NC 2025 | MMCLKin: 通过对比学习提升激酶抑制剂活性与选择性预测
今天介绍的是发表在Nature Communications的一项研究,其核心成果为提出MMCLKin这一全新的激酶抑制剂活性与选择性预测框架。激酶因结构高度保守而成为药物研发中最具挑战性的靶点之一,尤其在筛选高选择性抑制剂时更是困难重重。该研究团队通过引入多模态与多尺度的对比学习策略,并结合几何图网络、蛋白语言模型以及多头注意力机制,实现了对激酶–抑制剂互作特征的深度整合与准确建模。MMCLKin在两个高质量三维激酶–药物数据集上均取得领先表现,并在多类具有结构多样性的蛋白–小分子数据集上展现出卓越的泛化能力。此外,模型兼具可解释性,能够定位关键残基与功能基团,并在后续的ADP-Glo实验中成功识别出多种能够抑制LRRK2 G2019S突变体的候选化合物。总体而言,MMCLKin为高效与高选择性激酶抑制剂的发现提供了先进而实用的计算工具。

获取详情及资源:
0 摘要
开发选择性激酶抑制剂始终面临困难,主要源于激酶结构高度保守以及全激酶组实验成本高昂,这凸显了精准预测激酶–抑制剂亲和力与特异性的必要性。此处提出的MMCLKin是一种由注意力一致性引导的对比学习框架,将几何图网络与序列网络相结合,并通过多头注意力以及多模态、多尺度对比学习实现对激酶–抑制剂活性与选择性的准确且可解释预测。MMCLKin在两个三维激酶–药物数据集上均优于已有方法,并在十个不同类型的蛋白–药物数据集与一个突变感知数据集上展现出强泛化能力,在已知与未知激酶结构上的筛选表现亦十分稳健。进一步对注意力系数的分析表明,MMCLKin能够识别决定激酶–抑制剂结合的重要残基与分子功能基团。此外,ADP-Glo实验验证显示,在MMCLKin识别的20个化合物中有5个能够抑制致病性的LRRK2 G2019S突变体,其中4个具有纳摩尔级效力。总体而言,MMCLKin展现出作为发现高效且选择性激酶抑制剂的有力工具的潜力。
1 引言
激酶在多种生命过程中发挥关键作用,其异常调控与多类进行性疾病密切相关,包括自身免疫性疾病、癌症以及神经系统疾病。因此,蛋白激酶逐渐成为本世纪最重要的药物靶点之一。然而,选择性强且高效的激酶抑制剂研发依旧面临巨大挑战,核心原因在于激酶结构具有高度进化保守性,尤其是ATP结合位点。许多在早期表现良好的化合物往往因为选择性不足导致的脱靶效应而在临床前或临床试验中失败。虽然体内外激酶组测定能够提供跨全激酶组的多维结构–活性信息,但其成本高昂且实验劳动量巨大,使其通常只能用于评估极少量化合物。因此,构建精准的全激酶组生物活性预测方法对于发现同时具备高选择性与高亲和力的激酶抑制剂具有重要意义。
在当前研究中,深度学习方法在预测激酶–抑制剂亲和力方面愈发受到重视,其分子表征主要分为基于序列和基于图的两类。序列方法依赖SMILES表示药物并使用激酶序列,这些数据具有易得且丰富的优势。然而由于深度学习本质上依赖模式匹配,序列方法常因过大的自由度而产生错误的训练模式,从而引发过拟合。与此相比,图方法可将生物分子表示为二维或三维图,二维图刻画原子特征、键连接与分子邻接关系,而三维图则通过融入空间构象特征更贴近真实的分子结构。尽管如此,三维激酶结构的获取成本极高,样本数量因此受限而影响模型表现,同时图结构的固有稀疏性也可能妨碍模型捕获复杂的分子关系。
另一方面,激酶–药物亲和力预测方法亦可按照互作粒度划分为全局互作方法与局部互作方法。全局方法强调药物特征、完整的蛋白激酶信息以及二者之间整体的作用,若结合三维图表示可更充分捕获三维空间相互作用信息,但也可能引入大量噪声,从而削弱对关键识别因素的关注。局部方法则聚焦于关键结合位点及其与药物之间的局部互作,但过分强调局部可能忽略诸如蛋白折叠状态等与激酶功能密切相关的全局结构特征。
基于上述不同表征与粒度之间的互补性,提出了一种节点级的多模态、多尺度注意力一致性对比学习方法MMCLAC,以整合多种异质信息,并充分考虑序列与图表征的差异性以及局部互作嵌套于全局结构中的层级特征。该方法建立在一个前提之上,即每个化合物–激酶系统都是客观存在的实体,其内部蕴含自身的交互信息以及结构化且可学习的注意力分布。MMCLAC通过构建层级式对比学习框架,对来自序列与三维图两类模态以及局部原子级与全局结构级两种尺度的注意力系数进行对比,以同时捕获激酶与药物的序列信息、上下文特征与空间结构特征,并减轻激酶序列高度自由度与图结构稀疏性对模型性能的负面影响。局部与全局互作注意力的对比进一步促使模型在保持整体激酶结构认知的同时,更加聚焦于关键的药物–口袋互作。值得注意的是,MMCLAC通过节点级对比学习强制保持注意力一致性,不仅对齐同一体系中不同模态与尺度的注意力分布,也提升了区分不同体系之间细微差异的能力,从而增强激酶–药物互作特异性与选择性的捕获效果。
基于MMCLAC方法,构建了完整的MMCLKin框架,旨在通过有效整合多模态与多尺度互作信息提升激酶抑制剂选择性与亲和力的预测能力。首先构建了两个高质量的三维激酶–药物数据集,以降低噪声并精确反映结构特征。随后,MMCLKin结合几何图网络与基于大语言模型的序列网络,用以捕获蛋白激酶的空间结构与进化特征、激酶抑制剂的三维构象与化学特征,以及二者的局部和全局互作,并通过多头注意力机制量化激酶–药物结合中各组成部分的注意力分布。在注意力一致性原则指导下,MMCLKin利用MMCLAC方法进一步提炼同一系统中不同模态与尺度下的关键特征,同时识别不同体系之间的细微差异。
在两个构建的数据集上采用三种划分策略进行评估,并扩展至十个结构多样的蛋白–药物数据集及一个突变感知数据集。结果显示,MMCLKin在激酶抑制剂选择性与亲和力预测上始终保持高精度,并在更广泛的蛋白–药物互作场景中展现出强泛化能力。随后从三类情形进一步评估其性能:具有已解析结构的激酶、缺乏结构数据的激酶以及特定突变激酶。无论激酶结构是否已知或存在突变,MMCLKin均展现出可靠的虚拟筛选能力与预测精度,体现出方法的稳健性、泛化性与灵活性。
ADP-Glo实验进一步验证了这一能力。在MMCLKin筛选出的20个化合物中,有5个能够有效抑制致病性的LRRK2 G2019S突变体,其IC50分别为468 nM、2.081 nM、1384 nM、8.694 nM、130.3 nM。可视化分析显示,MMCLKin能够识别关键互作特征,包括与特异性结合密切相关的激酶hinge区残基,以及能够与口袋残基产生极性互作的药物中的极性原子或功能基团。
总体而言,MMCLKin不仅展现出强大的预测能力,并且由于不依赖晶体结构,在实验结构数据有限的情况下对其他保守蛋白家族与突变激酶亦具有广阔的应用前景。
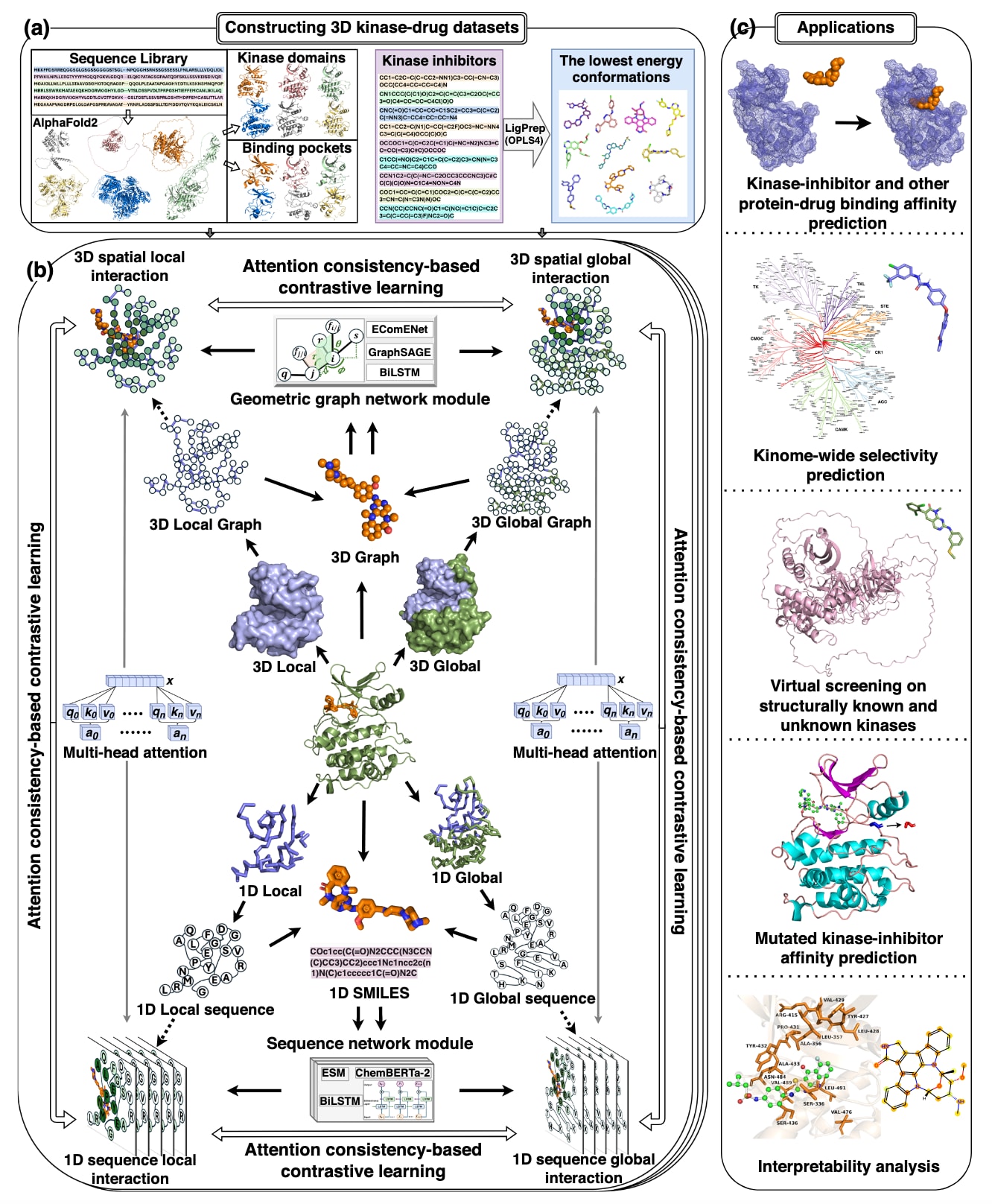
图1|展示了MMCLKin的整体框架。 首先在部分a中,通过从AlphaFold2预测的三维结构中提取高置信度的激酶结构域与关键结合口袋,并结合OPLS4力场下LigPrep模块生成的小分子最低能量构象,构建了两个三维激酶–抑制剂数据集。部分b中,MMCLKin利用几何图网络与序列网络模块,分别从激酶–药物的三维结构与一维序列中提取局部与全局的互作特征,随后借助多头注意力机制更全面地学习激酶–药物之间复杂的互作模式。最终,模型融入多模态与多尺度的注意力一致性对比学习方法MMCLAC,以有效融合并学习这些互作特征,从而实现精确预测。部分c展示了MMCLKin在四类不同应用场景中的表现以及相关的可解释性分析。
2 结果
2.1 MMCLKin的整体框架
MMCLKin是一种深度学习模型,旨在通过跨模态、跨尺度提取并整合激酶与抑制剂之间的关键信息,从而精确预测其选择性与活性。为减少数据噪声,构建了两个高质量的三维激酶–药物数据集3DKDavis与3DKKIBA,其中激酶结构来源于AlphaFold2预测的高置信激酶结构域与关键结合位点,小分子构象通过LigPrep生成最低能量构象。随后,框架采用几何图网络模块以捕获激酶的空间结构特征以及小分子的三维构象特征,该模块同时兼顾局部结构的完整性与全局空间布局。与此并行的序列网络模块基于蛋白语言模型与化学语言模型,用于抽取激酶序列中的进化信息与小分子中的化学细节特征。
接下来,MMCLKin利用多头注意力机制自动学习激酶–抑制剂跨不同范围的依赖关系,并量化系统中各组成成分对预测任务的贡献。与此同时,提出的MMCLAC方法通过在节点级别对齐空间结构、序列、局部互作与全局互作四类特征的注意力分布,确保信息整合的充分性与有效性。这一机制不仅使模型能够全面捕获复合物中的重要互作特征,也能够区分不同激酶–抑制剂系统中的结合模式差异,从而增强模型的可解释性与泛化能力。
在最终的预测阶段,MMCLKin整合不同模态与不同尺度的互作信息以生成预测结果。在多种应用场景中,MMCLKin均展现出具有竞争力的表现,包括激酶–抑制剂以及其他蛋白–药物的亲和力预测、激酶抑制剂选择性分析、对已知结构、未知结构与突变激酶的虚拟筛选以及可解释性分析。ADP-Glo实验测试进一步验证了模型的筛选能力,在MMCLKin筛选出的20个化合物中有5个能够有效抑制LRRK2 G2019S突变体,其中4个达到纳摩尔级别的抑制活性。这些结果表明MMCLKin具备较高的预测精度,尤其在处理结构未解析的激酶以及临床相关突变方面表现出显著潜力。
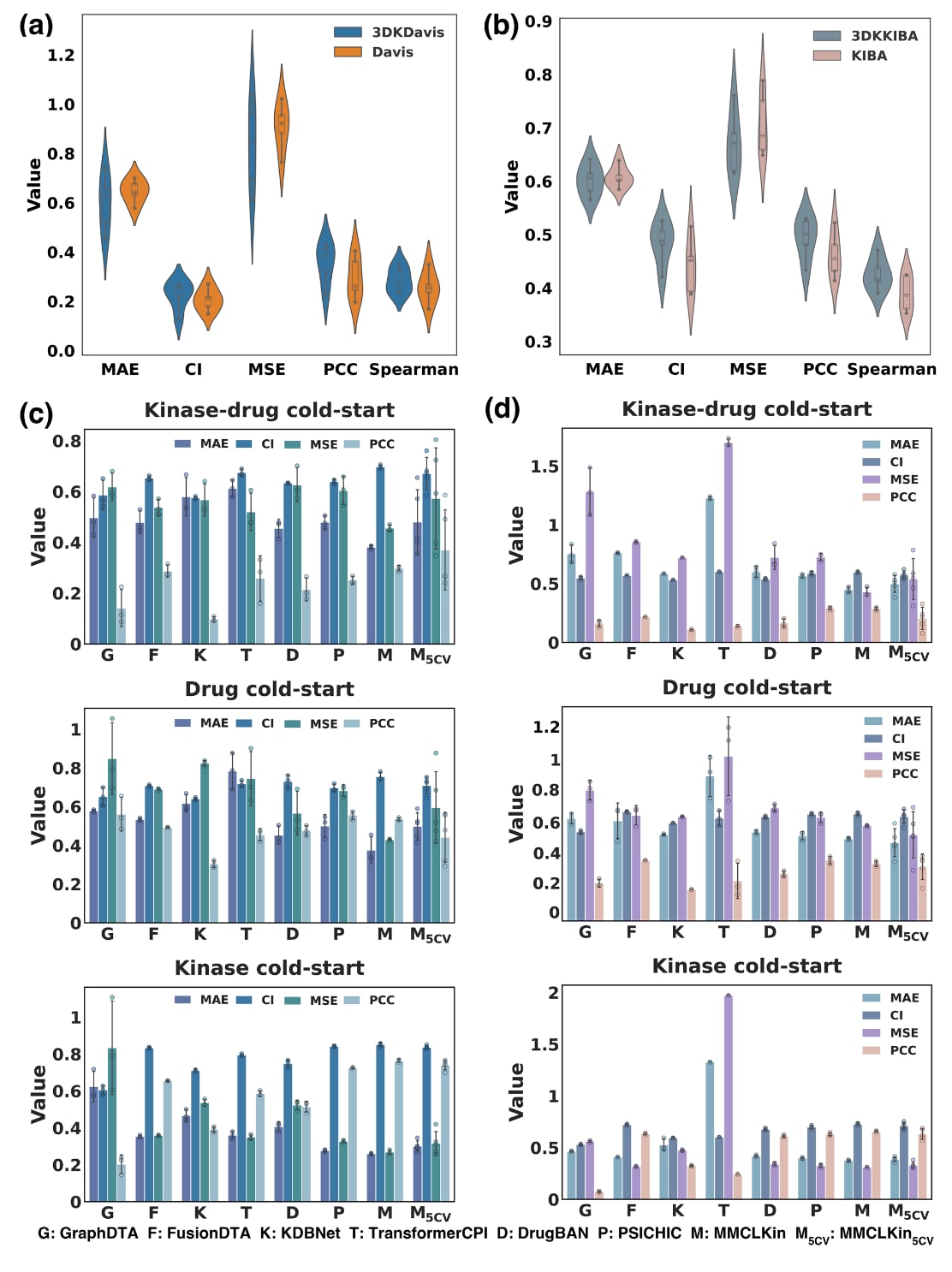
图2|展示了MMCLKin在两个构建的三维数据集上,在药物冷启动、激酶冷启动以及激酶–药物冷启动三种划分策略下的亲和力预测表现。 部分a比较了ConPLex在构建的3DKDavis数据集与原始Davis数据集上的表现,每种方法均进行了五次独立重复(n=5)。箱线图以中位数为中心线,箱体上缘与下缘表示四分位数范围,须线对应最大值与最小值,散点表示各次独立运行的数据点。部分b呈现了ConPLex在构建的3DKKIBA数据集与原始KIBA数据集上的性能对比,同样基于五次独立重复,n=5,箱线图元素的含义一致。部分c比较了MMCLKin与其他模型在3DKDavis数据集的三种划分策略下的激酶–药物亲和力预测性能,每种方法均进行了三次独立重复(n=3),结果以均值±标准差形式给出。部分d进一步展示了在低药物相似度的LSKIBA数据集上,MMCLKin与其他模型在三种划分方式下的预测表现,同样基于三次独立重复(n=3),以均值±标准差表达。所有模型均通过多个性能指标进行严格评估,包括CI、MAE、PCC、MSE与Spearman系数。源数据已作为Source Data文件提供。
2.2 MMCLKin在激酶–药物亲和力预测中展现稳健表现
首先利用仅包含激酶序列与药物SMILES的原始Davis与KIBA数据集,通过序列模型ConPLex对比评估构建的3DKDavis与3DKKIBA数据集的实用性与质量。ConPLex基于预训练蛋白语言模型ProtBert对蛋白序列进行编码,并使用分子指纹表示药物,随后通过protein-anchor对比共嵌入策略将蛋白与药物映射至共享潜在空间,从而区分真实互作与非互作样本。图2a与2b展示了ConPLex在四个数据集上采用激酶–药物冷启动划分进行五次独立运行后的结果分布。结果显示,与原始数据集相比,ConPLex在3DKKIBA与3DKDavis上的五个指标均得到提升,尤其是在CI、PCC与Spearman系数上的显著改善,说明构建的数据集能够帮助模型更好捕获激酶–抑制剂的关键互作信息。
随后对MMCLKin在这两个构建数据集上的亲和力预测性能进行评估,并采用三种数据划分方式。比较对象涵盖了来自三类输入模态的六种代表性基线模型,包括基于序列的TransformerCPI、FusionDTA、PSICHIC,基于二维图的GraphDTA与DrugBAN,以及基于三维结构的KDBNet。GraphDTA是药物–靶点亲和力预测的经典基线;TransformerCPI以抗数据偏差能力与可解释性见长;FusionDTA利用全局序列特征,在Davis与KIBA上均表现出较高准确性;KDBNet通过几何图网络刻画激酶–药物局部空间与拓扑结构;DrugBAN通过条件域对抗学习对齐异质数据中的互作表示,增强对新药物–新靶点的泛化能力;PSICHIC则整合结构约束以捕获蛋白–药物结合的物化机制。所有基线模型的细节见补充材料。
针对MMCLKin与对比模型的超参数进行了多轮调优,并基于三次独立运行的均值、标准差与分布进行比较。在3DKDavis数据集中,MMCLKin在激酶冷启动与激酶–药物冷启动下,在四项指标上均显著优于所有对比模型且具有更低的预测波动。在激酶冷启动下,MMCLKin相较表现最佳的序列方法PSICHIC,实现了17.74%的MSE降低(0.269对0.327)与6.14%的MAE降低(0.260对0.277)。在更具挑战性的激酶–药物冷启动中,MMCLKin的MAE相较二维图基线DrugBAN降低了16.26%(0.381对0.455)。在药物冷启动划分中,MMCLKin同样在MAE、MSE与CI上取得最佳表现,MSE与MAE较DrugBAN分别减少0.137与0.077。
为严格评估泛化能力,构建了LSKIBA基准集,这是由3DKKIBA中Tanimoto相似度低于0.4的化合物对组成的低相似度子集,旨在确保训练与测试化合物在结构上的显著差异。在图2d中,MMCLKin在三种划分策略下的MAE与MSE均优于其他方法。在激酶冷启动中,MSE与MAE分别达到0.310与0.376;在药物冷启动中,相较三维几何基线KDBNet的MSE降低了8.42%。同时,MMCLKin在激酶冷启动中取得最高的PCC与CI,在另外两种划分中也保持了可比表现。
进一步使用五折交叉验证评估模型稳健性。在3DKDavis的激酶–药物冷启动划分中,五折版本MMCLKin5CV的PCC相比MMCLKin提升0.072;在LSKIBA的药物冷启动中,其MSE与MAE分别较MMCLKin降低0.057与0.024。尽管MMCLKin5CV在部分指标上未超过MMCLKin,但全面超越所有基线模型。例如在LSKIBA中,MMCLKin5CV在激酶与激酶–药物冷启动划分下的MAE均低于所有对比模型;在3DKDavis的激酶冷启动中其MSE与PCC也优于所有基线。
随后在3DKDavis上量化了MMCLKin在三种划分下的预测不确定性,并分析其与MAE的Spearman相关性及其校准性能。Spearman系数越高表明不确定性与预测误差的匹配度越高,校准曲线越接近理想对角线则说明置信输出越可靠。结果显示,在激酶冷启动(ρSpearman=0.786)与激酶–药物冷启动(ρSpearman=0.675)中均呈显著正相关,尤其在低不确定性区间中,不确定性较低对应更高预测准确性,校准表现也较佳。然而在药物冷启动中,相关性明显较低,且MMCLKin倾向低估真实误差,这可能与3DKDavis中药物数量有限(共68个)导致不确定性学习受限有关。
综上所述,构建的3DKDavis与3DKKIBA数据集显著提升了激酶–药物关键信息的学习效果。在这些数据集下,MMCLKin在三种数据划分中以及五折验证中均展现稳定且高精度的亲和力预测性能,体现其强泛化性与先进性。同时,相比单一模态模型,序列与三维图信息的联合输入展现明显优势;相比仅关注局部互作(KDBNet)或仅关注全局互作(DrugBAN、FusionDTA)的模型,MMCLKin联合建模局部与全局互作表现出更加全面的能力。
为系统分析不同表征的贡献,构建了四个子模型:MMCLKSeque、MMCLK3DGraph、MMCLKLocal与MMCLKGlobal,并保持主模型的架构与超参数不变。结果显示四个子模型的所有指标均不及MMCLKin,其中MMCLK3DGraph与MMCLKLocal的MAE分别上升29.76%与22.18%,MSE上升6.05%与17.09%;PCC与CI也均显著下降。此外,构建去除ESM特征的变体MMCLKNOESM,将ESM嵌入替换为简单的索引编码,其在四项指标上均全面下降,表明ESM嵌入对于捕获蛋白序列关键信息至关重要。
作为该研究的核心方法创新之一,对比分析了是否使用MMCLAC对模型性能的影响。在3DKDavis激酶–药物冷启动划分中,加入MMCLAC使MAE与MSE分别降低15.35%与12.76%,CI与PCC分别提升5.26%与17.92%。对注意力相关的对比损失进行分析后发现,不含MMCLAC的模型在四类注意力损失上波动显著,而加入MMCLAC后损失收敛性大幅提升,说明MMCLAC能够有效约束不同模态与尺度下的注意力权重,增强系统一致性。
总体来看,ESM嵌入增强了MMCLKin对蛋白序列关键信息的捕获能力;四类独立表征的整合与MMCLAC带来的跨模态、跨尺度特征融合机制共同促成了MMCLKin在激酶–药物亲和力预测上的高精度、稳定性与泛化性。各表征的差异贡献也为未来多模态、多尺度整合方法提供了有价值的思路。这些结果整体印证了模型设计的初衷。
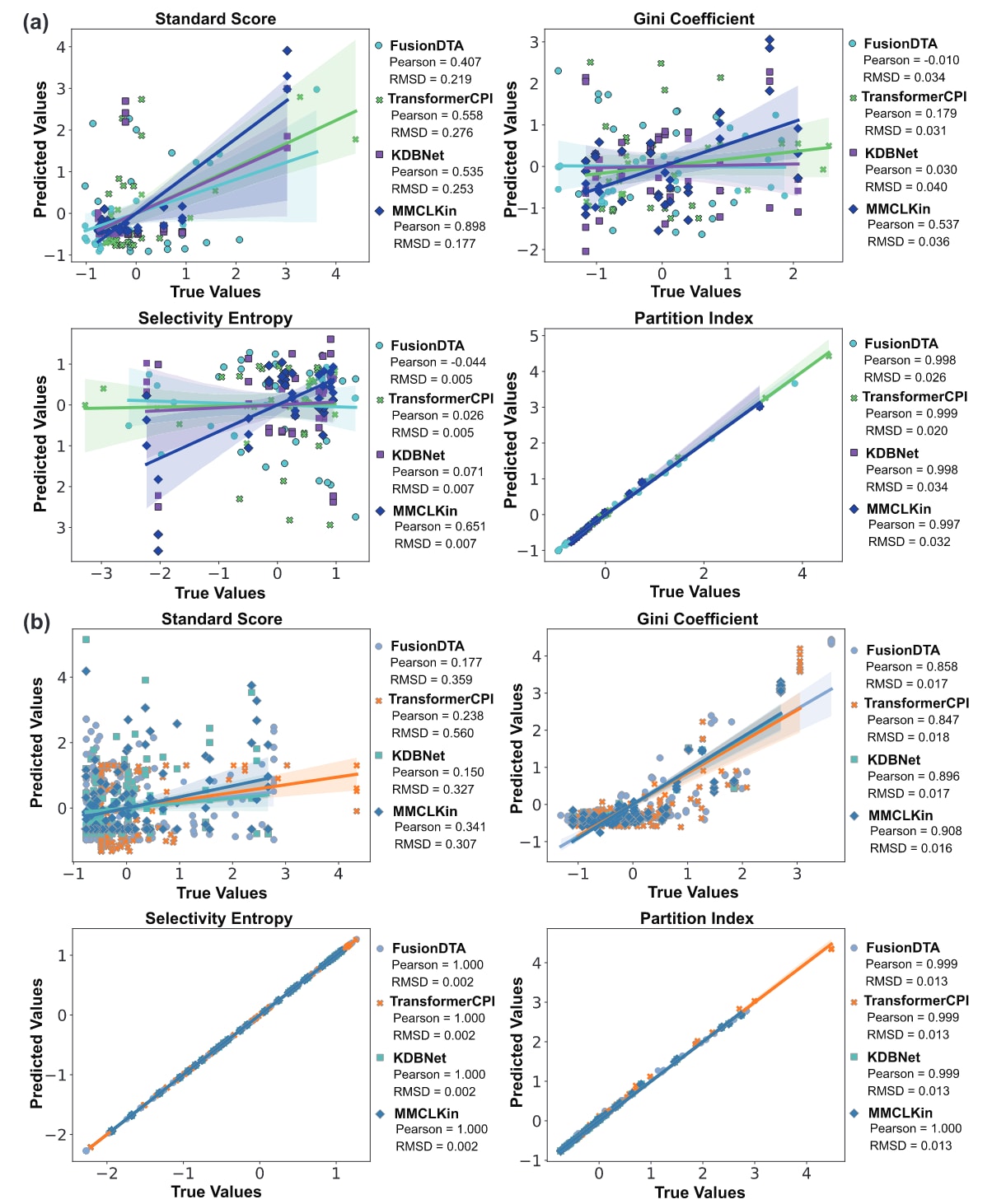
图3|展示了MMCLKin在全激酶组范围内的激酶抑制剂选择性预测表现。 部分a比较了MMCLKin与其他多种模型在3DKDavis数据集上的选择性预测性能(n=39),部分b则比较了在低药物相似度的LSKIBA数据集上的表现(n=156)。Pearson用于衡量每项指标中预测选择性分布与实验选择性分布之间的线性相关性,RMSD用于评估二者整体偏差程度。图中的阴影区域表示95%置信区间。源数据已作为Source Data文件提供。
2.3 MMCLKin在全激酶组范围内展现出良好的激酶抑制剂选择性预测能力
高选择性的激酶抑制剂通常与较低的脱靶互作高度相关,因此选择性被视为激酶靶向药物研发中的关键决定因素。基于此,进一步在3DKDavis与低相似度的LSKIBA数据集上评估了MMCLKin的激酶抑制剂选择性预测能力。为覆盖更广泛的人类激酶组,采用药物冷启动划分策略。同时引入一组常用的选择性评价指标,包括standard score、Gini系数、选择性熵与partition index(定义见补充材料),用于全面评估MMCLKin的选择性预测表现。standard score用于衡量化合物在多少个激酶上达到超过指定阈值的亲和力;Gini系数刻画化合物在不同激酶间亲和力分布的不均匀程度,Gini越高代表越倾向于少数激酶,即选择性更强;选择性熵则反映亲和力分布的离散程度,熵值越低,选择性越高;partition index基于结合常数衡量化合物相对于参考激酶的偏好性结合。
虽然这四类指标能够从全局层面对化合物的选择性进行多维度评价,但它们并不能直接反映预测选择性的准确度。因此进一步分析预测选择性与实验选择性在每个指标上的相关性,通过相关系数来更直接衡量模型的选择性预测性能。相关性越高,说明预测与实验之间的一致性越强。
图3展示了MMCLKin、FusionDTA、TransformerCPI与KDBNet在两组数据集上预测与真实选择性指标的Pearson相关性。在3DKDavis数据集上,MMCLKin在standard score、Gini系数与选择性熵方面均显著优于所有基线模型,Pearson相关系数分别达到0.898、0.537与0.651,表现出更高的预测–实验一致性。在低相似度的LSKIBA数据集中,MMCLKin在所有选择性指标上均获得最高或与最高持平的Pearson相关性,显示其在高结构差异场景下依然具备稳健的泛化能力。
值得注意的是,在partition index指标上,所有模型在两个数据集上几乎都呈现接近完美的相关性,这可能是因为该指标强调的是相对亲和力排序,而相对排序比绝对值更易被预测。同样的情况也出现在LSKIBA数据集的选择性熵上,其Pearson相关性也普遍接近1.0。然而在包含仅68个化合物的3DKDavis数据集中,选择性熵对局部预测误差更为敏感,导致不同模型间的表现差异更为明显。
总体来看,MMCLKin能够更准确地预测全激酶组范围内的激酶抑制剂选择性,这不仅有助于发现高选择性的激酶抑制剂,也间接证明了MMCLKin能够识别不同激酶靶点间细微的结合差异,从而为高选择性抑制剂的理性设计提供重要参考。此外,不同选择性指标之间的差异性也为后续选择性建模研究提供了有价值的启示,例如standard score与Gini系数对模型性能差异更为敏感,可能更适合作为评估模型选择性预测能力的指标。
表1|展示了MMCLKin与多种已报道方法在CASF-2016测试集上的性能对比。
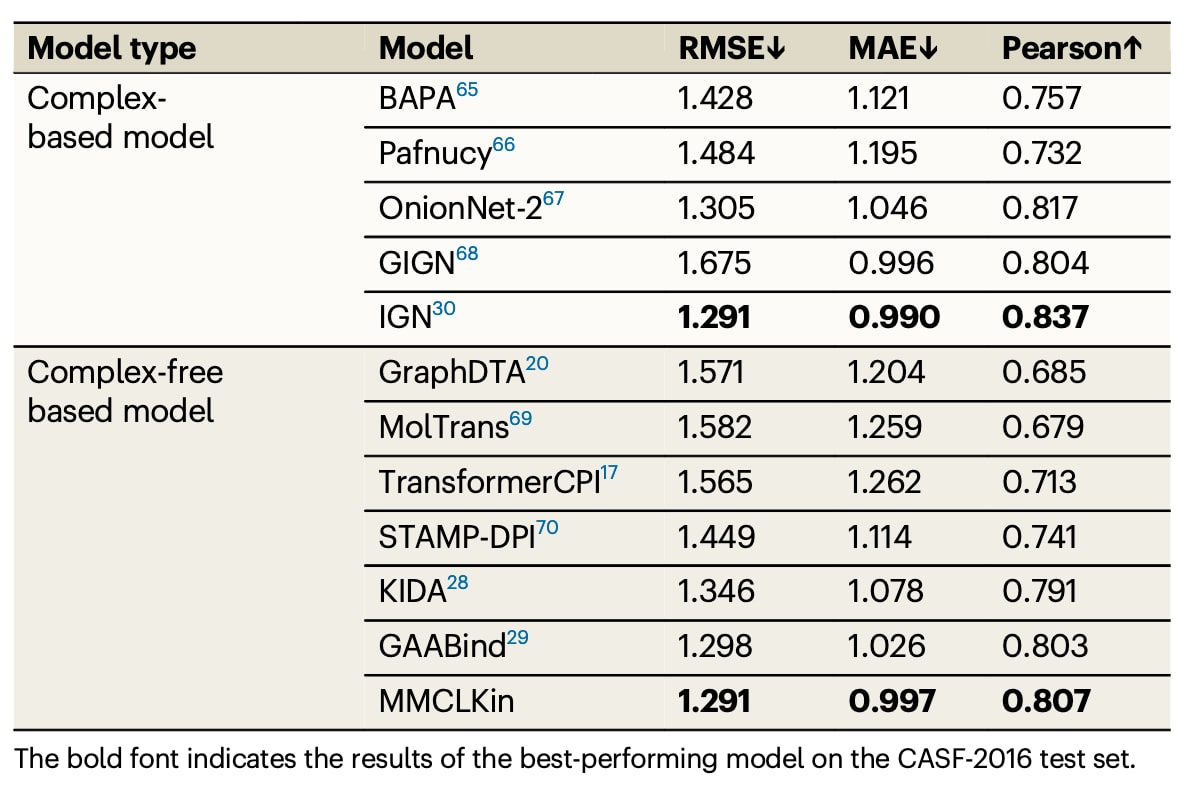
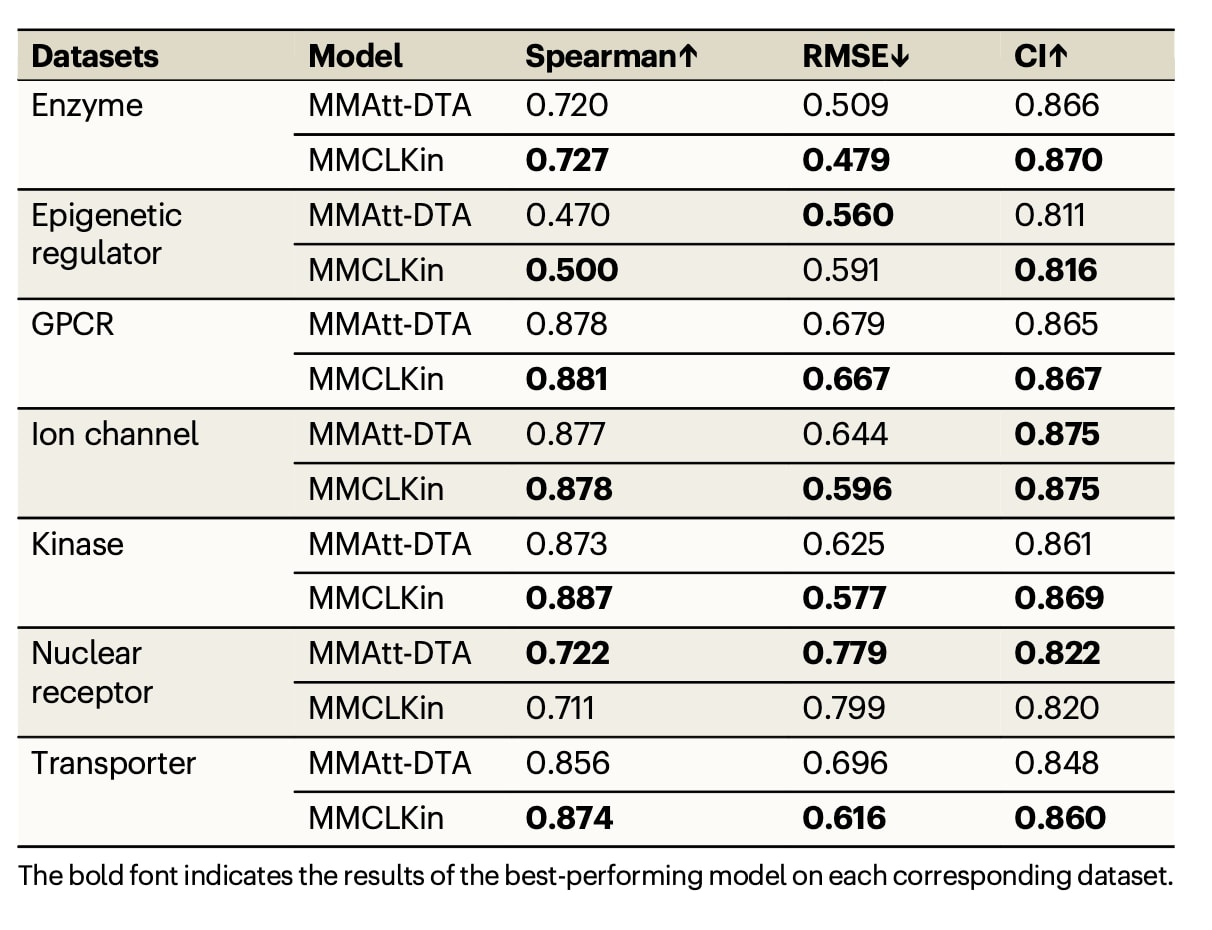
表2|展示了MMCLKin与MMAtt-DTA在七个靶点超家族上的性能对比。
2.4 MMCLKin在多样化蛋白结构中展现出良好的泛化能力
除了在保守的激酶结构上评估MMCLKin的预测性能外,还系统考察了其在十个结构多样的蛋白质数据集上的泛化能力。这些数据集包括PDBbind(PDBbind v2020与CASF-2016)、七个靶点超家族以及IDG-DREAM Drug–Kinase Binding Prediction Challenge中的两类激酶数据集。对于PDBbind数据集,以CASF-2016基准集作为测试集,并将所有与CASF-2016重叠的复合物从PDBbind v2020的general与refined集合中剔除以避免数据泄漏。随后从refined集合中随机选取500个样本作为验证集,其余样本与general集合合并组成训练集。需要指出的是,尽管MMCLKin并不依赖实验复合物结构,该研究仍将其与依赖复合物结构的模型及非结构依赖模型进行比较,以实现更全面的评估,相关对比模型的实验结果来自已发表文献。
对于其余九个数据集,蛋白三维结构与小分子构象均按照构建3DKDavis与3DKKIBA数据集所采用的流程生成。表1展示了MMCLKin与以往模型在CASF-2016测试集上的性能对比。MMCLKin在三项指标上均全面优于所有不依赖复合物结构的模型,特别是在MAE上达到0.997,比几何感知模型GAABind低0.029。同时,MMCLKin获得1.291的RMSE与0.807的PCC,优于基于序列(MolTrans、TransformerCPI)、基于图(GraphDTA、GAABind)或基于三维点云(KIDA)的模型。在与依赖复合物结构的模型比较时,MMCLKin也展现出高度竞争力,RMSE与MAE均与表现最好的模型IGN接近。
表2总结了MMCLKin与MMAtt-DTA在七个靶点超家族上的对比结果。MMCLKin在七个数据集中的六个上优于MMAtt-DTA,并在Enzyme、GPCR、Ion channel、Kinase与Transporter数据集上同时获得RMSE、CI与Spearman三项指标的最佳表现。例如在Kinase、Transporter、Enzyme与Ion channel数据集上,MMCLKin的RMSE分别较MMAtt-DTA降低7.68%、11.49%、5.89%与7.45%;在Spearman相关性上,在Kinase与Transporter数据集上分别提高0.014与0.018。此外,在Epigenetic regulator数据集上,MMCLKin也获得最高的CI与Spearman系数,Spearman的提升幅度达到6.38%。
进一步在IDG-DREAM挑战赛的Round 1与Round 2数据集中对MMCLKin进行了系统性比较。为构建更高质量的训练集,剔除了3DKDavis中所有亲和力标注为5的样本,因为此类值多作为缺乏结合证据的默认标记,而非真实的亲和力标签。结果显示,MMCLKin在两个轮次中均取得了具有竞争力的表现。在Round 1中,Spearman为0.430、RMSE为1.147,仅次于表现最好的模型。在Round 2中,其Spearman达到0.482、RMSE为1.068,与最佳模型表现相当。
这些结果说明MMCLKin在结构多样的蛋白质数据集上具有更高的预测精度与出色的泛化能力。同时,其在GPCR与Transporter等蛋白家族上依旧保持稳定的高质量预测,进一步展示其在非激酶的保守蛋白家族中的潜在适用性。更为重要的是,MMCLKin在无需依赖实验晶体结构的条件下仍能保持优秀表现,这一特性可能成为其显著优势,有望加速针对结构未解析蛋白的创新药物研发。
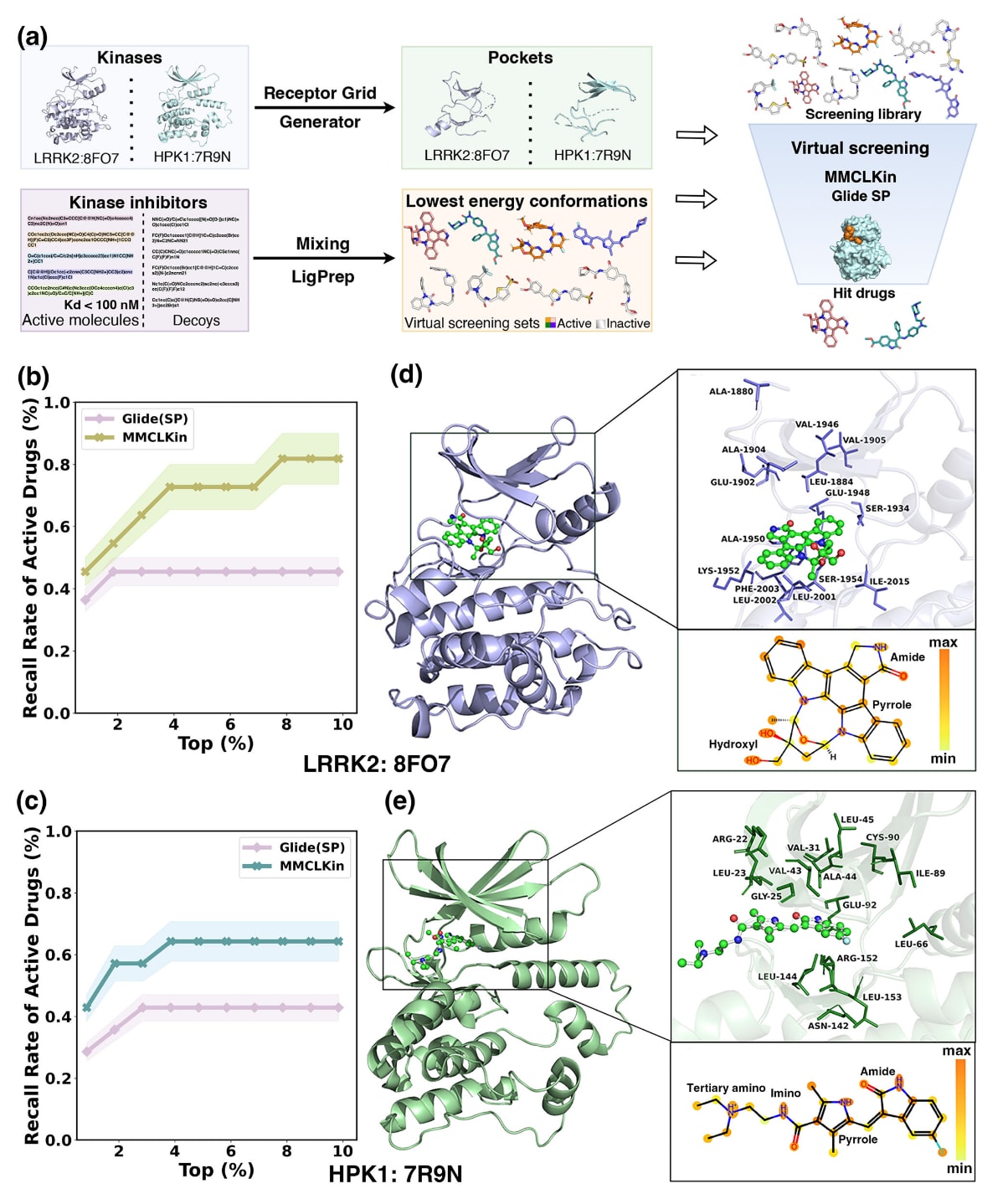
图4|展示了MMCLKin在具有实验结构的两个激酶靶点LRRK2与HPK1上的虚拟筛选表现与可解释性分析。 部分a显示了MMCLKin与Glide SP在LRRK2与HPK1上的虚拟筛选流程。部分b比较了两种方法在LRRK2(PDB ID: 8FO7)活性分子召回率方面的表现。部分c展示了它们在HPK1(PDB ID: 7R9N)上的活性分子召回率对比。部分d展示了MMCLKin在8FO7-BDBM50308060复合体系中识别的关键残基与功能基团。部分e展示了MMCLKin在7R9N-BDBM4814复合体系中识别的关键残基与功能基团。为增强结构展示的直观性,两个复合物均通过Glide SP生成。源数据已作为Source Data文件提供。
2.5 MMCLKin在具有实验三维结构的两个激酶靶点上展现出强虚拟筛选能力与良好可解释性
为验证MMCLKin在真实药物研发场景中的适用性,进一步对其在两个具有实验结构的激酶靶点(LRRK2与HPK1,详细信息见补充材料)上的虚拟筛选性能进行了系统评估。在虚拟筛选实验中,选取了在激酶冷启动划分下表现最接近平均水平的MMCLKin模型,并将其与业界广泛使用的高精度对接工具Schrödinger Glide SP(2023)进行对比。筛选所用的受体结构来自高分辨率的野生型PDB晶体结构,LRRK2使用8FO7,HPK1使用7R9N。活性化合物来源于BindingDB,筛选条件为实验测定的Kd低于100 nM,具体列表见补充表。通过DUD-E构建对应的decoy集合,并与活性分子合并形成最终筛选集。随后利用Receptor Grid Generator与LigPrep模块完成结合口袋准备以及小分子最低能量构象生成。虚拟筛选效果通过活性化合物的召回率进行评估,计算方式见补充材料。
结果清晰展示了MMCLKin相比Glide SP拥有显著优势。在LRRK2筛选中,Glide SP在排名前1%的化合物中召回36.36%的活性分子,而MMCLKin达到45.45%;在前5%与前10%的阈值下,MMCLKin的召回率分别为72.73%与81.82%,均明显高于Glide的45.45%。在HPK1筛选中,MMCLKin同样表现突出,在前1%、2%、10%中分别达到42.86%、57.14%、64.29%的召回率,显著优于Glide的28.57%、35.71%与42.86%。
为进一步研究模型的可解释性,将注意力系数映射至激酶靶点及代表性抑制剂的残基上,并以注意力权重最高的前15个残基作为展示区域。在LRRK2中,Val1946、Glu1948、Ala1950、Lys1952与Ser1954位于hinge区域,这一区域对ATP稳定结合以及多种上市激酶抑制剂的特异性至关重要。其中Glu1948与Ala1950已被证明是形成氢键的核心残基。其他残基如Glu1902、Ala1904与Val1905位于N端小结构域的β折叠区域,而Leu2001、Leu2002、Phe2003、Ile2015等则位于C端结构域的β折叠区域。在HPK1中,Glu92位于保守激酶结构域的hinge区域,Arg22、Leu23、Gly25、Val31、Val43、Ala44、Leu45、Ile89、Cys90分布在小结构域的β折叠区域,而Leu144、Asn142、Arg152、Leu153位于大结构域的β折叠区域。这些区域均被文献广泛报道为决定激酶抑制剂结合特异性与结合稳定性的关键区域,与MMCLKin识别结果高度一致。
MMCLKin同时对抑制剂中的极性基团表现出显著关注,如羟基、氨基、亚氨基、叔氨基、吡咯与酰胺基,这些功能基团普遍能够与激酶结合口袋中的残基形成强极性作用,从而提升结合亲和力与选择性。
综上所述,这些结果不仅验证了MMCLKin在虚拟筛选中的优异能力,其在识别活性激酶抑制剂方面明显优于行业领先的对接工具,还体现了其高级的可解释性。MMCLKin能够有效捕获激酶–药物系统中的关键原子、功能基团与残基,体现其基于深度学习的蛋白–配体相互作用理解能力,进一步强化其在潜在激酶抑制剂发现中的价值。
2.6 MMCLKin在缺乏实验三维结构的两个激酶靶点上仍保持良好虚拟筛选性能与可解释性
由于MMCLKin不依赖实验解析的结构,进一步在两个缺少实验三维结构的激酶靶点NUAK2与CRK12上评估其稳健性。对于这两个靶点(图5a),使用AlphaFold2预测的激酶结构域作为受体,并利用P2Rank预测活性位点中心,进而选取14 Å范围内的残基定义结合口袋。由于CRK12已知的抑制剂数量有限,根据文献报道的化合物被视为活性分子。NUAK2与CRK12的筛选集合依据LRRK2与HPK1相同的标准流程构建并处理。
结果显示,即便在没有实验解析结构的情况下,MMCLKin依然保持强劲甚至更具竞争力的筛选能力。在NUAK2筛选中(图5b),MMCLKin在排名前2%、5%与10%的化合物中分别达到了41.67%、50%与66.67%的召回率,显著高于Glide SP的25%、33.33%与41.67%。在CRK12筛选中(图5c),优势更加明显,MMCLKin在前5%与前10%阈值下分别达到53.85%与84.62%的召回率,远高于Glide SP的15.38%与23.08%。这些结果既证明了MMCLKin在不同激酶靶点上的强筛选能力,也展示了其在结构未解析激酶上的潜力。
在应用于预测结构时,MMCLKin同样表现出良好的可解释性。在NUAK2中(图5d),MMCLKin优先关注了位于hinge区域的Glu130、Tyr131、Ala132、Arg134与Asp136等残基,这些残基属于关键的前15个高注意力残基。在CRK12中(图5e),Pro431、Tyr432与Ala433等残基同样被突出识别。其他被识别的关键残基大多分布于N端与C端结构域的β折叠区域,显示MMCLKin对预测三维结构具有良好的适应能力。
对激酶抑制剂的分析表明,MMCLKin始终关注氨基、醚键、酮羰基、吡咯与酰胺等极性功能基团,这些基团易与结合口袋残基形成关键氢键或静电相互作用,从而增强激酶–药物结合的特异性与强度。
综上,MMCLKin在实验解析与预测结构的激酶上均展现出稳定且具竞争力的虚拟筛选性能;同时其能够从原始激酶–药物数据中自主捕获关键原子、功能基团与残基,体现其自学习能力与可解释性,有助于加深对模型预测行为的理解。MMCLKin在不同激酶体系中呈现的差异性识别模式进一步证明了其区分不同蛋白结构的能力,为解析特定激酶–抑制剂互作奠定了基础。综合来看,MMCLKin是面向已解析与结构未知蛋白药物研发的极具潜力的工具。
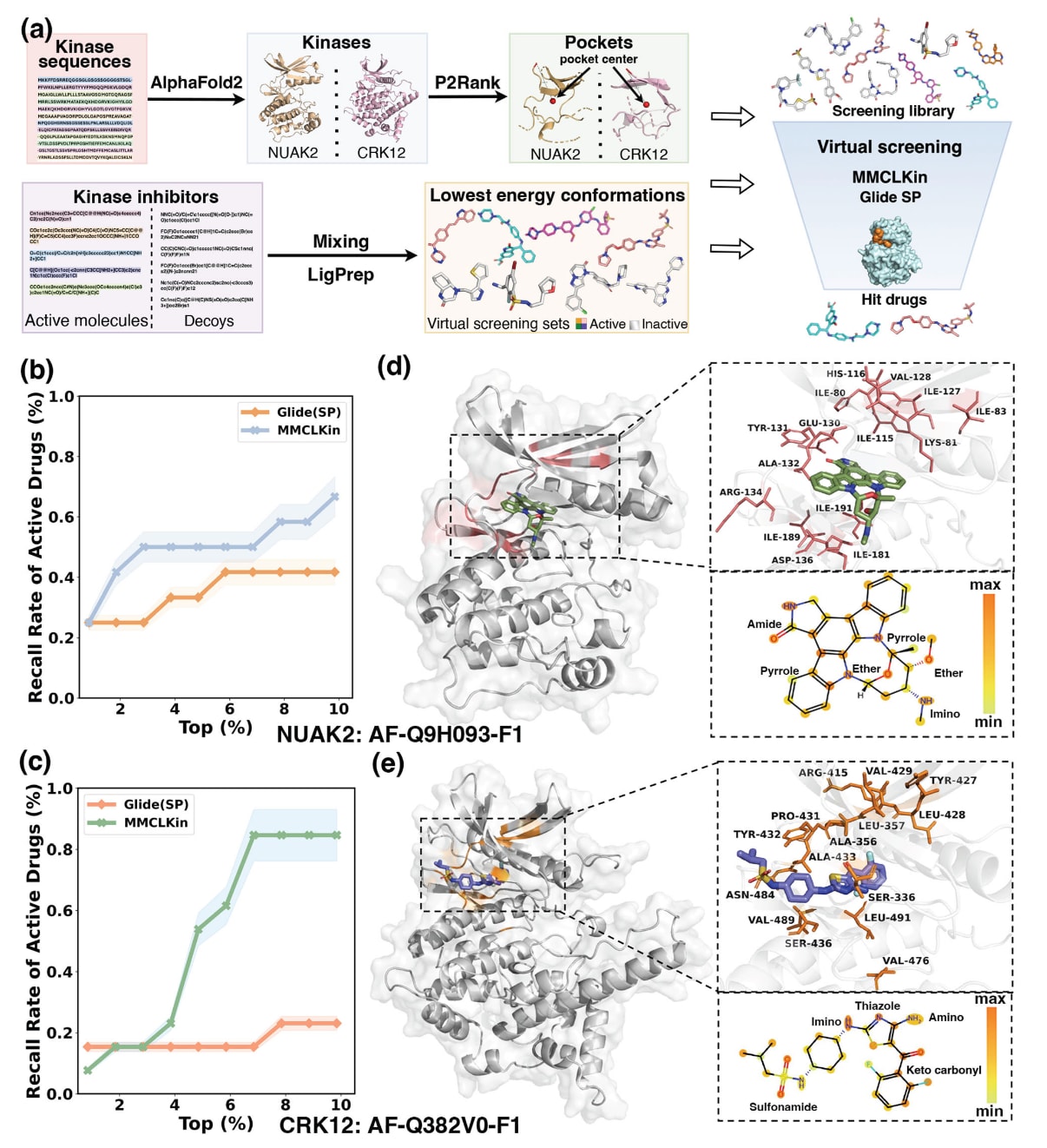
图5|展示了MMCLKin在无实验解析结构的NUAK2与CRK12激酶靶点上的虚拟筛选性能与可解释性分析。 部分a显示了MMCLKin与Glide SP在NUAK2与CRK12上的虚拟筛选流程。部分b比较了两种方法在NUAK2活性分子召回率方面的表现。部分c展示了它们在CRK12上的活性分子召回率对比。部分d展示了MMCLKin在NUAK2复合体系中识别的关键残基与功能基团。部分e展示了MMCLKin在CRK12复合体系中识别的关键残基与功能基团。为增强结构展示的直观性,两个复合物均通过Glide SP生成。源数据已作为Source Data文件提供。
2.7 基于MMCLKin的LRRK2 G2019S抑制剂发现及其生物活性验证
激酶残基突变常与多种疾病及耐药性的产生密切相关。针对突变激酶进行靶向筛选有助于发现具有突变特异性活性的先导化合物。因此,在评估MMCLKin对野生型激酶的能力之外,还从三个方面系统分析了其对突变激酶靶点的预测能力:其一,评价MMCLKin对LRRK2野生型与G2019S突变型两者抑制剂活性的预测准确度;其二,在包含3082对野生型–突变型激酶数据的集合上进行全面评估;其三,基于虚拟筛选并结合实验验证,寻找潜在的LRRK2 G2019S突变体抑制剂。
首先评估了MMCLKin区分LRRK2野生型与G2019S突变体之间细微差异的能力。选取高分辨率实验结构(野生型为8FO7,突变体G2019S为8TZC)作为受体,并构建来自BindingDB的平衡激酶–抑制剂数据集,用于微调在3DKDavis上训练的MMCLKin模型。随后利用微调后的模型预测此前工作中鉴定的四个活性化合物的pIC50值。补充表S6的结果表明,MMCLKin对LRRK2野生型和G2019S突变体的预测结果与实验数据高度一致。例如LY2023-24对G2019S突变体的预测pIC50为6.747,实验值为6.661;L R R K2-IN-1对野生型LRRK2的预测pIC50为8.315,与实验值8.509高度接近。此外,无论针对野生型还是突变型,实验pIC50越高的抑制剂,其预测值也越高。上述结果表明MMCLKin能够准确预测针对LRRK2野生型与G2019S突变体的药物活性,具备分辨突变引起的细微结合变化的能力,为发现对激酶突变体具有高选择性与亲和力的抑制剂提供可靠依据。
为系统评估MMCLKin对野生型与突变型激酶的预测能力,进一步构建了包含3082对野生型–突变型激酶数据的突变感知数据集3DKinMW,其中涉及五个不同激酶靶点:E2BBR(4对)、JAK2(1对)、RET(2343对)、LRRK2(591对)与MET(143对)。其三维结构的获取流程与3DKDavis和3DKKIBA一致。在药物冷启动条件下对模型进行训练与评估,使模型能够学习同一化合物分别与野生型与突变型激酶的互作模式,并预测测试集中未见化合物的双重pIC50值。三次独立实验的平均结果显示,MMCLKin在该数据集上仍保持较强性能,CI为0.766,MSE为0.350,PCC为0.728,MAE为0.459。
进一步将MMCLKin预测与ADP-Glo生物实验结合,验证其在突变激酶药物发现中的实际应用潜力。利用不含任何LRRK2相关数据训练得到的MMCLKin模型,在ChemDiv库中约18万个化合物上针对LRRK2 G2019S突变体(PDB ID: 8TZC)进行筛选。得分高于5.9的前5000个化合物进入进一步对接分析,使用Glide高精度模式进行分子对接。对接得分低于−8.0的化合物被保留,并采用k-means聚类以保证结构多样性;最终基于其结合构象的人工筛选选出二十个代表性化合物,重点关注与hinge区域关键残基Glu1948与Ala1950形成相互作用的构象。随后对这20个化合物在10 μM浓度下使用ADP-Glo激酶活性实验进行初筛(此浓度已被相关研究采用),LRRK2-IN-1作为阳性对照。其中五个化合物(LY2025-01至LY2025-05)表现出超过50%的抑制率。之后通过10点三倍梯度稀释法测定上述五个化合物的IC50值。
如图6a所示,五个候选化合物在拓扑结构上十分多样,其核心骨架与取代基存在明显差异。值得注意的是,LY2025-04(FN-1501)在10 μM时表现出100%的抑制率,略高于阳性对照LRRK2-IN-1的99.72%(图6b)。后续的生化实验进一步验证LY2025-04具有接近LRRK2-IN-1的高效活性,IC50分别为8.694 nM与7.001 nM(图6c)。另外,LY2025-01、LY2025-02与LY2025-05在10 μM时的抑制率均超过90%。其中LY2025-01此前未被报道为激酶抑制剂,其IC50达468 nM,表现出良好活性并显示其作为G2019S突变LRRK2功能调节剂的潜在价值。LY2025-02(sbp-7455)此前报道可抑制ULK1/2,而其对LRRK2 G2019S的IC50为2.081 nM,甚至优于参考抑制剂LRRK2-IN-1,显示出更高的靶点选择性与结构兼容性。LY2025-05(Befotertinib)是一种已获批用于非小细胞肺癌的药物,其对G2019S突变体的IC50为130.3 nM,展现出在帕金森病及其他LRRK2突变相关疾病中的再利用潜力。LY2025-03(IC50为1384 nM)虽然活性较弱,但仍可作为进一步优化的起点。
综上所述,MMCLKin在突变激酶靶向药物发现中表现出高预测精度与可靠筛选能力。这些结果验证了其在复杂激酶靶点场景中的稳健性,也表明其是开展突变感知药物研发的极具潜力的工具。
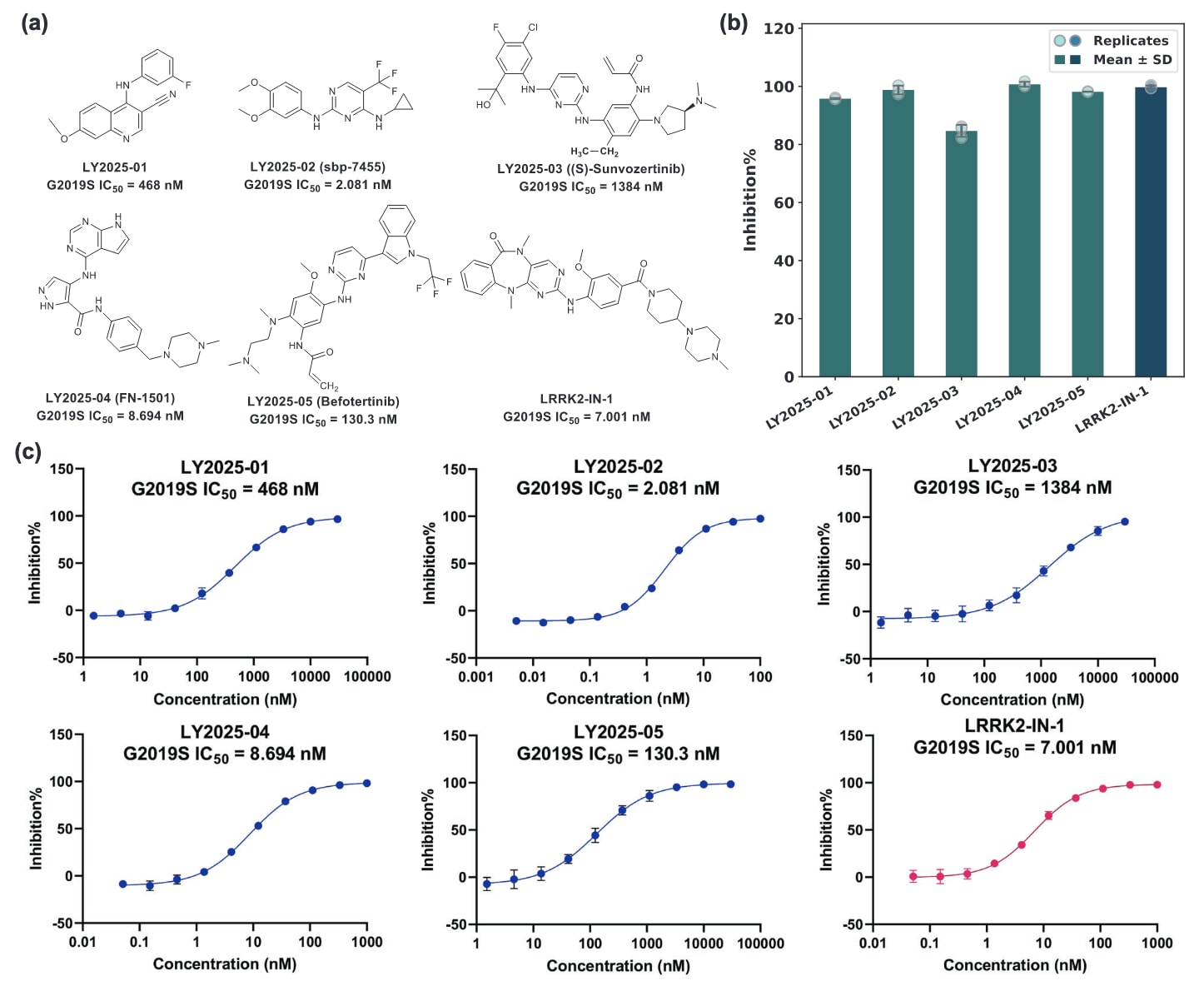
图6|展示了MMCLKin识别的五个化合物及阳性对照的化学结构,以及它们在10 μM条件下的抑制率与IC50值。 部分a显示了在10 μM浓度下抑制率超过50%的五个候选化合物及阳性对照的化学结构。部分b给出了五个候选化合物与阳性对照在10 μM下的抑制比例,每种方法均进行了三次独立重复(n=3),数据以均值±标准差表示。部分c展示了五个候选化合物及阳性对照针对LRRK2 G2019S突变体的IC50值,同样基于三次独立重复(n=3),数据以均值±标准差表示。源数据已作为Source Data文件提供。
3 讨论
由于激酶结构高度保守,高效且高选择性的激酶抑制剂发现依然是当前生物医学研究中极具挑战性的任务。全激酶组实验测定的高成本进一步凸显了发展高精度预测方法以评估激酶–抑制剂结合亲和力与选择性的迫切性。该研究提出了MMCLKin框架,用于预测不同激酶上的抑制剂活性与选择性。该框架利用几何图网络捕获空间结构特征,通过基于大语言模型的序列网络提取进化与化学信息,并采用多头注意力机制建模复杂的激酶–药物互作,同时量化系统中各要素对预测任务的贡献。此外,提出的多模态与多尺度注意力一致性对比学习方法能够有效整合多类互作特征。
在两个构建的高质量三维激酶–药物数据集上,综合评估结果表明MMCLKin在抑制剂活性与选择性预测方面优于多种对比方法。其在十个结构多样的蛋白数据集以及一个突变感知数据集上的强预测表现进一步展示了模型的泛化性与适应性。同时,MMCLKin在结构已知、结构未知以及具有挑战性的突变激酶上的虚拟筛选能力均表现良好;注意力系数分析显示模型能够从原始数据中自动识别关键残基与分子功能基团,证明其可解释性与自主学习能力。
最终,通过ADP-Glo实验验证,在MMCLKin筛选出的20个化合物中有5个能够有效抑制LRRK2 G2019S突变体,其中4个呈现纳摩尔级活性,进一步证明MMCLKin在发现高效突变激酶抑制剂方面的实用潜力。
总的来说,MMCLKin是推动高选择性与高亲和力激酶抑制剂发现的稳健且通用的框架。其在结构多样化数据集上的优异表现也暗示其在其他非激酶蛋白家族中的广泛应用可能性。尽管多尺度与多模态信息的整合显著增强了模型的表达能力,但与基于序列的轻量模型相比,其在训练与推理阶段的计算需求有所增加。未来的关键挑战在于如何从这些异质表征中更加高效地提取核心信息,并开发更简化的融合策略,以在不牺牲预测性能的前提下提升计算效率。