳性对照处理后,MALAT1 RNA水平的变化情况。g,通过划痕愈合实验评估Mol14处理后细胞迁移能力的变化。数据以三次独立实验的均值±标准差表示,统计显著性采用单侧t检验评估,图中标注了具体的P值。DMSO表示二甲基亚砜。
3 讨论
GerNA-Bind旨在对RNA–配体相互作用进行建模并预测其结合特异性,从而加速RNA靶向虚拟筛选和药物发现。传统方法高度依赖高通量筛选和高分辨率结构数据,不仅成本高昂,而且在不同RNA类型之间的泛化能力有限。近年来提出的深度学习方法在一定程度上提升了预测性能,但多侧重于结合结果本身,往往未能显式建模RNA–配体相互作用过程,使得预测结果的可解释性受到限制。GerNA-Bind通过在碱基与原子层面构建成对接触矩阵,在提升结合预测性能的同时,显著增强了模型的泛化能力与解释性,有效弥补了现有方法的不足。
作为一种几何深度学习框架,GerNA-Bind整合了一维序列、二维图结构和三维空间构象等多状态RNA–配体表示,并引入基于证据的学习策略与不确定性估计模块,从而提升预测结果的准确性、可靠性和可解释性。在计算层面,该方法在公开数据集上的RNA–配体结合特异性预测任务中取得了当前最优性能,并进一步扩展至RNA结合位点的精确预测。在实验层面,通过湿实验验证,GerNA-Bind成功识别出18种结构多样的候选小分子以及一种能够选择性靶向致癌相关MALAT1三股螺旋结构的代表性化合物,并获得了功能层面的验证结果。这些发现凸显了GerNA-Bind在加速虚拟筛选流程和推动RNA靶向药物发现方面的实际应用潜力。
尽管GerNA-Bind在RNA–配体结合特异性预测中展现出领先性能,该方法仍存在一定局限性。当前模型专注于RNA–配体相互作用,尚不支持对更复杂体系的建模,例如RNA–蛋白相互作用、多聚物之间的相互作用或含有化学修饰碱基的RNA。此外,模型性能在一定程度上受限于高通量RNA–配体相互作用数据的可获得性。未来可通过整合来自结构分析工具的更多实验数据,或借助蛋白–小分子相互作用数据集进行迁移学习,以缓解这一限制。由于GerNA-Bind依赖预测得到的RNA三级结构,结构误差在所难免,为降低其影响,模型在设计中融合了序列、二级结构和三级结构特征以增强整体鲁棒性。后续研究可进一步引入结构构象集合或置信度评分机制,以持续提升预测可靠性。此外,尽管GerNA-Bind能够提供稳健的预测结果,其应用效果在与生成式模型结合时将得到进一步增强。例如,RFDiffusionAA或AlphaFold3等模型在生成复合物结构后,仍需要进行质量筛选和特异性评分,而GerNA-Bind在这一过程中可发挥重要作用。未来的研究方向还包括将该框架扩展至其他大分子相互作用体系,整合更多实验与计算数据,并优化其与生成式建模框架的协同方式,以进一步推动RNA靶向药物发现的发展。目前,RhoFold仅能输出单一的静态三维构象,而在相互作用建模中引入RNA构象柔性仍是一个关键且具有挑战性的研究方向。
总体而言,GerNA-Bind构建了一个稳健的RNA–配体结合特异性预测计算框架,有效应对了RNA靶向治疗中虚拟筛选和候选分子选择所面临的重要挑战。其多模态架构将实验解析与计算预测的RNA结构有机结合,在保证预测精度的同时兼顾可解释性和泛化能力。随着生成式建模方法和多大分子相互作用建模技术的不断发展,GerNA-Bind有望在计算驱动的结构生物学领域奠定基础,为疾病相关RNA靶点的治疗干预提供新的研究路径