NC 2025 | TAPB: 一种干预式去偏框架,用于缓解药物-靶点相互作用预测中的靶点先验偏差
今天介绍的这项工作来自 Nature Communications。靶标先验偏差框架——TAPB,通过一系列创新方法有效缓解了药物-靶点相互作用(DTI)预测中的“靶点先验偏差”。该框架的核心思想是识别并减少由靶点标签分布不平衡引起的虚假关联,从而避免模型过度依赖数据中的偏差。具体来说,TAPB引入了氨基酸随机化、混杂变量对齐模块(CAM)和干预式训练,这些方法共同作用,增强了模型的泛化能力和预测性能。氨基酸随机化通过扰动输入序列,防止模型记住靶点特征,而CAM和后门调整则帮助模型消除与靶点相关的虚假偏差。TAPB框架能够有效捕捉真实的药物-靶点相互作用,避免了传统模型中因依赖不正确的靶点-标签关系而导致的性能下降。TAPB在多个数据集上的表现证明了其强大的泛化能力,特别是在跨领域和冷拆分的场景中,展现了比传统方法更优的预测能力。此外,TAPB提供了一种zero-shot预测方法,即仅使用源领域的训练集进行训练,避免了重复构建目标领域数据集和重新训练的计算负担,从而提供了一种更通用且便捷的解决方案。通过对比实验,TAPB不仅在传统的DTI预测任务中表现优异,也能有效应对药物偏差数据集,显示出其广泛的适用性和稳健性。

获取详情及资源:
0 摘要
药物-靶点相互作用(DTI)预测 对于药物重定位至关重要。以往在 BioSNAP 和 BindingDB 数据集上的DTI研究,通常将偏差预测归因于“药物偏差”,然而研究表明,“靶点先验偏差”才是主要问题。该偏差来源于“先验倾向”,即训练数据中靶点标签分布的不平衡。从因果推断的角度来看,靶点的“先验倾向”是一个混杂变量,导致基于
1 引言
药物-靶点相互作用(DTI)预测在探索现有药物潜在应用中至关重要,能够显著加速药物从实验阶段到临床应用的转化。药物发现的计算方法涵盖了不同的策略。一方面,虚拟对接模拟从结构角度探索生物分子之间的相互作用;另一方面,开发了多种基于数据的方法用于DTI预测,包括传统的机器学习和深度学习方法,这些方法主要集中在两种类型:一种是利用图数据进行推荐,另一种则是使用序列数据进行预测。特别地,基于序列的预测方法提供了标记级的可解释性,能够为药物重定位提供有价值的见解。这些方法通常采用双塔架构以适应不同的输入格式,如SMILES、氨基酸序列、指纹或分子图等。这些输入数据会被编码为嵌入表示,然后进行聚合,用于二分类任务,估算药物
尽管在模型架构和特征工程方面取得了显著进展,但在序列数据集上训练的DTI模型,如BioSNAP和BindingDB,往往会表现出对特定输入的偏向,而不是捕捉药物-靶点相互作用的真实机制。这限制了模型的泛化能力,成为药物重定位应用中的一大障碍。先前的研究将这一问题归因于“隐藏模式偏差”,即“药物偏差”。例如,UdanDTI采用了不对称的架构和注意力聚合机制,强化靶点分支并减弱药物分支,从而减轻了“药物偏差”。然而,研究结果表明,“靶点先验偏差”即“靶点偏差”在BioSNAP和BindingDB数据集的领域内和跨领域拆分中对偏差预测起到了更重要的作用。这一偏差反映了模型在进行预测时,主要依赖于靶点特征,而不是药物-靶点之间的真实相互作用。造成“靶点先验偏差”的原因是“先验倾向”。
直观来说,“先验倾向”描述了在DTI训练集中,各个药物或靶点的正负交互标签的不平衡。模型可以通过简单地捕捉药物或靶点的标签倾向,而非真实的相互作用机制,来最小化损失。例如,在图1b中,训练集中靶点
为了验证“先验倾向”是否导致序列DTI数据集(如BindingDB和BioSNAP)上的偏差预测,构建了两个反先验数据集:一个具有较高药物“先验倾向”,另一个则具有平衡的“先验倾向”。“先验倾向导致偏差预测”部分的研究结果支持了这一假设。需要特别指出的是,所识别出的“靶点先验偏差”来源于BioSNAP和BindingDB数据集,而其他采用不同数据集和模型架构的DTI预测方法,如NRLMF,可能会表现出不同的偏差。
在该研究中,研究团队提出了一种用于缓解靶点先验偏差的干预式去偏框架(TAPB)。从因果推断的角度来看,DTI序列数据集中的靶点“先验倾向”是一个混杂变量,它为靶点和预测之间打开了后门路径,使得DTI模型难以通过
该研究的贡献如下:首先,重新评估了BioSNAP和BindingDB数据集(包括领域内和跨领域拆分),并确认“靶点先验偏差”是这些数据集预测偏差的主要来源,这一原因与先前认定的“药物偏差”不同。统计分析支持了这一结论,反倾向实验也证实了“先验倾向”在基于序列数据集训练的DTI模型中的“药物偏差”和“靶点偏差”中发挥作用。其次,重新从因果角度框定了DTI预测,并提出了TAPB这一干预式去偏框架,该框架集成了氨基酸随机化(通过残基删除(70%)和突变(20%)破坏虚假关联)和后门调整,通过混杂变量字典
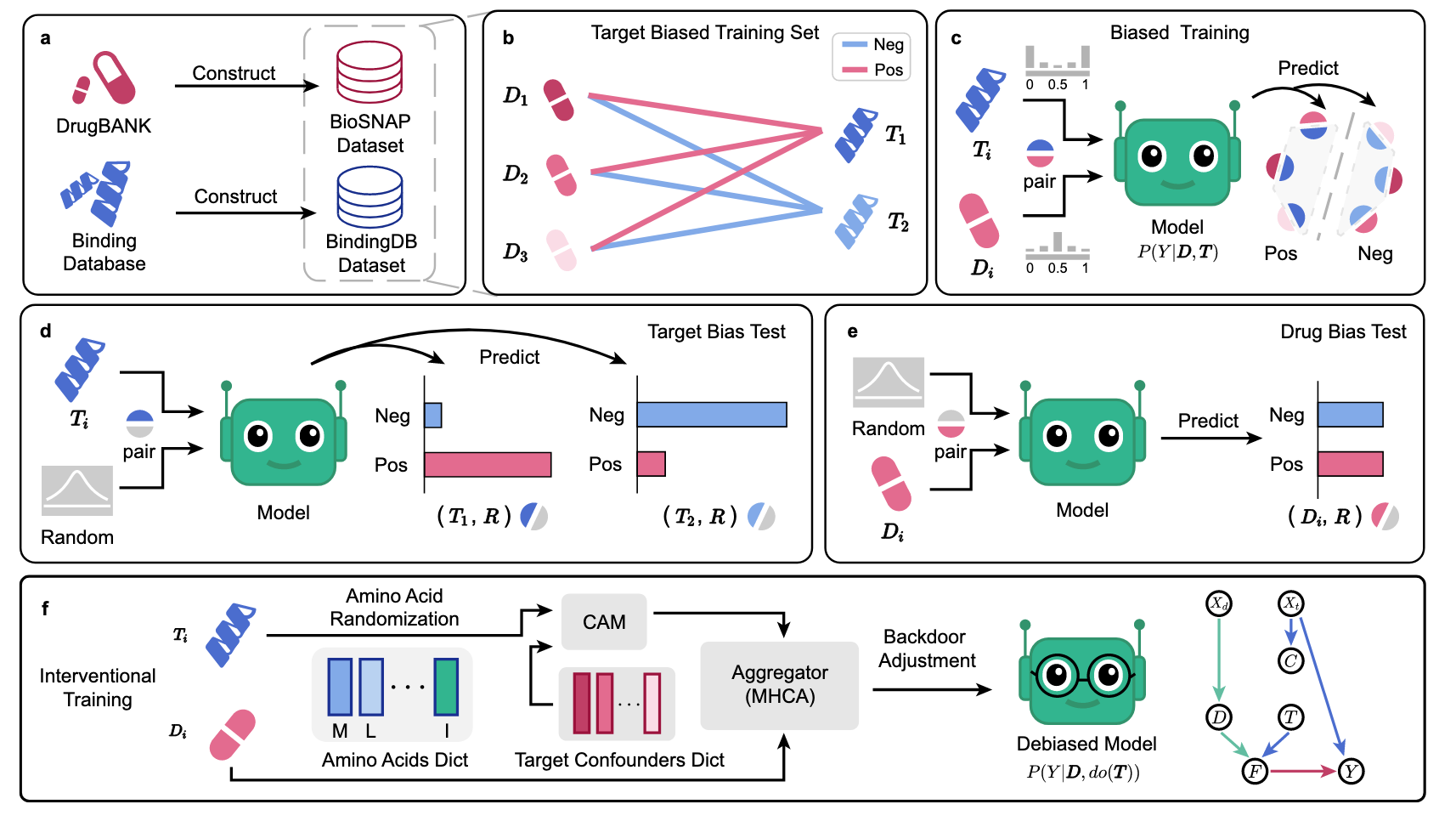
图1 | 展示了DTI偏差分析与框架概述。 a BioSNAP和BindingDB数据集的构建。b 靶点偏向训练集示意图,其中某些靶点(T1和T2)分别表现出正负“先验倾向”,而药物Di的偏差则较不明显。c 偏差训练过程示意图,模型从具有内在“靶点偏差”的数据集中学习。d “靶点偏差”测试表明,将随机生成的特征R与靶点T1配对时,得到较高的正向评分,而T2则观察到相反的结果。e “药物偏差”测试显示,当药物D与随机生成的特征R配对时,得分相对平衡,表明对预测的影响较小。f 该研究团队的TAPB干预式训练,结合氨基酸随机化、混杂变量对齐模块(CAM)和多头跨注意力机制(MHCA),在该研究团队的SCM假设下计算
2 结果
2.1 序列数据集的 DTI 预测公式
由于在DTI序列数据集中缺乏标记级的真实值,先前的研究通常将DTI预测重新定义为二分类任务。例如,MolTrans、TransformerCPI和DrugBAN等研究将DTI预测公式化为二分类问题。设
在此框架的基础上,先前的研究主要通过优化特征编码器和聚合器,或引入额外特征,来提升模型的性能。
2.2 药物偏差与靶点偏差:哪种更严重?
先前的研究,如TransformerCPI、DrugBAN和UdanDTI,基于DTI序列数据集进行了训练,并确认了偏差预测的存在,普遍假设“药物偏差”导致模型更多依赖药物特征。然而,是否“药物偏差”是影响DTI序列数据集(如BindingDB和BioSNAP)中偏差预测的主导因素仍值得探讨。为了测试药物或靶点特征的影响力,采用了t-SNE可视化分类特征。具体来说,对于训练集中的每对药物-靶点
如果预测没有偏差,那么对
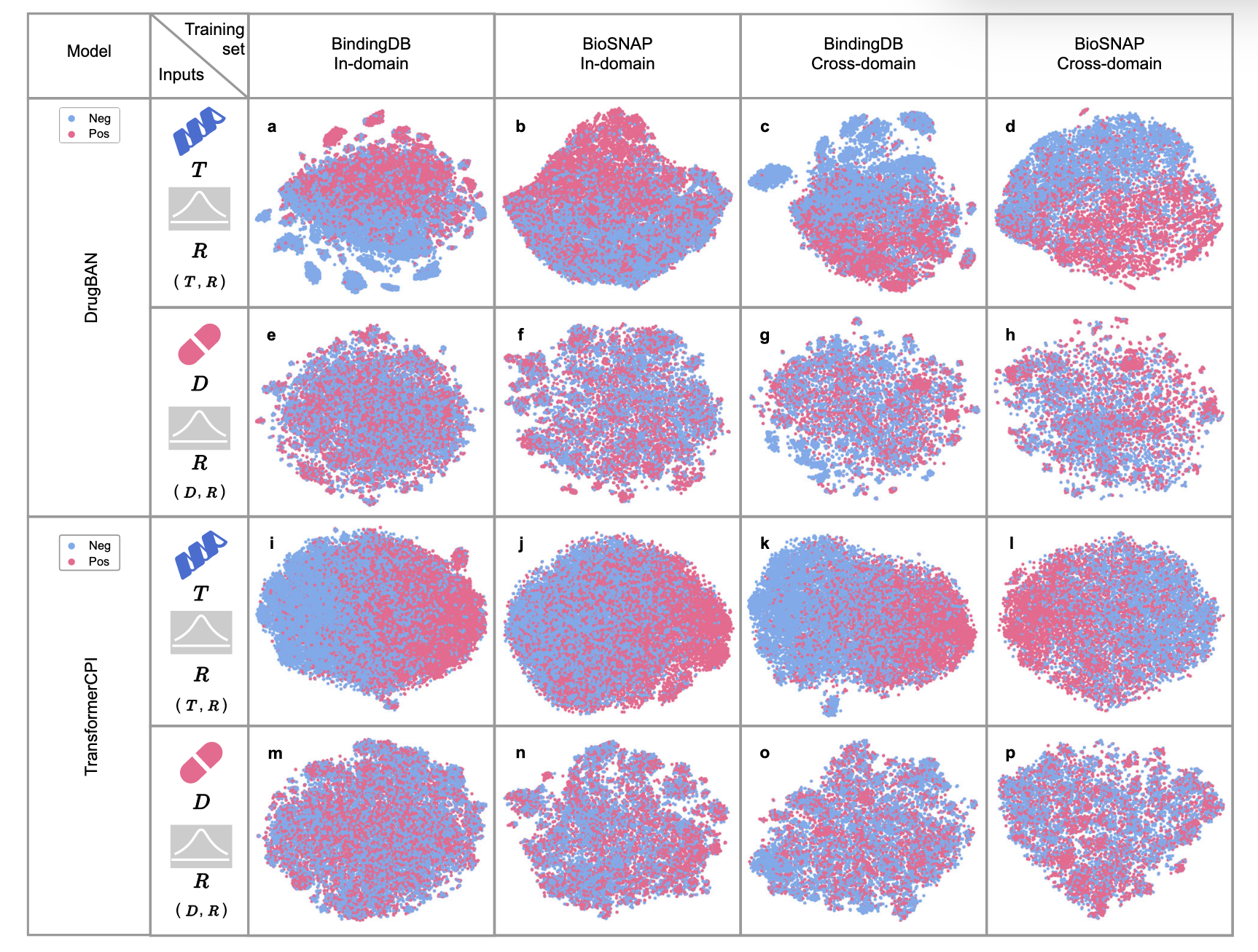
图2 | 展示了DrugBAN和TransformerCPI在BindingDB和BioSNAP数据集上对分类特征(D, R)和(T, R)的t-SNE可视化。 a DrugBAN在BindingDB数据集的领域内拆分上训练,输入为(T, R)。b DrugBAN在BioSNAP数据集的领域内拆分上训练,输入为(T, R)。c DrugBAN在BindingDB数据集的跨领域拆分上训练,输入为(T, R)。d DrugBAN在BioSNAP数据集的跨领域拆分上训练,输入为(T, R)。e DrugBAN在BindingDB数据集的领域内拆分上训练,输入为(D, R)。f DrugBAN在BioSNAP数据集的领域内拆分上训练,输入为(D, R)。g DrugBAN在BindingDB数据集的跨领域拆分上训练,输入为(D, R)。h DrugBAN在BioSNAP数据集的跨领域拆分上训练,输入为(D, R)。i TransformerCPI在BindingDB数据集的领域内拆分上训练,输入为(T, R)。j TransformerCPI在BioSNAP数据集的领域内拆分上训练,输入为(T, R)。k TransformerCPI在BindingDB数据集的跨领域拆分上训练,输入为(T, R)。l TransformerCPI在BioSNAP数据集的跨领域拆分上训练,输入为(T, R)。m TransformerCPI在BindingDB数据集的领域内拆分上训练,输入为(D, R)。n TransformerCPI在BioSNAP数据集的领域内拆分上训练,输入为(D, R)。o TransformerCPI在BindingDB数据集的跨领域拆分上训练,输入为(D, R)。p TransformerCPI在BioSNAP数据集的跨领域拆分上训练,输入为(D, R)。
2.3 先验倾向导致偏差预测
假设偏差预测是由“先验倾向”引起的,先验倾向被正式定义为DTI序列数据集中每个药物或靶点固有的系统性标签分布偏差。具体来说,这指的是不同药物(药物级别先验)或靶点(靶点级别先验)之间正负样本比率的统计显著偏离,这些偏离会导致虚假关联,模型可以利用这些关联来最小化损失,而不学习真实的相互作用机制。为了量化不同DTI数据集中的“先验倾向”,设计了以下标签测试:
其中,
其中,
其中,
其中,
计算了每个“先验倾向”、整体“先验倾向”和相应的统计显著性,即药物的
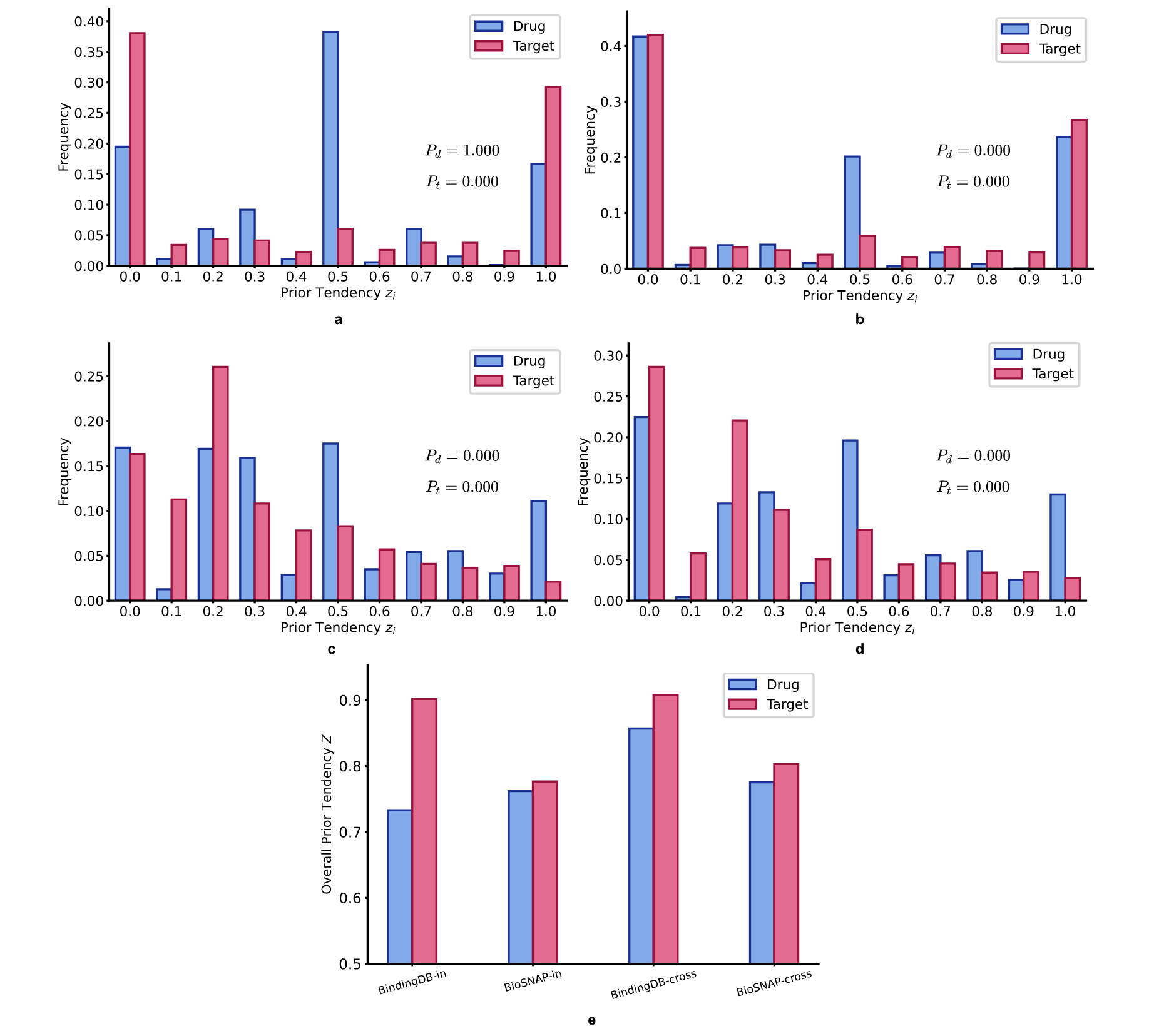
图3 | 展示了BindingDB和BioSNAP数据集中药物和靶点的“先验倾向”统计可视化。 a BindingDB领域内拆分训练集药物和靶点的“先验倾向”频率分布以及
为了验证究竟是“先验倾向”导致了偏差预测,对BindingDB数据集进行了重新拆分:药物偏差训练集:如图4a所示,首先从BindingDB领域内训练集选择所有正样本对,形成正样本集
平衡训练集:如图4d所示,首先从BindingDB领域内训练集选择所有正样本对,形成正样本集
在这两个反先验训练集上使用相同的超参数训练DrugBAN,并进行偏差测试。图4b和4c中的分类特征可视化显示,与图2中明显的(
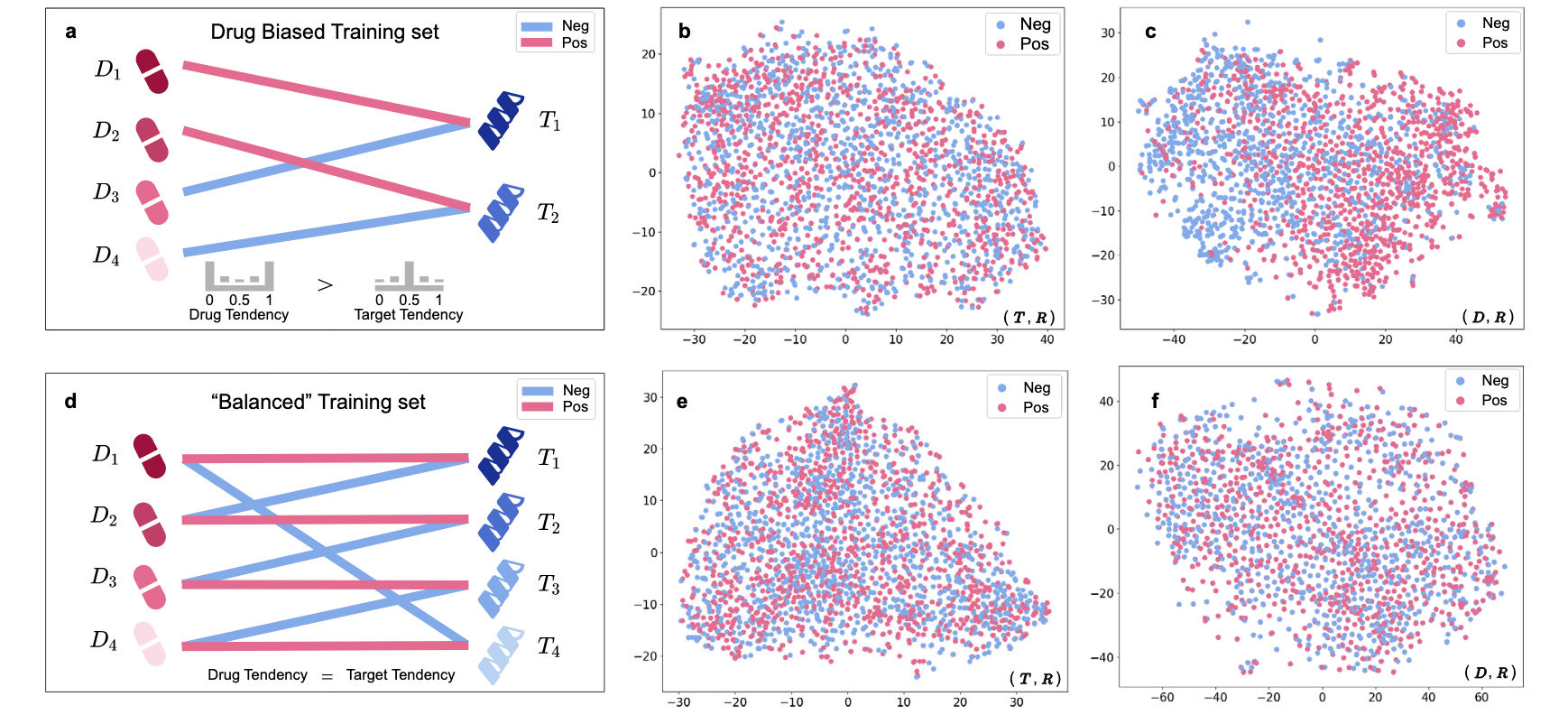
图4 | 展示了构建药物偏差和“平衡”训练集,以及DrugBAN分类特征(D, R)和(T, R)的t-SNE可视化。 a 药物偏差训练集构建示意图。b 在药物偏差训练集中使用(T, R)测试“靶点偏差”。c 在药物偏差训练集中使用(D, R)测试“药物偏差”。d “平衡”训练集构建示意图。e 在“平衡”训练集中使用(T, R)测试“靶点偏差”。f 在“平衡”训练集中使用(D, R)测试“药物偏差”。
2.4 先验倾向导致偏差预测
该研究,介绍了一种用于缓解药物-靶点相互作用预测中靶点先验偏差的干预式去偏框架(TAPB),如图5所示。TAPB框架与传统的DTI模型在本质上有所不同,主要体现在它集成了氨基酸随机化、混杂变量对齐模块和干预式训练,通过后门调整估算
图5c中的氨基酸随机化用于正则化输入序列。首先,随机删除ESM-2特征中70%的残基,以减少计算量并破坏序列模式。随后,每个残基特征以20%的概率进行独立突变,通过从氨基酸字典中随机采样替代。此双重随机化通过破坏标签特定的模式,防止学习虚假的关联。未使用无监督领域适应(UDA)技术,如CDAN,但在跨领域设置下取得了更好的结果,表明TAPB具有很强的泛化能力。需要注意的是,TAPB是一个去偏框架,替换编码器或聚合器可以进一步提升性能。TAPB的各个组成部分是通用的,后门调整的计算需要满足特定的假设。方法的伪代码在补充算法1中提供。
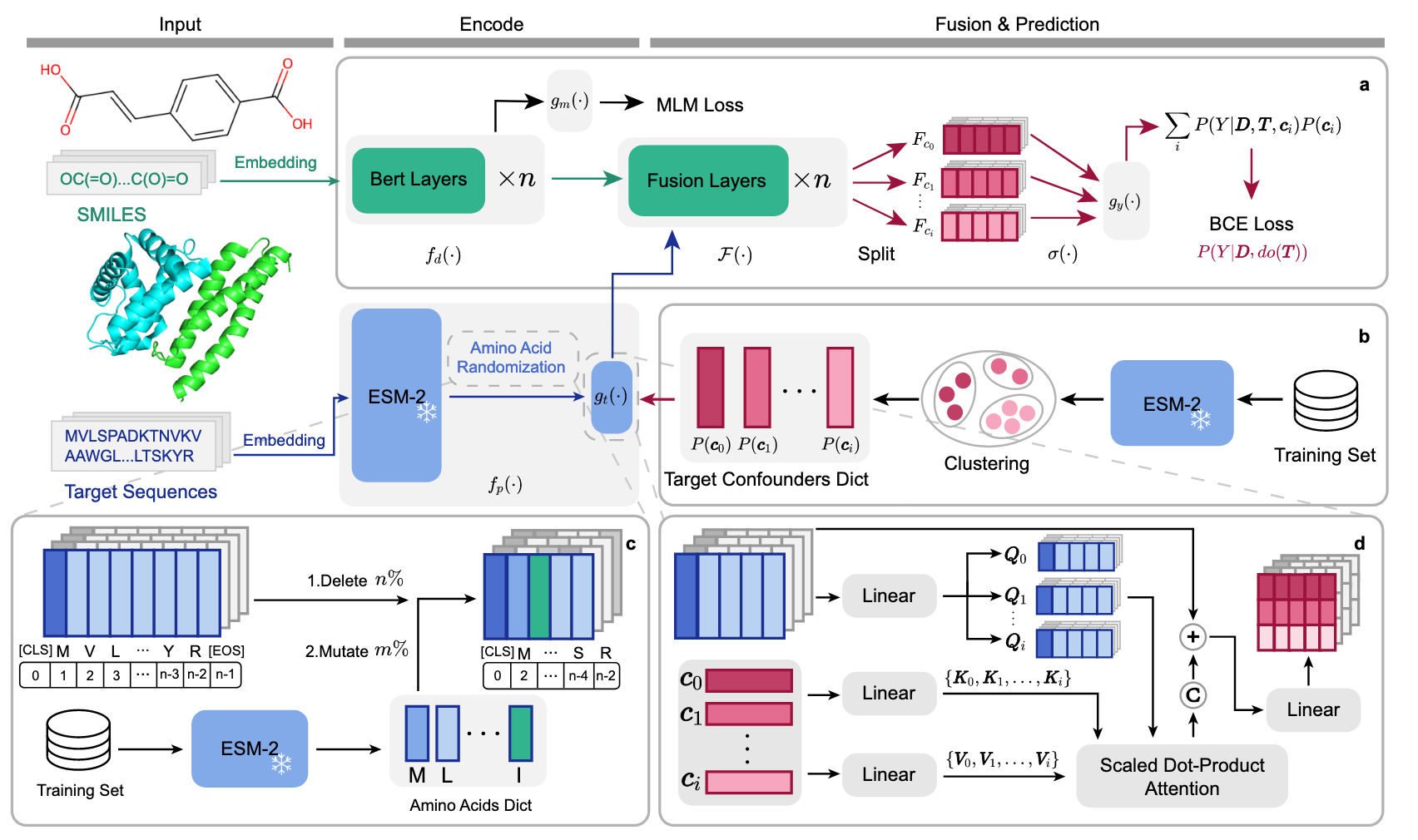
图5 | 展示了TAPB 框架的架构。 a TAPB干预式训练:药物编码器BERT
2.5 数据集和评估协议
为了确保严格和全面的评估,模型的分类性能在四个公开可用的数据集上进行评估,涉及六种设置:BindingDB和BioSNAP数据集的领域内和跨领域拆分,Davis数据集的领域内拆分,以及Human数据集的冷拆分。数据集的概述见补充说明1和补充表格1。此外,补充说明5和补充图3揭示了Davis数据集中的“靶点先验偏差”和Human数据集中的“药物先验偏差”。
对于领域内拆分,数据集被随机分为训练集、验证集和测试集,比例为7:1:2。值得注意的是,在这些领域内的场景中,靶点在训练集、验证集和测试集之间的重叠远高于药物。相比之下,由DrugBAN构建的跨领域拆分包含一个源领域训练集、一个目标领域训练集和一个目标领域测试集,源领域的药物/靶点与目标领域数据没有重叠(CVS4)。
对于所有数据集,进行了五次独立运行,并使用不同的随机种子,报告了接收操作特征曲线下面积(AUROC)、精确召回曲线下面积(AUPRC)、准确率、敏感性和特异性。使用Youden指数来调整最优阈值,以在敏感性和特异性之间提供更有效的平衡。对于领域内拆分,选择了验证集AUROC最高的模型检查点,并报告了测试集的性能。遵循DrugBAN的跨领域数据集协议,模型在源领域上进行训练,并直接在目标领域的测试集上进行评估,报告了相应的指标。
TAPB与五个基线模型——MolTrans、TransformerCPI、DrugBAN、PSICHIC和MlanDTI进行比较。与这些依赖
2.6 领域内比较
在BioSNAP数据集的领域内拆分上,TAPB在所有基线模型中表现出全面的优势,如图6a所示,其在每个评估指标上的多边形面积更大。根据补充表格3,与表现次好的基线模型PSICHIC相比,TAPB显示了显著的提升:AUROC提高了2.3%;AUPRC提高了2.2%;准确率提高了2.7%;敏感性提高了3.4%;特异性提高了1.9%。这些具有统计显著性的提升凸显了TAPB在缓解靶点先验偏差方面的有效性。尽管在BindingDB数据集中存在严重的“靶点偏差”,在该数据集中模型通过仅仅记住靶点即可获得高性能,导致所有基线模型都取得了强劲的结果,但仍然进行了公平比较。如图6b所示,TAPB保持了强劲的竞争力,在AUROC上仅比表现最好的方法DrugBAN落后0.2%,在AUPRC上落后0.3%,补充表格3显示了这一点,表明其具有竞争力的性能。
值得注意的是,TAPB在Davis数据集上的每个指标上都超过了所有基线模型,如图6c所示。补充表格3确认,TAPB在AUROC上超过了最强的基线模型PSICHIC 2.1%,在AUPRC上超过了7.4%——这是所有数据集中观察到的最大性能差距。这凸显了TAPB在捕捉复杂相互作用模式方面的卓越能力。在Human数据集的冷拆分场景中,“药物偏差”是主导偏差,该研究团队也在这种相反条件下评估了TAPB的表现。如图6f所示,TAPB保持了竞争力的表现,在AUROC上超过了DrugBAN 2.0%,在AUPRC上超过了2.8%,并且在五个基线模型中超越了三者。这个结果表明,尽管TAPB是为了缓解“靶点偏差”而设计的,但它在药物偏差数据集上也取得了强劲的表现,突显了其在预期应用场景之外的稳健性和泛化能力。
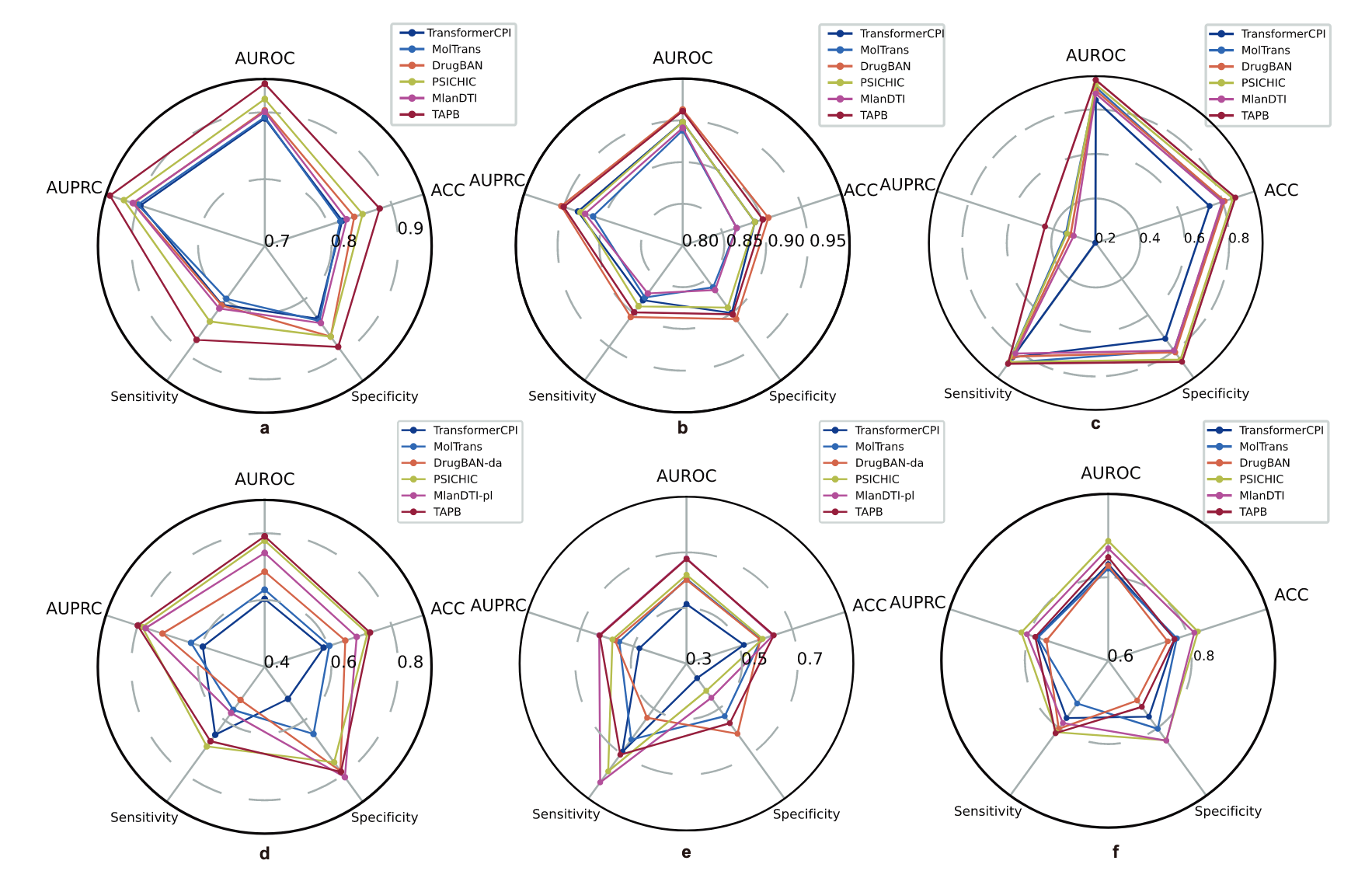
图6 | 展示了在BioSNAP、BindingDB、Davis和Human上的性能雷达图比较。 a BioSNAP的领域内评估。b BindingDB的领域内评估。c Davis的领域内评估。d BioSNAP的跨领域评估。e BindingDB的跨领域评估。f Human的冷拆分评估。所有数据集均进行了五个不同随机种子的实验。源数据已提供为源数据文件。
2.7 跨领域比较
如图6d和6e所示,TAPB展示了出色的跨领域泛化能力。补充表格4提供了全面的性能比较。在BindingDB数据集的跨领域拆分中,TAPB保持了强劲的竞争力,取得了显著的结果,AUROC为0.676,准确率为0.630,特异性为0.565,同时在AUROC上超过DrugBAN-da(使用CDAN)7.5%,在AUPRC上超过5.8%,在准确率上超过5.1%。值得注意的是,即使没有使用领域适应技术,TAPB仍然优于DrugBAN-da,表现出更强的泛化能力,这验证了去偏框架的有效性。图6d展示了BioSNAP数据集跨领域拆分的结果,供有兴趣的读者参考。
TAPB强大的跨领域泛化能力源于其缓解靶点先验偏差的核心方法,从而避免了对虚假靶点-标签相关性的依赖。传统模型依赖
2.8 消融实验
在三个数据集上进行了消融研究:Davis数据集的领域内拆分,以及BioSNAP和BindingDB数据集的跨领域拆分。使用了七个TAPB变体,这些研究旨在全面评估关键组件的影响:(1)TAPB-CNN:用未经训练的CNN替换ESM-2编码器;(2)TAPB-Base:基线双塔架构,使用ESM-2编码器、二分类损失和平均池化(没有干预式训练);(3)TAPB-R:在TAPB-Base的基础上增强了氨基酸随机化;(4)TAPB-RM:在TAPB-R的基础上增加了掩蔽语言建模(MLM)损失;(5)TAPB-RMBA:在TAPB-RM的基础上加上了后门调整(没有CAM,省略了
鉴于ESM-2强大的表示能力,通过将其在TAPB-Base架构中的使用去除,并用随机初始化的CNN编码器替代(即TAPB-CNN),进行消融测试。如图7a–c所示,TAPB-Base显著优于TAPB-CNN,证明了预训练蛋白质编码器的优势。同时,为了确认ESM-2特征不会引发“靶点偏差”,对前面章节中引入的“平衡”数据集进行了“靶点偏差”和“药物偏差”测试,详细内容见补充说明3。如补充图2所示,两个测试均未表现出类似于图2中的聚类现象,表明纳入ESM-2编码器增强了靶点表示能力,并且不会引发“靶点偏差”,这种偏差主要是由数据引发的。
氨基酸随机化显著提高了模型性能,并作为防止模型记住靶点的最直接方法,从而避免了对相互作用模式的学习不足。如图7a–c所示,TAPB-R在所有三个数据集上持续取得比TAPB-Base更高的AUROC和AUPRC,尤其在Davis数据集上,验证了随机化策略的有效性,并证明了防止靶点记忆的合理性。TAPB-RM在所有三个数据集上略微优于TAPB-R和TAPB-Base。尽管相较于氨基酸随机化,性能提升较不显著,但药物MLM损失有效加强了药物在靶点偏向数据集中的表示,从而减少了靶点的影响。
根据理论分析,TAPB需要CAM和后门调整来计算
为了测试该研究团队方法的泛化性,将残基随机删除集成到DrugBAN(Non_DA)和TransformerCPI中,分别使用补充表格16和表格9中指定的超参数。由于该研究团队的残基突变和干预式训练需要预训练的编码器和基于MHCA的聚合,因此仅选择了残基随机删除。图7d展示了BioSNAP数据集跨领域拆分上的结果,使用随机删除的TransformerCPI在AUROC和AUPRC上分别有近10%的显著提升,而使用随机删除的DrugBAN在AUROC上相较基线有1%的提高。这一差异可能源于TransformerCPI的聚合器架构更适合在该修改下建模DTI。结果确认了该研究团队的残基随机删除是一种通用、模型无关的设计。
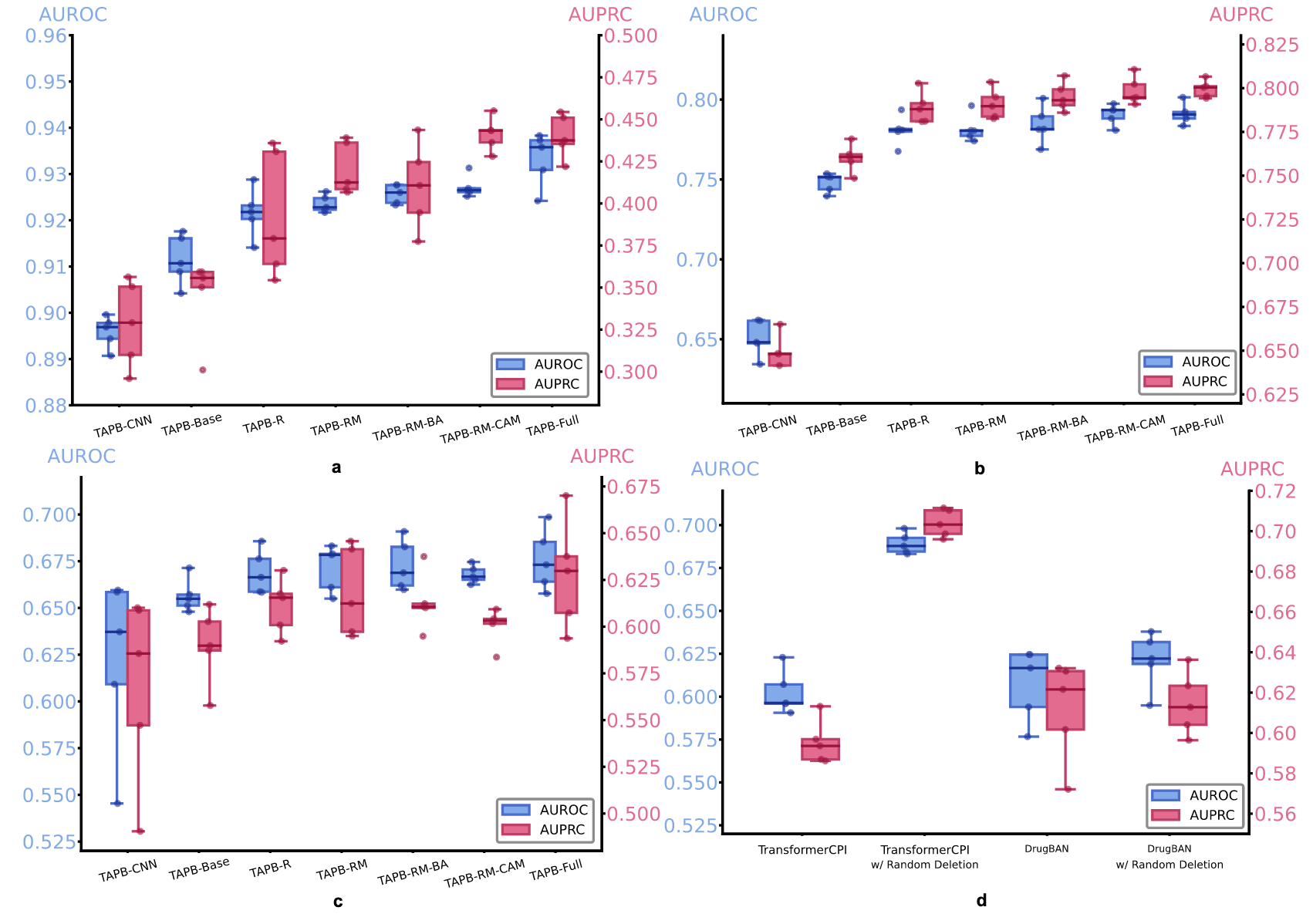
图7 | 展示了消融研究结果。 a TAPB关键组件在Davis数据集上的消融研究。b TAPB关键组件在BioSNAP数据集跨领域拆分上的消融研究。c TAPB关键组件在BindingDB数据集跨领域拆分上的消融研究。d TransformerCPI、使用随机删除的TransformerCPI、DrugBAN和使用随机删除的DrugBAN在BioSNAP数据集跨领域拆分上的AUROC和AUPRC。消融研究在所有数据集上使用五个不同的随机种子进行。箱形图显示中位数(中心)、25-75百分位数(箱体边界)和3倍IQR内的最小值和最大值(须)。单个数据点(n = 5)叠加展示。源数据已提供为源数据文件。
2.9 TAPB的可解释性
TAPB在分子和氨基酸层面提供了有价值的见解,为药物重定位提供了有用的信息。该模型在聚合器的最后一层使用了八个注意力头,每个注意力头捕捉到不同的相互作用模式。这些注意力图被可视化,以解释模型的关注点。为了突出潜在的结合位点,该研究团队通过对多个注意力头进行平均,聚合了多头注意力图,从而为药物和靶点生成了单独的注意力得分。这些得分与通过X射线晶体学获得的真实配体-蛋白相互作用图进行比较,配体的相互作用在5 Å半径内的接触被可视化。
对于靶点,基于注意力图,重点突出结合位点周围的关键区域,重要的氨基酸用不同颜色标记。模型预测的与真实相互作用匹配的区域用红框标出。此外,药物注意力图中排名前五的原子被可视化,表示它们对结合的预测贡献,使用RDKit进行可视化。对接计算使用AutoDock Vina(v1.2.5)进行。二维配体-蛋白相互作用图使用Maestro(v13.5,Schrödinger LLC)中的配体相互作用图模块生成,三维相互作用图使用PyMOL(v2.5,Schrödinger)准备,其他残基、二级结构元素和表面图以灰色显示。选择了来自Protein Data Bank(PDB)的两个正向共结晶结构:Aloisine A(PDB ID:1UNG)和Elaidamide(PDB ID:1KQU),来自BioSNAP领域内测试集。
对于PDB ID:1UNG,Aloisine A(RP107)是一个有效的周期蛋白依赖性激酶(CDK)抑制剂。TAPB的药物注意力图识别了这些氢键和相互作用位点,分别在二维和三维对接图中显示,如图8a0、a1、a3、a5和a7、b、d所示。模型捕捉到一个氮原子作为氢键受体,作用于CYS83的主链,另一个氮原子作为氢键供体与同一残基相互作用,如图8a0和a7所示。此外,图8c0、c1、c4、c5、c6和c7强调了CYS83在配体-蛋白结合中的作用,进一步验证了TAPB在精确检测真实结合位点方面的能力。
对于PDB ID:1KQU(与底物类似物复合的人类磷脂酶A2),Elaidamide是一种脂肪酸酰胺,已在睡眠不足的猫的脑脊液中发现,并抑制人类滑膜磷脂酶A2(PLA2)。TAPB准确识别了这些相互作用位点:羟基作为氢键供体与GLY29的主链相互作用,氨基作为氢键供体与HIS47和ASP48相互作用,如图8e0、e7、g0、g1、g4和g5所示。靶点注意力图正确地突出了GLY29、HIS47和ASP48在配体-蛋白结合中的重要性,如图8g0、g1、g4和g5所示。该研究团队再次展示了三维和二维对接图,显示了两个氢键在5 Å内,如图8f和h所示。
尽管先前研究中的DTI模型提供了可解释性并能够揭示隐藏的相互作用,但它们是在偏差数据上训练的,容易受到“靶点先验偏差”的影响,从而可能导致对真实相互作用的错误预测。TAPB有效地识别并缓解了这种偏差,显著提高了相互作用检测的准确性。因此,TAPB为下游的计算筛选和实验验证提供了更可靠的预测。
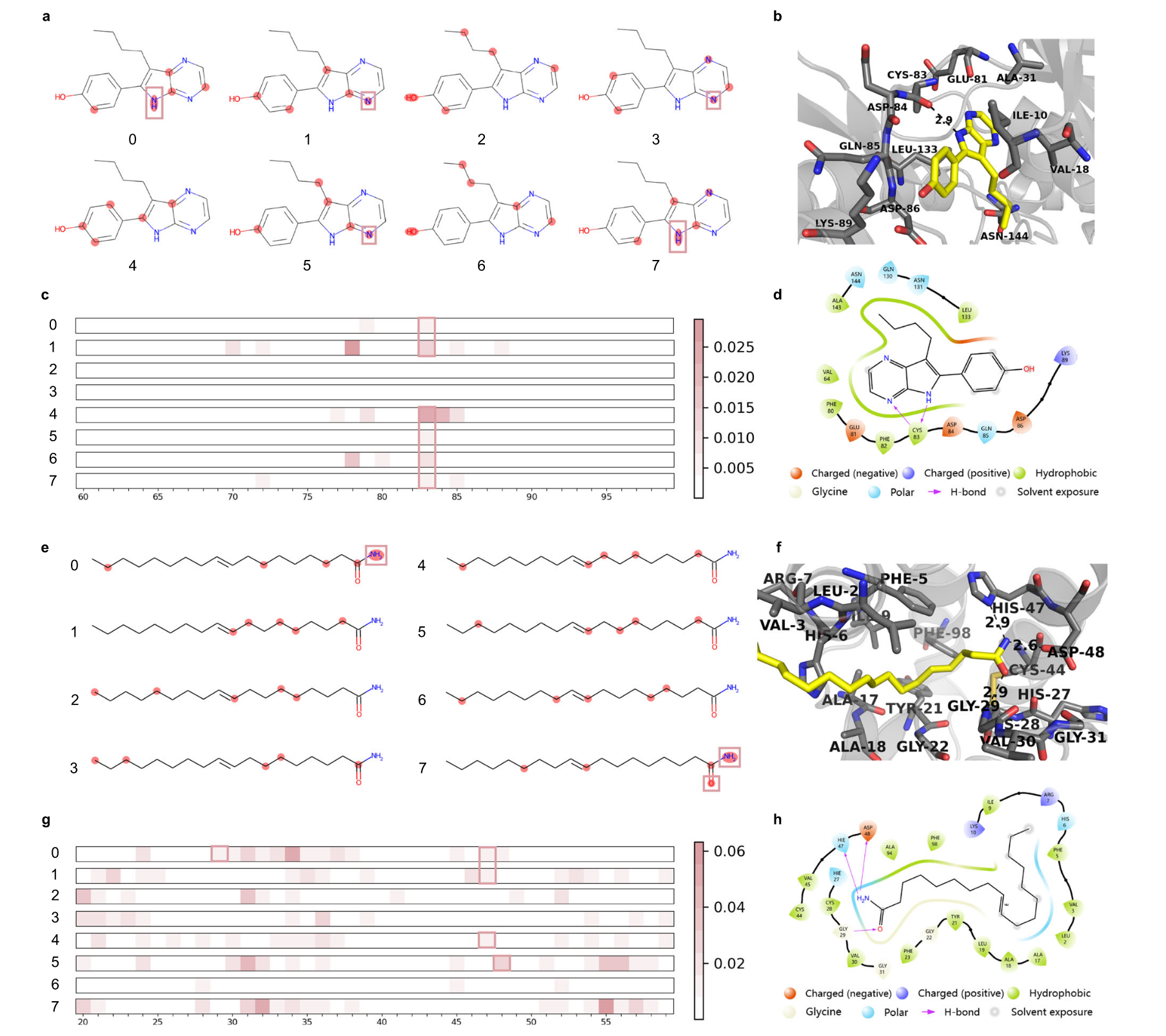
图8 | 展示了TAPB的注意力图可视化及与实际配体-蛋白结合位点的比较。 a 药物Aloisine A的二维结构,基于注意力图突出的前五个原子。b Aloisine A在配体-蛋白复合物结构中的相互作用和真实结合位点(PDB ID:1UNG)。c 1UNG的靶点注意力图,突出显示蛋白质结构中的氨基酸。d Aloisine A在配体-蛋白复合物结构中的相互作用和真实结合位点(PDB ID:1UNG)。e 药物Elaidamide的二维结构,基于注意力图突出的前五个原子。f Elaidamide在配体-蛋白复合物结构中的相互作用和真实结合位点(PDB ID:1KQU)。g 1KQU的靶点注意力图,突出显示蛋白质结构中的氨基酸。h Elaidamide在配体-蛋白复合物结构中的相互作用和真实结合位点(PDB ID:1KQU)。
三维相互作用图中的虚线表示氢键。
3 总结
该研究成功识别并缓解了“靶点先验偏差”,这一现象在以往的研究中未受到足够重视。通过一系列实验,确认了“先验倾向”,即靶点标签分布的不平衡,是一个混杂变量,导致DTI预测中靶点与预测之间的虚假关联。提出的TAPB框架通过氨基酸随机化、混杂变量对齐模块(CAM)和干预式训练有效解决了这一偏差。这些方法不仅提高了模型的泛化能力,还增强了预测能力,最终使模型变得更加稳健和可靠。
该研究中的“先验倾向”概念不仅仅局限于数据集中标签的分布。它涵盖了可能来源于多种因素的更广泛偏差,包括药物中的特定功能团、靶点中的子序列,甚至其他非序列特征。这种偏差可能导致模型捕捉到虚假的关联,而非真实的药物-靶点相互作用,从而损害了模型的泛化能力。缓解偏差的方法不仅限于后门调整。包括多模态框架中的对比学习方法(例如CLIP和ConPLex)也能有效应对这种偏差。然而,必须认识到,消除先验偏差并不能保证完全去除所有偏差,因为其他形式的偏差可能依然存在。要实现真正准确和可靠的DTI预测,仍然是一个持续的挑战,亟需持续的研究努力和方法创新。未来的DTI模型应在尽可能没有这种偏差的数据集上进行训练,并应基于生物学度量进行评估,而不仅仅是算法性能。这些生物学度量可能与训练数据中的标签不同,从而迫使模型揭示更真实的相互作用。
尽管TAPB准确预测了Aloisine A与1UNG、Elaidamide与1KQU的结合位点,但它也产生了大量噪声。例如,图8a和8e中的仅少数几个注意力头探讨了真实的结合位点,且这些头在预测这些位点时的一致性较低。同样,对于靶点,如图8c和8g所示,每个氨基酸的注意力权重相对较小,这可能是由于序列较长以及Softmax归一化的原因。此外,专注于药物与靶点相互作用的注意力头是不同的,这表明模型可能没有完全同步相关的注意力机制。这种不一致意味着TAPB的预测并不完全稳定,可能会受到潜在偏差的影响,类似于“靶点先验偏差”。
无监督领域适应(UDA)技术(例如DrugBAN中使用的CDAN和UdanDTI中的MCD)要求同时访问源领域和目标领域数据进行模型适应,这通常会改善跨领域的泛化性能。相比之下,该研究旨在探索一种更通用且便捷的零-shot预测范式,其中TAPB仅使用源领域训练集。该方法避免了重复构建目标领域数据集和针对新药物或靶点重新训练的计算负担和应用复杂性。与UdanDTI的比较见补充说明6和补充表格17、18。
尽管已有许多努力在构建无偏数据集方面取得进展,但创建一个完全无偏的DTI数据集仍然具有挑战性。在该研究中,提供了从因果角度解决偏差的新见解。该研究团队的发现不仅仅局限于DTI预测,因为“先验倾向”现象可能在其他领域也普遍存在。未来的研究可以探索TAPB在其他领域的应用,例如DTA、多视角融合或VQA等,这些领域也可能存在类似的偏差。此外,从因果角度进一步探讨“先验倾向”的机制,可能会促进开发出更稳健的模型,减少对虚假关联的敏感性。随着DTI预测的不断发展,因果推断技术的整合将在确保模型捕捉到真实相互作用并有效泛化到新数据方面起到关键作用。
由于混杂变量是不可观察的,该研究团队尝试实现基于代理变量的混杂调整方法(参考文献44)。然而,当将其集成到深度学习管道中时,遇到了显著的计算挑战,特别是在矩阵求逆过程中分布的可靠估计和数值不稳定性方面。值得注意的是,目前的生物学数据集限制了代理变量仅能使用序列衍生的特征,这可能不足以捕捉到完整的混杂生物学机制的范围。未来引入多模态数据(如结构或功能注释)可能通过提供正交信息源来增强代理变量的质量,从而更好地逼近潜在的混杂因素。尽管因果推断理论为不可观察的混杂因素提供了有原则的解决方案(例如参考文献44–46),但适应深度学习框架仍然不是一项简单的任务。该研究团队在讨论中明确承认这些局限性,并将在未来的工作中优先弥合这一方法论差距,特别是关注多模态代理变量的优化。
尽管该研究团队对