PNAS 2025 | SiteAF3: 基于AlphaFold3的条件扩散实现结合位点特异性精准折叠

获取详情及资源:
0 摘要
准确预测生物大分子复合物的结构对于理解生命过程并推动药物研发至关重要。尽管AlphaFold3已取得重要突破,但在特定结合位点提升预测精度仍具挑战。此处提出的SiteAF3是一种基于条件扩散、可实现结合位点精准折叠的方法,构建于AlphaFold3框架之上。SiteAF3通过固定受体结构并可选地引入结合口袋与热点残基信息,从而优化扩散过程。针对蛋白–小分子、蛋白–肽段以及蛋白–核酸等数据集的系统评估显示,SiteAF3在复合物结构预测的准确性上始终优于AlphaFold3,尤其在孤儿蛋白与变构配体情境中表现更为突出,同时显著降低了计算成本。SiteAF3以易用插件形式兼容AlphaFold3,为更精确刻画生物分子相互作用提供了高价值工具。
1 引言
准确预测生物大分子复合物的结构是理解生命过程的基础,也是推动合理药物设计与开发的关键环节。人工智能的革命性进展,尤其是AlphaFold2的问世,使人类在预测单个蛋白质三维结构方面取得前所未有的突破。在此基础上,AlphaFold3的推出成为又一重要里程碑,其能力扩展至更广泛的生物分子类型及其相互作用。然而,AF3仍然存在一定局限。其性能高度依赖多序列比对(MSA),当孤儿蛋白缺乏序列同源性或生成的MSA存在错误时,预测可能出现偏差。此外,共折叠模型难以准确预测变构配体的结合,界面以及整体复合物结构的预测精度仍有提升空间。在计算成本方面,构象采样的运行速度明显慢于传统对接方法,大体系计算还可能受到GPU显存限制。同时,AF3的口袋引导预测功能尚未公开,限制了其在特定应用中的可获取性与可定制性。
为克服这些局限并进一步提升结合位点特异性的复合物结构预测精度,提出了SiteAF3,这是一种基于AF3并采用条件扩散策略实现位点特异折叠的新方法。SiteAF3在AF3的基础上进行了两项重要改进。首先,对原始扩散模块进行微调,通过固定受体结构实现条件扩散,使配体在预定义受体背景下探索构象空间更为聚焦。其次,SiteAF3可选择通过MSA模块引入结合口袋或热点残基信息,从而显式引导预测过程。依托实验解析或高可信度预测的受体结构与口袋信息,SiteAF3能够生成高精度的配体–受体复合物结构。
SiteAF3具有若干突出的优势。其一是兼容性高,可作为轻量级插件无缝集成至AF3之中,并保持AF3对蛋白、小分子、核酸与离子等多类型复合物的广泛支持,同时可处理多条链的体系。其二是计算成本低,在大体系中固定链数量增加时显著降低GPU显存占用,并在不使用遗传搜索的情况下大幅加快预测速度,适用于高通量虚拟筛选。
此项研究呈现了系统的基准测试与案例分析,显示SiteAF3在蛋白–小分子、蛋白–肽段、双链DNA与RNA的复合物预测中均稳定优于AF3。这些结果验证了条件扩散策略的有效性以及结合位点信息的重要作用。
总体而言,SiteAF3在生物大分子复合物的位点特异结构预测中迈出了关键一步,相较于AF3在准确性、效率与应用灵活性方面均实现显著提升,并为共折叠时代的传统对接开辟了新的可能性。
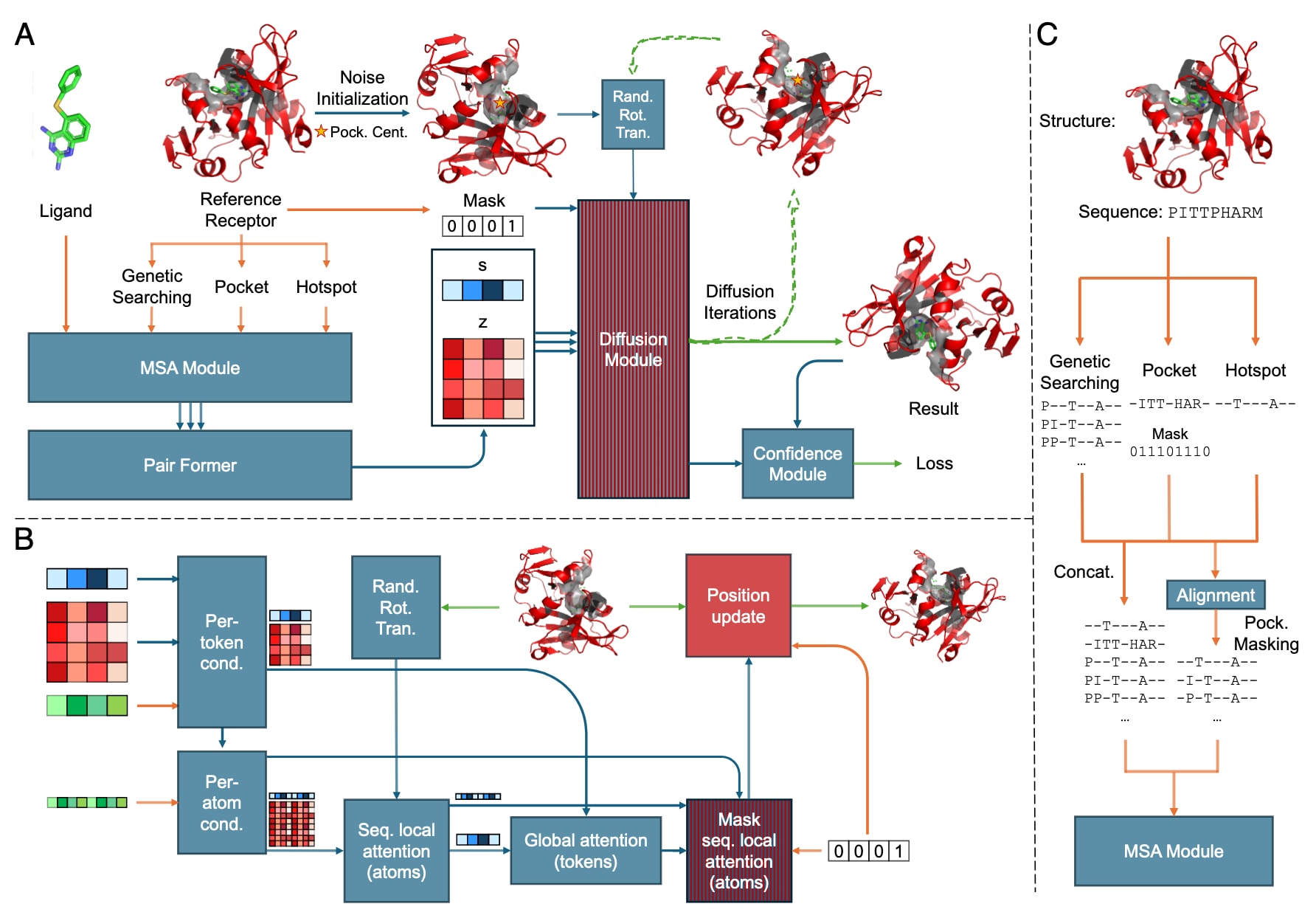
图1|SiteAF3的架构细节。 矩形表示处理模块,箭头表示数据流向。矩形颜色含义如下:红色表示新增模块;蓝红条纹表示经过微调的模块;蓝色表示与AlphaFold3相同的模块。箭头颜色含义如下:橙色表示输入数据;蓝色表示抽象网络激活;绿色表示输出数据。口袋周围的绿色点表示物理原子坐标。(A) 推理流程的整体架构。黄色星形表示口袋中心。(B) 条件扩散模块,其中粗粒度数组表示每个token的表征(绿色为输入,蓝色为pair,红色为single,01为mask),细粒度数组表示每个原子的表征。(C) MSA输入准备。左图为口袋与热点序列与遗传搜索得到的序列拼接;右图为利用口袋信息对遗传搜索序列进行mask处理。缩写说明:Pock. Cent.-口袋中心;Cond.-条件化;Rand. Rot. Trans.-随机旋转和平移;Seq.-序列。
2 结果
2.1 SiteAF3的网络结构
SiteAF3的整体架构与AF3保持一致,包括输入准备、表征学习、结构预测与置信度评估四个主要组成部分。与AF3相比,其核心差异体现在两方面:其一,结构预测模块采用条件扩散模型;其二,表征学习阶段通过MSA模块引入额外的结合口袋与热点残基信息。此类改动旨在提升复合物结构预测的准确性,尤其是在受体结构预测较差或需要强调特定结合位点的情境下。
与AF3一致,SiteAF3中的扩散模块直接作用于以点云形式表示的原子坐标,并同时结合抽象的粗粒度token表征,而不依赖旋转框架或显式等变处理。然而,其噪声初始化方式不同。SiteAF3中,配体的原子坐标以口袋中心为均值、给定半径范围内的高斯分布进行初始化,而受体的相对原子坐标则保持固定。为确保初始状态的多样性,口袋中心还会在2 Å半径球内轻微扰动。随后,为增强输出结构的鲁棒性,初始化后的点云将经历随机平移与旋转。
第二项差异出现在第二个序列局部注意力模块中,SiteAF3引入一个只更新配体坐标的mask。该mask不仅保证了扩散过程中受体结构的固定,还显著降低了GPU显存占用,从而提升了模型在大体系中的适用性。第三项改动为加入“Position update”模块,用以将固定受体坐标与更新后的配体结构无缝组合生成完整复合物。
在基础模型的初期测试中,即便配体的噪声初始化围绕口袋进行,其预测的结合位置仍偶尔偏离目标位点。为解决此问题,首先利用AF3中的遗传搜索工具为MSA模块生成模板,以评估模型能够学习多少口袋信息;随后进一步尝试直接通过MSA嵌入指定的热点与口袋残基信息。这些策略显著提升了预测精度,使配体更趋近目标结合区域。然而也观察到MSA可能引入错误的位点偏置。为尽可能降低误导影响,采用口袋信息对比对后的全长度MSA模板进行mask处理。经口袋mask的AF3 MSA后,SiteAF3在口袋定义明确的蛋白–配体数据集上展现出显著性能提升。需强调的是,该方法不同于AF3中未公开的口袋引导折叠功能,因此两者的模型表现并不等价。
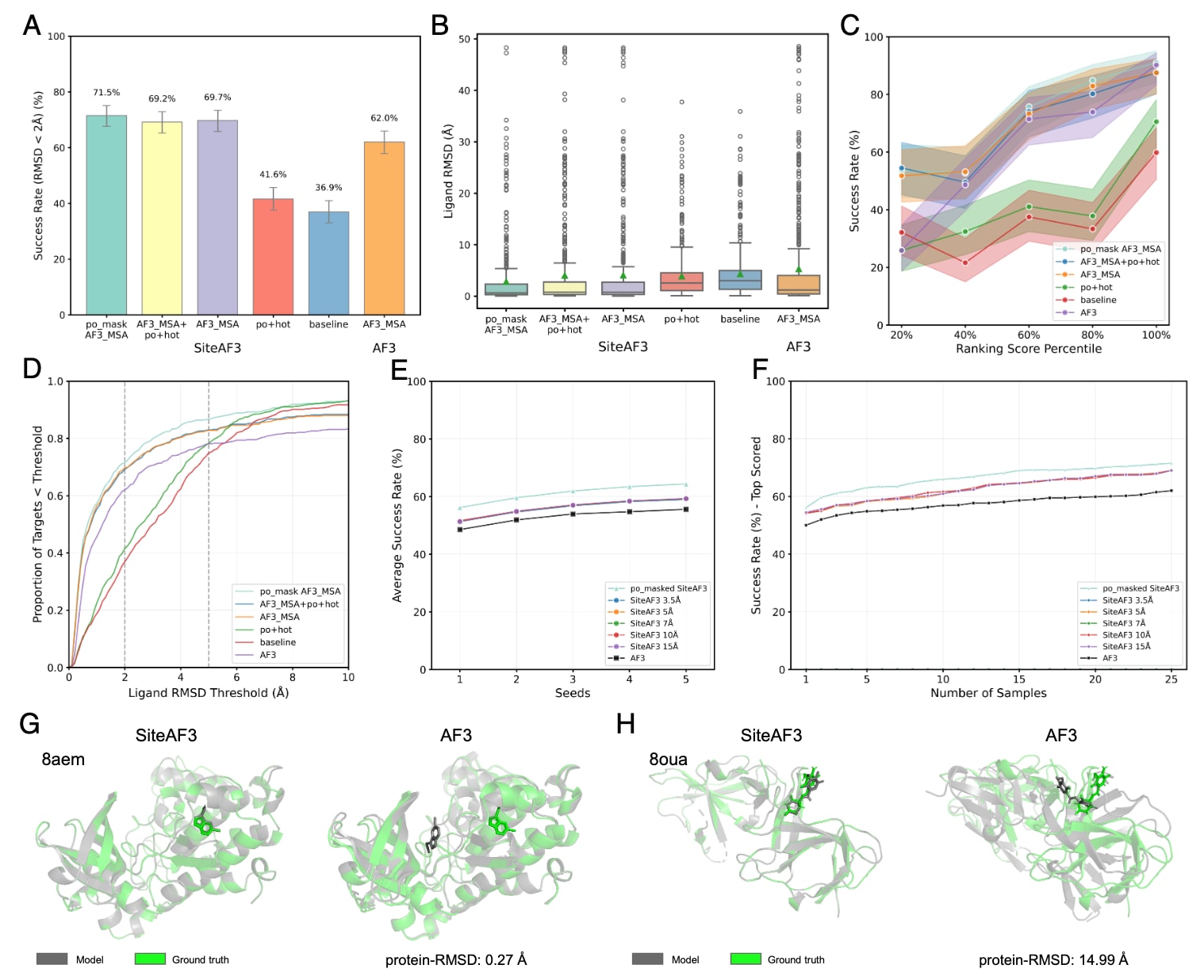
图2|FoldBench蛋白–配体数据集上的蛋白–小分子复合物结果。 每个样本被分配五个随机数,每个随机数对应五个采样,共得到25个结构,统计分析取其中最佳结果。(A) 不同模式下的成功率,成功率定义为LRMSD小于2 Å的案例占比。(B) SiteAF3与AF3的LRMSD比较。(C) SiteAF3与AF3的成功率与置信度相关评分之间的关系。评分按数量分位数进行分段,每一段均匀划分测试样本。(D) LRMSD在0到10 Å范围内的累积分布,灰色虚线从左到右分别代表2 Å与5 Å的阈值。(E和F) 不同模式与超参数对LRMSD平均成功率的影响。展示每个seed下从最差到最佳的采样数量分布对应的成功率,以及25个采样中最高评分结构的成功率。SiteAF3默认使用AF3 MSA。(G) 案例分析:变构化合物LVF结合于CK2alpha(PDB 8aem)。SiteAF3使用AF3遗传搜索、口袋与热点信息。SiteAF3识别了正确的结合位点,而AF3虽成功折叠蛋白,却将配体放置在错误口袋。(H) 案例分析:cereblon isoform 4与11F复合物(PDB 8oua)。SiteAF3使用AF3遗传搜索、口袋与热点信息。SiteAF3成功预测了结合构象,而AF3生成的蛋白结构错误,导致对接失败。
2.2 蛋白–小分子复合物的结果
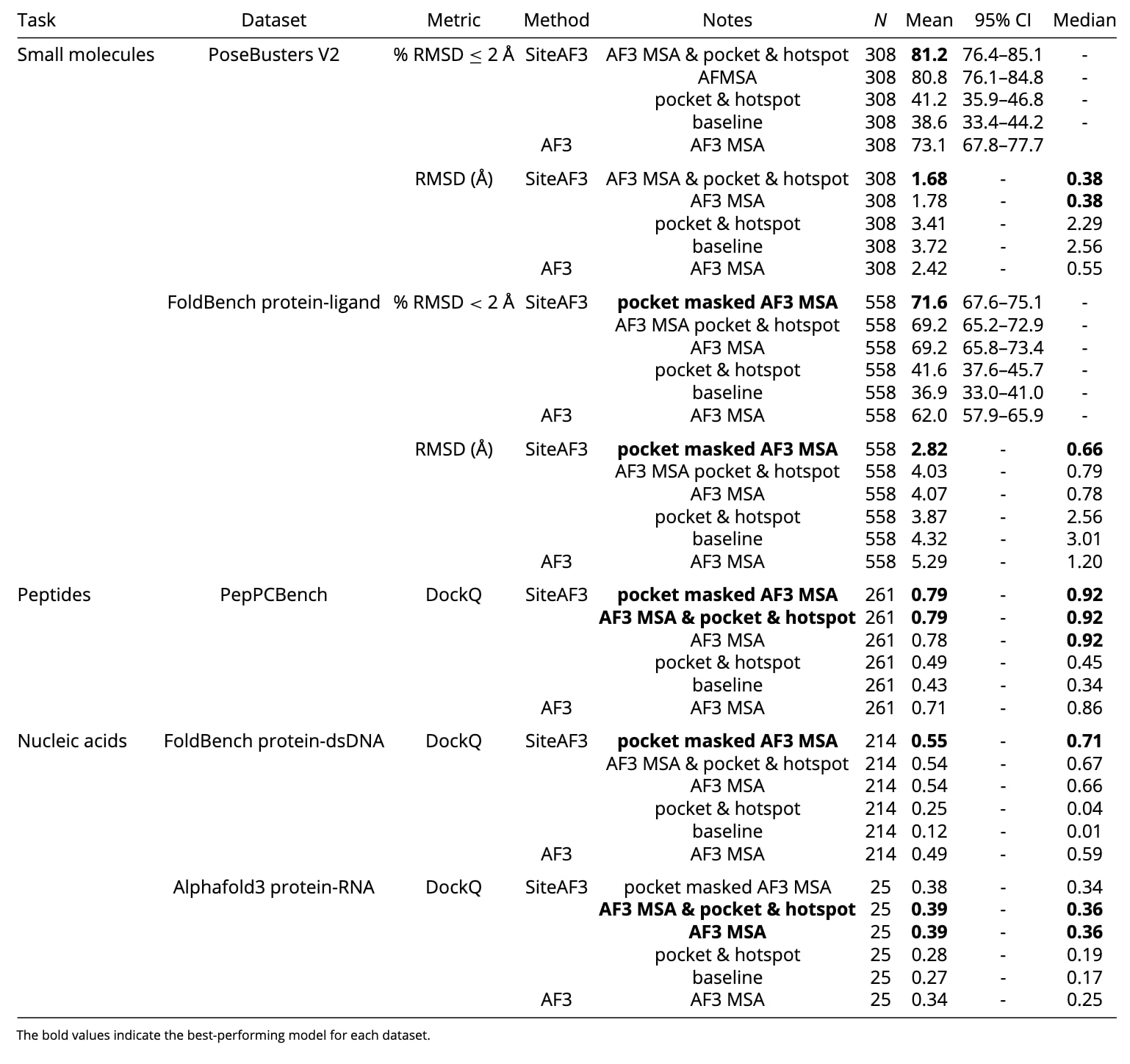
精准预测蛋白–小分子复合物结构在药物设计领域具有核心意义。尽管AF3在该任务上已显著优于传统对接工具与其他深度学习方法,SiteAF3仍进一步将预测准确性推向更高水平。为全面评估SiteAF3的性能并与AF3进行对比,选择了两个基准数据集:FoldBench蛋白–配体数据集(图2与表1)以及PoseBustersV2数据集。FoldBench数据未包含在AF3的训练集中,而PoseBustersV2整体用于AF3训练。
评估中采用了SiteAF3的五种配置模式:模式1(口袋mask的AF3 MSA)使用口袋信息对比对后的全长遗传搜索模板进行mask;模式2(AF3_MSA+po+hot)结合遗传搜索、口袋与热点残基信息;模式3(AF3_MSA)仅依赖AF3的遗传搜索;模式4(po+hot)仅使用口袋与热点信息;模式5(baseline)在表征学习阶段不添加任何额外信息。在FoldBench数据集上成功复现了AF3报告的62.0%成功率,落在其原论文的95%置信区间内(64.9%)。值得注意的是,最佳的SiteAF3模型达到71.6%的准确率,显著高于AF3。相比基础模型,仅加入口袋与热点信息(不依赖遗传搜索)也带来明显提升。
从LRMSD来看,使用AF3 MSA+口袋+热点信息的SiteAF3模式相较AF3使中位数降低30.9%,均值降低30.6%;而口袋mask的AF3 MSA在此基础上进一步降低中位数16.5%、均值30.0%。图2C显示,SiteAF3在排名分值较低的20%结构上显著提高准确率,在高分值部分则与AF3表现相当,说明其有效改善了低置信度结构的预测。
图2D展示了一个有趣现象:当使用AF3_MSA时,SiteAF3在0–10 Å范围内的累积准确率均优于AF3;但在未使用AF3_MSA时,SiteAF3在低精度区间略逊于AF3,但在5 Å之后po+hot与baseline模式又逐渐超过AF3,这表明AF3的MSA模板在未见过的蛋白–配体对中有时可能引入错误位点偏置。在PoseBustersV2中未观察到这一现象,提示对于AF3训练数据集,遗传搜索引入的表征可能存在过拟合。
最值得关注的是,口袋mask的AF3 MSA模式在0–10 Å范围内表现始终最佳,0–5 Å区间的趋势与使用全长MSA一致但斜率更大;在5–10 Å区间则始终优于未使用遗传搜索的模式,说明口袋引导可有效消除MSA模板中隐藏的偏差。
进一步模拟真实对接情境,即未知最优构象的情况下,SiteAF3在五个seed下对五个采样结构的平均成功率始终优于AF3;在25个采样中选最高分构象时亦保持优势(图2F)。此外,研究噪声初始化半径对性能的影响后发现,在3.5–15.0 Å范围内SiteAF3均优于AF3,且无显著差异,但较小半径(3.5 Å与5.0 Å)在推理中会导致原子重叠问题,因此最终选取7.0 Å作为模型半径。
在PoseBustersV2数据集中未能完全复现AF3原论文中AF3-2019cutoff的结果,可能因预处理或采样方法不同,但趋势仍然一致:SiteAF3显著优于AF3,且排名分值与成功率的线性关系更明确,而AF3在FoldBench上的排名分值表现似乎出现过拟合。
图2G与2H展示了两个SiteAF3优于AF3的实例。在8aem中,该结构同时含ATP正构位点与LVF变构位点,AF3错误地将LVF置于正构位点,而SiteAF3准确预测其变构结合。在PDB 8oua中,AF3甚至未能生成正确的受体结构,导致配体预测失败,而SiteAF3成功获得正确构象。
此外进一步评估了变构配体与含辅因子体系的表现。在变构配体数据集中(共14个,5个训练集内、9个未见),SiteAF3的RMSD显著优于AF3:在口袋mask AF3 MSA模式下,SiteAF3的平均RMSD不足AF3的三分之一,中位数仅为五分之一;25个采样中通常可得到多个正确构象,而AF3往往仅能得到一两个。辅因子体系(10个复合物)中,尽管受限于难以指定多个口袋,仅比较baseline与AF3-MSA模式,但SiteAF3仍普遍优于AF3。
综合而言,辅因子有时可改善AF3表现,但SiteAF3在变构配体与辅因子体系中展现了更稳健与更具普适性的预测能力。
表1|不同分子复合物的预测准确率
2.3 蛋白–肽段复合物的结果
肽段在多达40%的蛋白–蛋白相互作用中扮演关键角色,因此准确建模蛋白–肽段复合物对于理解生命过程以及推动肽类药物设计具有重要意义。AlphaFold3在该类复合物的预测中已相较AlphaFold2-Multimer取得明显提升,而SiteAF3进一步增强了这一预测能力。评估中采用PepPCBench的Full数据集作为主要基准(图3与表1)。测试包含与前述相同的四种SiteAF3配置模式。
需要注意的是,测试结果与原PepPCBench论文略有不同,主要由于该论文仅使用晶体结构对应的序列,而此处使用PDB网站提供的全长fasta序列进行评估。
图3A显示,所有结合AF3_MSA的SiteAF3模式均显著优于AF3,尤其是在依据置信度排名后位于底部40%的结构中,SiteAF3在DockQ得分上表现更佳。值得注意的是,口袋mask的AF3 MSA在该任务中并未带来进一步提升。仅使用口袋与热点信息的SiteAF3模式也明显优于基础模型。
从图3B与表1可见,最佳SiteAF3模型的平均DockQ比AF3高11.2%,中位数高7.0%。进一步考察native接触比例fnat、F1分数、界面RMSD(iRMSD)与LRMSD等指标,SiteAF3在捕捉蛋白–肽段界面相互作用、预测高柔性肽段构象方面均表现更为优越。
图3C说明,结合AF3_MSA的SiteAF3模式在整体成功率上高于AF3,且高质量DockQ预测比例比AF3高出超过10%,这对依赖高质量结构模型的肽段设计尤为关键。
鉴于噪声初始化策略的特点,进一步按肽段长度将DockQ结果分成三组:5-9、10-15、16-30个残基(图3D)。分析结果显示,SiteAF3在短肽段(5-9与10-15残基)上大幅优于AF3,而在较长肽段上提升幅度相对较小。这与噪声初始化策略相一致:短肽段空间分布紧凑,更能从口袋中心附近的局部初始化中获益;而长肽段通常具有更大界面,集中式初始化并非最佳。
图3E与附录图S3 E、F中的案例进一步展示了SiteAF3优于AF3的三种情况。在8tmz中,AF3在抗体结构预测阶段出现问题,导致口袋环境畸变,使其无法正确预测肽段结合位置;而SiteAF3表现出色。在8p0q与8ia5中,AF3虽能预测准确的蛋白结构,却完全错误判断肽段的结合位点,而SiteAF3成功预测正确构象。此外,在8s6n中,AF3将全长蛋白C端的一段无规卷曲误折叠成β-折叠,占据了原本的肽段结合口袋,从而导致预测失败。有趣的是,当不使用全长序列时,AF3反而能够成功预测该复合物;而SiteAF3并不会因序列长度与晶体结构不匹配而表现不稳。
这些结果共同表明,SiteAF3在蛋白–肽段复合物预测中具备更高的准确性、稳定性与适用性。
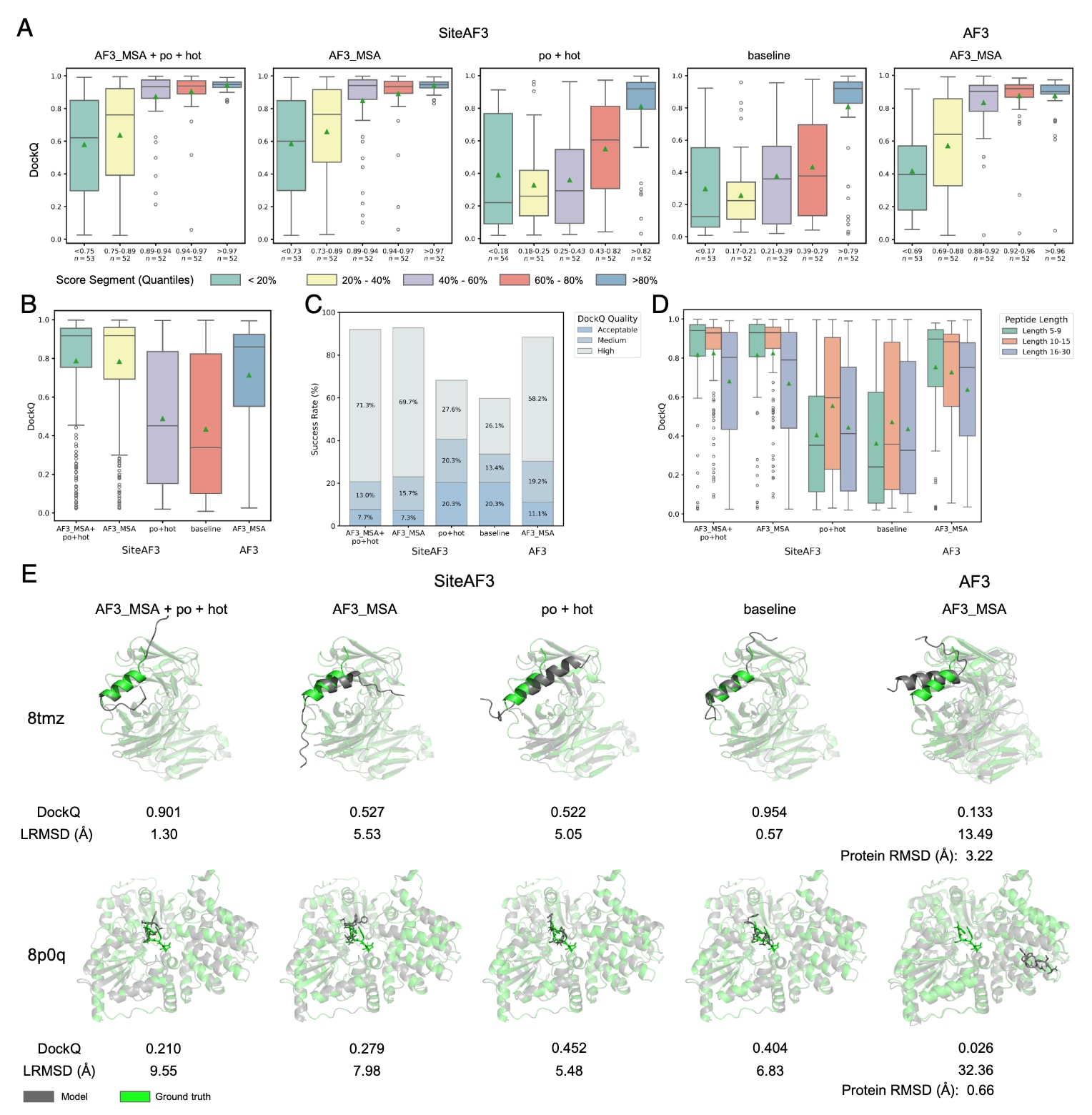
图3|PepPCBench数据集上的蛋白–肽段复合物结果。 每个样本被分配三个随机数,每个随机数对应五个采样,共得到15个结构,统计分析取其中最佳结果。(A) DockQ与置信度相关评分之间的关系。评分按数量分位数进行分段,每一段均匀划分测试样本。(B) SiteAF3与AF3的DockQ值比较。(C) 基于不同阈值的对接性能。(D) 三种不同肽段长度分类下SiteAF3与AF3的DockQ值比较。(E) 案例研究。上图:MERS-CoV刺突蛋白干螺旋肽段与抗体CHM-27复合物(PDB 8tmz)。SiteAF3成功对接,而AF3生成了错误的抗体结构。下图:AaNGT与肽段复合物(PDB 8p0q)。SiteAF3生成了正确构象,而AF3虽成功折叠蛋白,却找错了肽段结合位点。
2.4 蛋白–核酸复合物的结果
适配体是另一类具有高潜力的蛋白结合分子,阐明核酸与蛋白之间的精细相互作用对于合理设计高效适配体至关重要。SiteAF3能够进一步提升蛋白–核酸复合物的预测精度。与AF3原论文一致,此处主要关注蛋白与双链DNA(dsDNA)以及蛋白与RNA之间的相互作用。蛋白–dsDNA数据集来源于FoldBench中的蛋白–DNA数据集,而蛋白–RNA数据集则与AF3研究中使用的完全一致(图4与表1)。评估中采用与前述相同的四种SiteAF3配置模式。
图4A显示,在AF3_MSA模式下,SiteAF3在蛋白–dsDNA复合物中的预测质量显著提升,平均DockQ提高10.2%,中位数提高13.6%。采用口袋mask的AF3 MSA可进一步略微提升性能。值得强调的是,引入口袋与热点残基信息也能相较基础模型显著提高结构预测的准确性。
图4B显示蛋白–RNA复合物呈现相似趋势:在AF3_MSA模式下,SiteAF3的预测质量显著改善,平均DockQ提升14.7%,中位数更上升44.0%。口袋与热点信息同样在基础模型之上带来明显增益。
进一步分析排名分值与DockQ之间的关系(图S4A)。对于蛋白–dsDNA复合物,使用AF3_MSA的两种SiteAF3模式与AF3趋势一致,但平均DockQ始终更高;对于RNA复合物,由于样本量较小,难以观察到明确趋势。图S4 B与C则展示了SiteAF3在捕捉核酸分子构象以及其与蛋白界面复杂相互作用方面的增强能力。
需要指出的是,核酸分子通常远大于肽段,可能并不完全适配当前的噪声初始化策略,因此性能提升很大程度可能源自受体蛋白结构预测的改善。
图4C展示了PDB 7to1的案例,三个模型均由SiteAF3生成。baseline模式将dsDNA置于错误位置;加入口袋与热点残基信息后,尽管定位到正确结合位点,但碱基对发生偏移;只有AF3_MSA模式借助遗传搜索提供的完整模板信息,才能准确预测整个复合物结构。
图4D展示了另一个例子:在某蛋白区域的构象预测中AF3出现错误,导致DNA中部分碱基的取向偏差。口袋与热点信息足以帮助确定正确结合位点,但无法提供精确刻画核酸构象所需的结构细节;只有借助遗传搜索得到的完整模板信息才能精确修正核酸结构。
这些结果表明,SiteAF3不仅提升了蛋白–核酸复合物整体预测能力,还在捕捉界面关键相互作用方面展现出更高的准确性与稳健性。
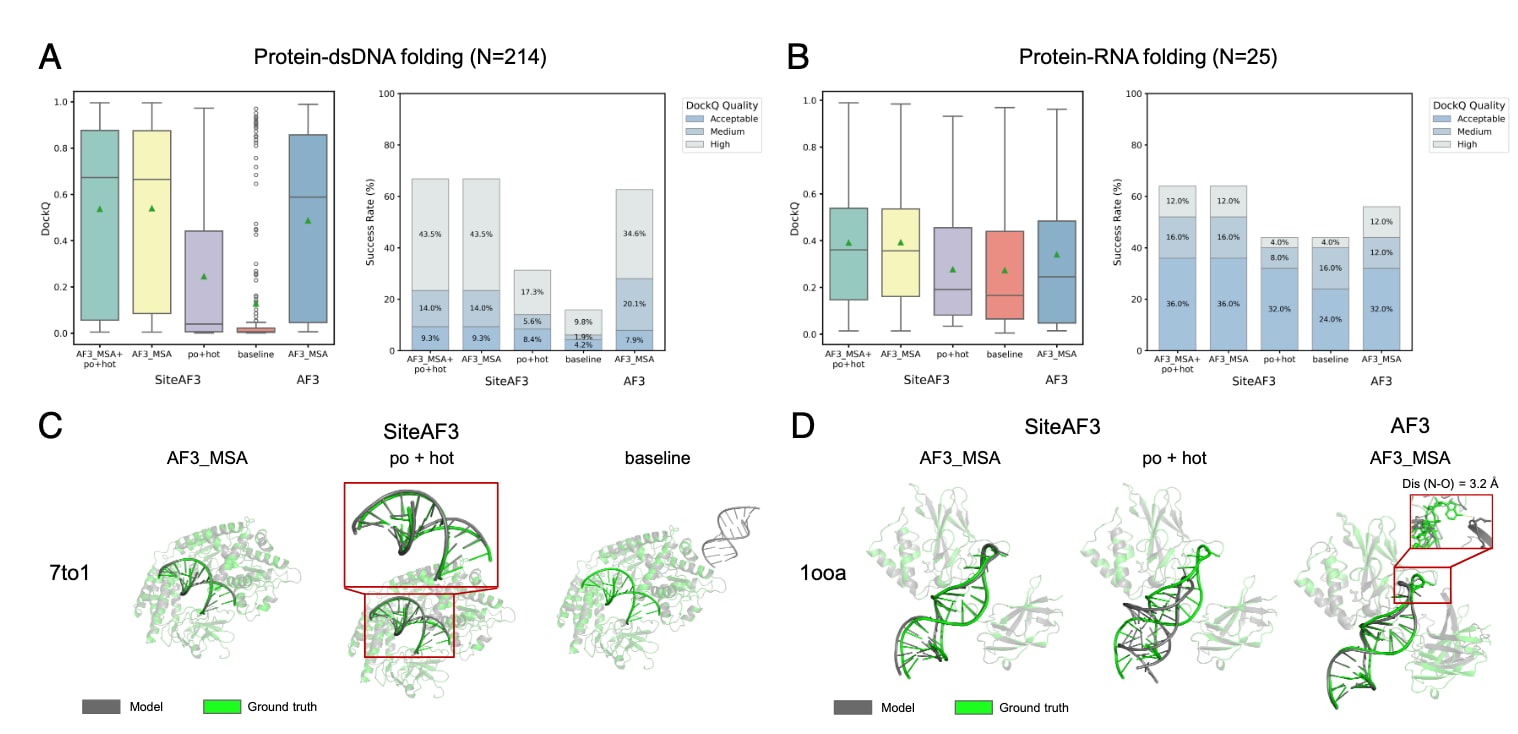
图4|蛋白–核酸复合物的结果。 每个样本被分配三个随机数,每个随机数对应五个采样,共得到15个结构,统计分析取其中最佳结果。(A) 蛋白–双链DNA(dsDNA)的对接性能。(B) 蛋白–RNA的对接性能。(C) 案例研究:RIG-I结合于p3SLR30末端(PDB 7to1)。三个模型均由SiteAF3预测:AF3_MSA模式(模式2)成功预测;口袋与热点信息模式(模式3)将dsDNA定位到正确位点但出现碱基对偏移;baseline模式(模式4)则将dsDNA放在错误位置。(D) 案例研究:NF-kappaB(p50)2与RNA适配体复合物(PDB 1ooa)。SiteAF3的AF3_MSA模式(模式2)成功对接,口袋与热点信息模式(模式3)有助于定位RNA,而AF3未能正确生成蛋白某一结构区域,导致DNA中碱基取向错误。
2.5 SiteAF3在计算成本上的优势
SiteAF3相比AF3在计算成本方面的优势主要体现在两个方面。第一点涉及GPU显存占用(SI附录表S1)。在训练过程中,使用显存为48 GB的RTX 6000 Ada显卡时观察到,当结构的token总数超过约3500时,AF3容易出现显存溢出错误。而SiteAF3通过在矩阵乘法阶段引入mask,使模型仅更新配体坐标并裁剪两两嵌入矩阵的规模,从而显著降低了此步骤的显存需求,使更大体系的计算成为可能。
第二点优势出现在未使用AF3_MSA时,即预测仅依赖口袋与热点信息。在某些情况下这一配置可能会降低精度,但其避免了计算代价极高的遗传搜索过程,因此显著加快了整体运行速度。相较于不使用AF3_MSA的AF3模型,SiteAF3在该模式下仍能保持可接受的准确性,因此这种配置在高通量筛选任务中具有潜在应用价值。
3 结论
SiteAF3是一种在AF3框架内基于条件扩散实现位点特异性折叠的方法,在多类生物大分子复合物结构预测中取得了显著提升,尤其在变构位点、高柔性结构以及已知结构但缺乏序列同源性的孤儿蛋白上表现突出。在蛋白–小分子相互作用中,对FoldBench与PoseBustersV2数据集的评估显示,SiteAF3在整体验证中均优于AF3,特别是在加入结合口袋残基信息时提升更为明显。值得注意的是,SiteAF3在低置信度预测中展现更高准确性,并显著降低了配体的RMSD。在蛋白–肽段复合物预测中(基于PepPCBench数据集),SiteAF3获得更高的DockQ得分,并更好地刻画界面相互作用及肽段构象,尤其对较短肽段效果更加突出。SiteAF3在蛋白–核酸复合物(dsDNA与RNA)的预测中同样表现优越,平均与中位DockQ得分均显著提升,在充分利用AF3的MSA信息时尤为明显。
除准确度提升外,SiteAF3还在计算成本上具备优势,包括更低的GPU显存占用以及在仅使用口袋与热点信息时的更快运行速度,这为高通量筛选提供了可能性。自AF3提出以来,出现了多种相似的共折叠模型,包括Boltz、Chai、Protenix、HelixFold等。其中AF3在FoldBench与PepPCBench等基准数据集上整体表现最佳,但其他框架也探索了不同方向。例如,Boltz系列以更开放的社区方式发展,Boltz-2则加入可直接预测结合自由能的亲和力模块。尽管SiteAF3的主要目标在于提升结构准确性,其方法学改进——如基于结合位点的条件化处理与扩散采样策略——具有普适性,可迁移至其他框架。如果与Boltz-2中的亲和力预测模块结合,有望进一步提升结合亲和力估计能力,从而体现该方法的广泛适用性。
尽管结果令人鼓舞,SiteAF3仍存在若干局限。首先,当前的配体噪声初始化策略采用围绕口袋中心的简化方式,并非所有结合情境的最优选择,尤其对于结合界面更为广泛或复杂的配体。未来可探索让噪声初始化具备可学习性或根据结合界面形状自适应调整,以提升采样效率。其次,当前版本缺乏处理共价对接的专门模块,若能扩展至准确建模共价作用,将显著拓展其在药物发现与酶机制研究中的应用范围。第三,SiteAF3目前将受体视为刚性结构,这限制了其捕捉结合口袋周围局部构象变化的能力。若引入局部柔性对接模块,使结合位点附近关键残基或环区能够随配体折叠协调运动,将有助于更好地模拟诱导契合效应,并提升在具有挑战性的结合场景中的预测准确性。