NC 2023 | VisNet: 基于等变向量–标量交互消息传递的分子几何表示增强方法
今天介绍的这项工作来自 Nature Communications。近年来几何深度学习在分子建模领域引起了显著关注。随着等变图神经网络的发展,模型在预测分子能量、力以及量子化学性质方面逐渐逼近从头算的精度。然而,这类模型在实际应用中仍面临两类主要挑战:一类是对几何信息的利用不足,导致模型难以充分捕捉分子结构中的关键空间关系;另一类则是高阶几何张量计算成本过高,使得模型难以扩展到更大、更复杂的分子体系。针对这些技术瓶颈,一种称为ViSNet的等变几何增强图神经网络方法被提出。该方法的核心在于设计了运行时几何计算策略,通过向量表示隐式获取角度、二面角和不正确扭转角等多类几何信息,使计算复杂度从常见显式方法的

获取详情及资源:
- 📄 论文: https://www.nature.com/articles/s41467-023-43720-2
- 💻 代码: https://github.com/microsoft/AI2BMD/tree/ViSNet
0 摘要
几何深度学习正持续推动分子建模领域的革新。尽管当前的神经网络模型在分子性质预测方面已逐渐逼近从头算精度,其在药物发现与分子动力学模拟等应用中的表现仍受到两类因素的限制:一是对几何信息的利用仍不充分,二是高计算成本带来的效率瓶颈。针对这些问题,一种名为ViSNet的等变几何增强图神经网络被提出。该方法能够以低计算开销有效提取几何特征,并对分子结构进行高效建模。ViSNet在多个分子动力学基准上表现优于已有方法,包括MD17、修订版MD17和MD22,并在QM9与Molecule3D数据集上展现出优异的化学性质预测能力。此外,通过一系列模拟与案例研究,ViSNet能够高效探索分子构象空间,并能在几何表征与分子结构之间提供合理的可解释性。
1 引言
分子建模在当代科学与工程领域中具有重要地位,有助于理解化学反应、推动新药研发,并促进科学与技术的发展。密度泛函理论是分子建模中常用的方法之一,能够精确计算分子的能量、力以及其他化学性质。然而,由于其计算需求极高,特别是在处理大规模分子体系或高精度计算时,常常需要大量计算资源与时间。机器学习提供了另一种选择,通过从参考数据中学习以接近从头算的精度,同时具有更高的计算效率。梯度域机器学习方法利用能量守恒与从头算分子动力学轨迹中的有限样本构建精确的分子力场,使得在保证精度的前提下实现高效模拟。其后基于对称性的扩展进一步将物理对称性引入力场构建,使结果能够达到处理柔性分子的高精度水准。迭代式的扩展方法又将这一思路推广到包含数百原子的体系,能够保持原子间关联并准确描述复杂分子与材料。
近年来,深度学习因其能从原始数据中自动学习特征而备受关注,但其对大量数据的依赖成为在更多场景中应用的限制。为了缓解深度学习势能模型对数据的依赖,一些研究将对称性的归纳偏置引入神经网络设计,形成几何深度学习的方向。对称性描述物理规律在平移与旋转等变换下保持不变,使得相关模型能够在有限数据场景中无需数据增强即可学习稳定的表示。
在几何深度学习框架中,等变图神经网络是一类重要方法,具备强大的分子几何建模能力。一类常见方法通过方向信息实现等变性,并将几何特征用于预测分子性质。例如,某些模型在已有的结构上加入二面角信息,通过显式的几何张量提取欧氏空间中的几何关系;也有方法采用向量嵌入并通过向量自身的内积隐式处理角度信息,从而降低显式几何提取的复杂度。另一类主流方法借助群表示论实现等变性,能达到更高精度,但计算成本相当高。这类方法利用高阶几何张量,在多个分子动力学数据集上获得领先性能,但由于需要计算繁重的Clebsch–Gordan积,虽然适用于具有周期性的系统与精心设计的模型结构,却难以在大规模非周期分子体系(如化学或生物分子)中实际应用。
针对计算成本与几何信息利用不足之间的矛盾,该研究提出ViSNet,即“Vector-Scalar interactive graph neural Network”。该方法通过运行时几何计算策略以线性时间隐式提取多种几何特征,包括角度、二面角与不正确扭转角,符合经典分子动力学力场中的几何构成,从而显著加速训练与推理并降低内存消耗。同时,为了扩展向量表示,模型引入球谐函数,并以内积取代计算代价高昂的Clebsch–Gordan积。进一步地,精心设计的向量–标量交互式等变消息传递机制能够在向量与标量表征之间高效交换几何信息,使模型能够充分利用结构中的空间信息。
在多个基准数据集的综合评估中,ViSNet在MD17、修订版MD17与MD22上的所有分子均优于现有方法,并在QM9与Molecule3D数据集上展现出卓越表现,说明其在分子几何表征中的强大能力。该模型还在一个大型量子化学竞赛的数据集上获得领先成绩。进一步的实验表明,当模型仅用950个样本训练后,用其驱动MD17中的分子进行分子动力学模拟时,所得的原子间距离分布及势能面与从头算模拟高度一致,显示出数据效率与高保真度。为进一步评估其在真实应用中的价值,研究中使用了一个包含约一万种构象的166原子小蛋白Chignolin数据集,构象由复制交换分子动力学得到,并经DFT计算。在该数据集上的评估结果显示,ViSNet显著优于经验力场,且其模拟所得到的受力计算与DFT非常接近。此外,模型在几何表征与分子结构之间展现出合理的可解释性。
该方法的贡献可概括为:通过RGC模块以线性时间隐式提取多类高阶几何特征;通过向量–标量交互机制充分挖掘几何信息;在多个任务中取得领先的能量、力及量子性质预测结果;在小分子与大体系如Chignolin上实现高保真的分子动力学模拟;并在几何表征与分子结构之间提供了合理的解释能力。
2 结果
2.1 ViSNet概述
ViSNet是一种灵活的等变图神经网络,可根据原子坐标与原子序数预测分子的势能、原子受力以及多种量子化学性质。如图1a所示,模型由嵌入模块、多层堆叠的ViSNet模块以及输出模块组成。原子序数与原子坐标首先输入嵌入模块,随后经过ViSNet模块以提取并编码几何表征,最终由输出模块根据几何表征预测分子性质。需要注意的是,ViSNet属于能量守恒型势能模型,即其预测的原子受力来自势能对坐标的负梯度。
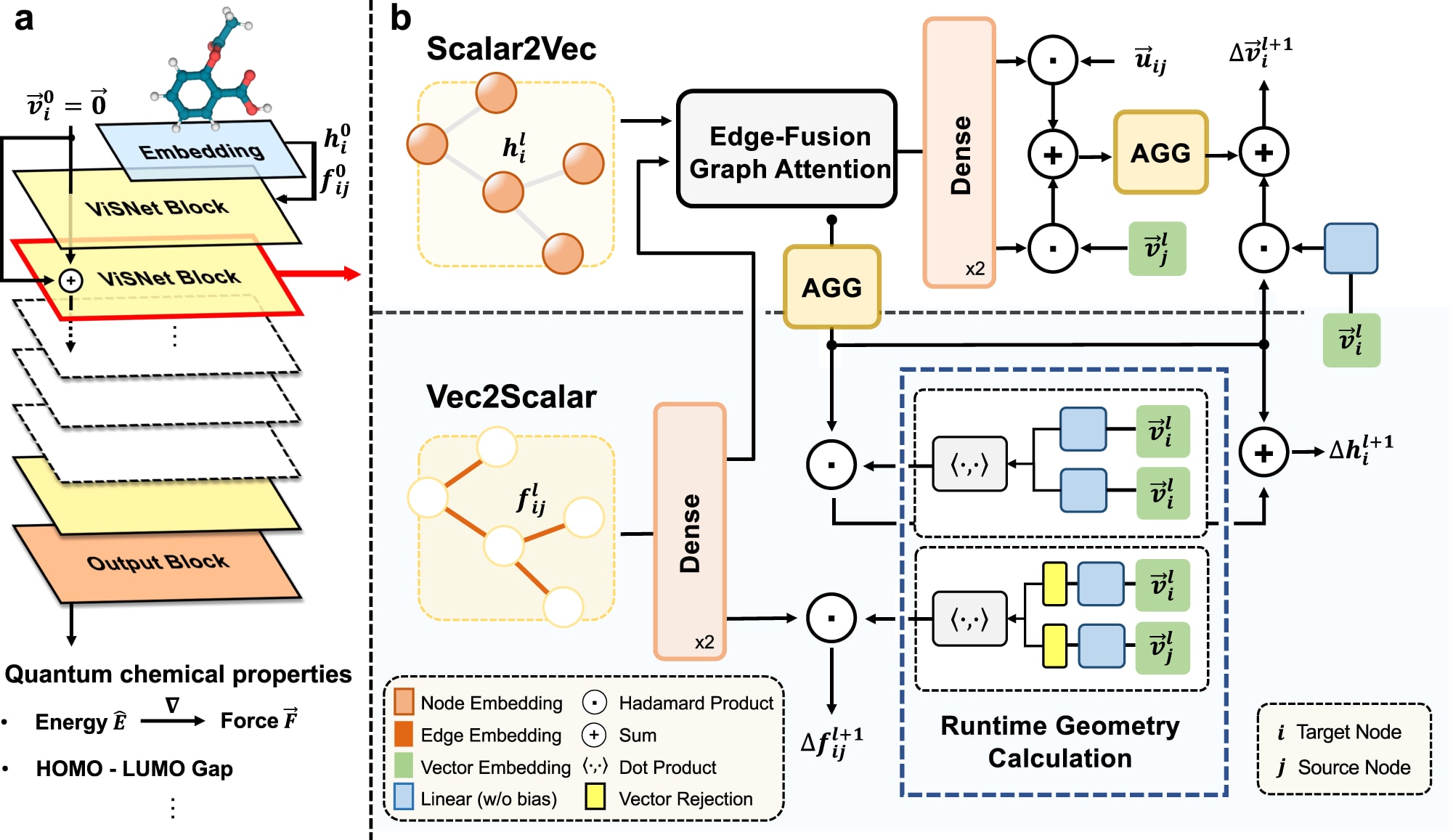
**图1 | ViSNet的整体架构a ViSNet的模型示意图。**ViSNet嵌入分子的三维结构,并通过一系列ViSNet模块提取几何信息,最终通过输出模块预测能量、力与HOMO–LUMO能隙等分子性质。b 单个ViSNet模块的流程图。一个ViSNet模块包含两个子模块:Scalar2Vec用于将标量嵌入附加到向量上;Vec2Scalar基于RGC策略更新标量嵌入。流程包括节点嵌入、边嵌入、方向单元、相对位置等输入,以及通过注意力与残差机制更新向量与标量嵌入。
经典力场的成功经验表明,诸如原子间距离、键角、二面角以及图2中的不正确扭转角等几何特征在决定分子总势能时具有重要作用。然而,以往研究对这些不变量几何特征的显式提取往往会在训练与推理过程中带来较高的时间或内存消耗。对于某一原子,其角度信息的计算复杂度随邻近原子数呈
以图2所示由四个原子组成的示例结构为例,目标节点
方向单元
其中
方向单元内积表示了从节点
与运行时角度计算类似,还可以计算节点
向量拒斥量定义如下:
其中
不正确扭转角来源于由四个节点形成的金字塔结构。在图2的示例中,节点
类似地,两者内积代表边
多项研究验证了高阶几何张量在分子建模中的有效性。然而,这些方法通常依赖Clebsch–Gordan积的计算,使得其计算成本过高,妨碍其在大体系中的应用。该研究在RGC思想基础上,将向量转换为球谐函数形式的高阶表示,同时以内积替代CG积。依据加法定理,扩展后的高阶张量仍可以以勒让德多项式的方式表示角度信息:
其中
为充分利用几何信息并增强标量与向量之间的交互,设计了向量–标量交互式消息传递机制,用于分别在节点与边的交互中处理角度与二面角信息。与以往主要更新节点特征的研究不同,该方法在消息传递中同时更新节点与边特征,使几何表示更加完整。该机制的关键操作如下:
其中
总而言之,RGC策略通过内积提取几何特征,而在ViS-MP中,标量嵌入与向量嵌入彼此循环更新,使模型能够从分子结构中学习到更为完整的几何表征。
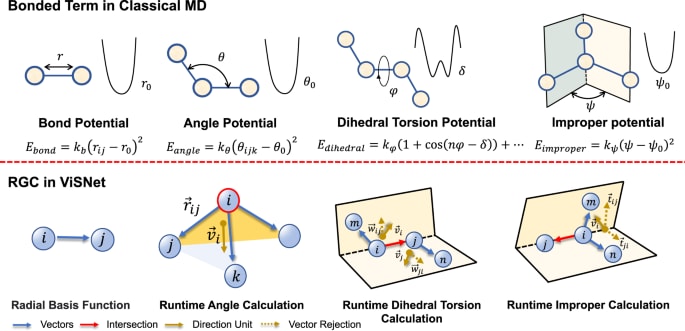
**图2 | 运行时几何计算(RGC)模块示意及其与经典分子动力学键合项的关系键合项包括键长、键角、二面角与不正确扭转角。**RGC模块以线性时间复杂度表示经典分子动力学的所有键合项。黄色箭头表示方向单元
2.2 精确的量子化学性质预测
为了评估ViSNet在量子化学性质预测中的表现,模型在多个主流基准数据集上进行了测试,包括MD17、修订版MD17、MD22、QM9、Molecule3D以及OGB-LSC PCQM4Mv2,这些任务涉及能量、力以及其他分子性质的预测。MD17包含七种小型有机分子的分子动力学轨迹,每种分子拥有从数十万到近百万不等的构象数。修订版MD17在数值精度上更为可靠。MD22是近年提出的更大规模的分子动力学数据集,其分子大小显著增加,范围从42个原子到370个原子,涵盖蛋白、脂质、碳水化合物、核酸以及超分子等复杂体系。QM9包含133385个小型有机分子,提供12类量子化学性质。Molecule3D则包含3899647个来自PubChemQC的数据,其基态结构与性质均基于DFT计算。ViSNet在此任务中侧重预测HOMO-LUMO能隙。PCQM4Mv2源自大型量子化学数据库,包含数百万分子的DFT计算HOMO-LUMO能隙,训练集中提供部分分子的三维结构,而验证与测试集不提供三维信息。
在这些任务中,ViSNet与多种先进方法进行了对比,包括DimeNet、PaiNN、SpookyNet、ET、GemNet、UNiTE、NequIP、SO3KRATES、Allegro、MACE等。结果显示,无论是小分子(MD17与修订版MD17)还是大分子体系(MD22),ViSNet在能量与力的预测上均取得最低的平均绝对误差。相较于采用较低几何阶数或未充分利用几何交互的模型,ViSNet通过强化几何特征的提取与交互获得性能提升;而相较于依赖高阶群表示且计算成本较高的模型,ViSNet通过球谐函数与内积替代Clebsch–Gordan积也展现出高效且准确的特性。
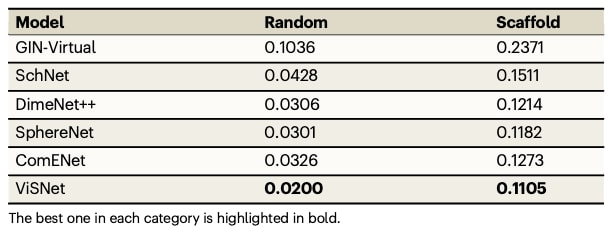
在QM9数据集上,ViSNet在12类量子化学性质中,有9项取得最佳表现,其他性质也达到了接近领先的结果。在Molecule3D的测试中,ViSNet在随机划分与骨架划分下均表现显著优于现有方法,尤其在随机划分中取得大幅性能优势。此外,ViSNet具有良好的可迁移性,可与其他多模态方法结合,例如Transformer-M,并在PCQM4Mv2数据集上取得突出成绩,成功获得NeurIPS 2022大赛PCQM4Mv2赛道的第一名。
为了进一步评估计算效率,ViSNet的推理时间也与多种模型进行了比较。延迟被定义为在一组给定坐标上计算力(即梯度)所需的时间。结果显示,在相同网络深度下,ViSNet显著快于部分高阶张量模型。例如,ViSNet在两层结构下比MACE节省约42.8%的时间;虽然Allegro通过高效的CG积实现了加速,但ViSNet在不同深度下仍保持更低的延迟,显示其在精度与效率上的平衡优势。
表1|MD17数据集中7种小型有机分子的能量(kcal/mol)与受力(kcal/mol/Å)平均绝对误差,与先进算法的对比
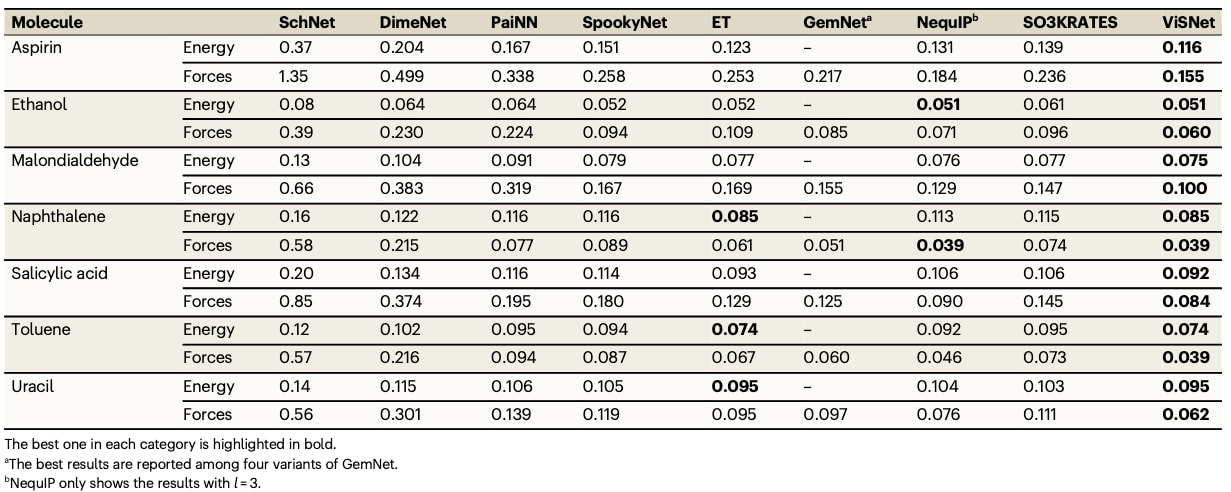
表2|rMD17数据集中10种小型有机分子的能量(kcal/mol)与受力(kcal/mol/Å)平均绝对误差,与先进算法的对比
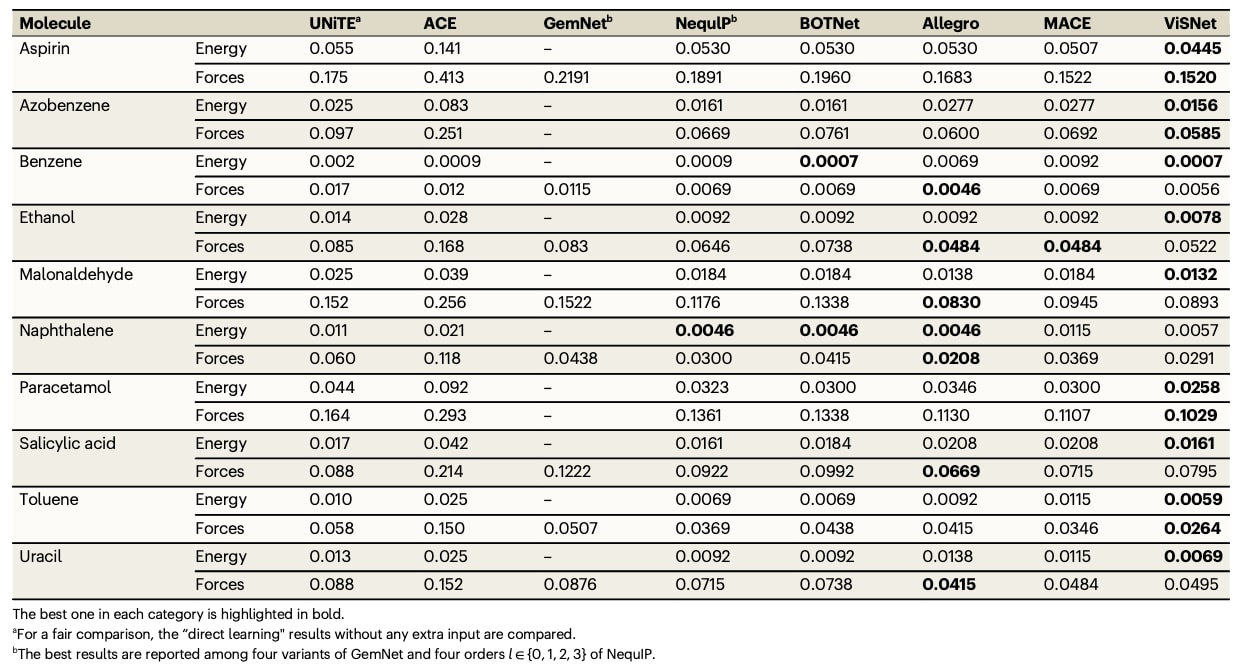
表3|MD22数据集中7种大规模分子的能量(kcal/mol)与受力(kcal/mol/Å)平均绝对误差
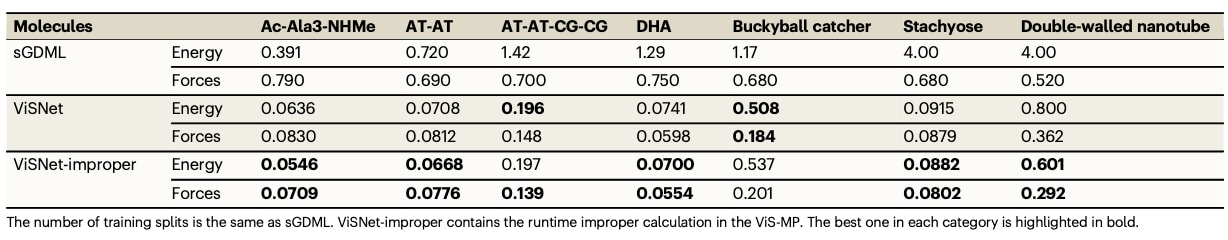
表4|QM9数据集中12类分子性质的平均绝对误差,与先进算法的对比
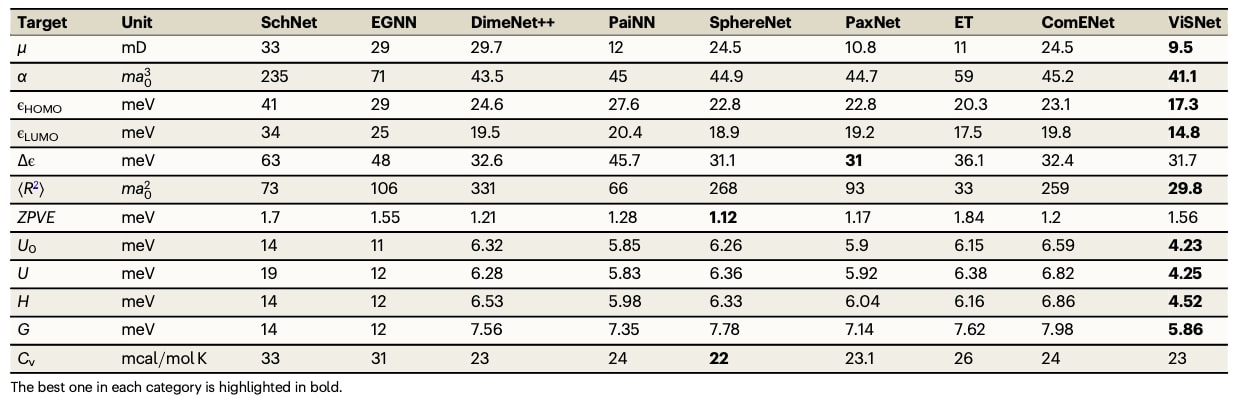
表5|Molecule3D测试集中HOMO–LUMO能隙(eV)在随机划分与骨架划分下的平均绝对误差,与先进算法的对比
2.3 高效的分子动力学模拟
为了评估ViSNet在分子动力学模拟中的潜力,该研究将仅使用950个样本训练得到的ViSNet模型引入ASE模拟框架,对MD17数据集中全部七种有机分子执行分子动力学模拟。模拟时间步长设为
相较于DFT,已有多项基于机器学习的力场方法,如sGDML、ANI、DPMD与PhysNet,已展示出在分子动力学模拟中显著的加速优势。ViSNet同样具备这一特性,其计算成本相对于DFT也大幅降低,相关结果可由补充材料中的数据与图表进一步说明。
为了进一步检验ViSNet在分子动力学性质方面的表现,在MD17数据集中以乙醇为对象进行了500 ps的NVE系综模拟,时间步长为
对于Ac-Ala3-NHMe,则通过Ramachandran图比较了三种方法的采样能力。自由能景观通过平均力势估计,
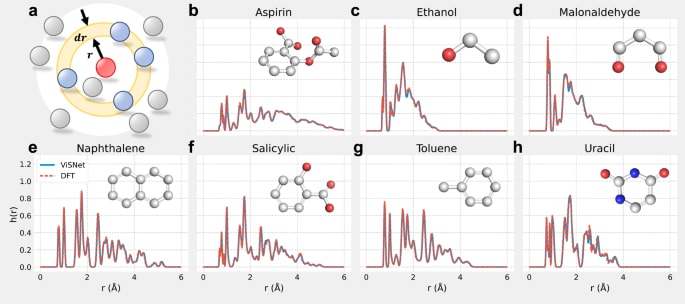
图 3|由ViSNet与DFT驱动的分子动力学模拟中原子间距离分布 a 示意图展示以任意原子为中心、半径为
2.4 全原子蛋白体系中的应用
为了检验ViSNet在真实场景中的适用性,研究中选用了包含166个原子的迷你蛋白Chignolin(如图4a所示)。该蛋白的数据集由复制交换分子动力学采样而来,包含约10000种不同构象,并通过DFT计算得到对应能量与受力信息。数据集按照8:1:1的比例划分为训练集、验证集与测试集。在这一数据集上,ViSNet与多种先进的机器学习力场进行了比较,包括ET、PaiNN、GemNet-OC、MACE、NequIP与Allegro,同时也与经验力场进行了比较,DFT结果作为基准。
图4b展示了Chignolin的自由能景观,以D3主链羰基氧与G7主链氮之间的距离
补充材料中的图S6展示了所有机器学习力场与经验力场在测试集上的预测能量与DFT能量的相关性。ViSNet取得更低的平均绝对误差与更高的
为了更全面地比较模型性能,还将模型的预测精度、训练耗时与模型规模结合进行分析,如图5所示。尽管ViSNet在训练速度上略慢于ET与PaiNN,但其通过引入更丰富的几何信息获得了更高的预测能力。与同样计算二面角的GemNet相比,ViSNet的运行时几何计算将显式二面角计算的复杂度从
在此基础上,还对Chignolin进行了分子动力学模拟。随机选取10种构象作为初始结构,对每一种构象运行100 ps的模拟。图4c展示了10条轨迹在模拟过程中相对于起始构象的平均RMSD,并以阴影表示标准差范围。图4d展示了模拟过程中由ViSNet计算的原子力与DFT力之间的逐分量平均绝对误差,并同样以阴影表示标准差范围。模拟结果显示,三维力的三个分量均保持较低误差,未出现偏向性。整体上,ViSNet驱动的分子动力学轨迹与量子化学计算之间高度一致,说明其在真实体系中的潜在应用价值。
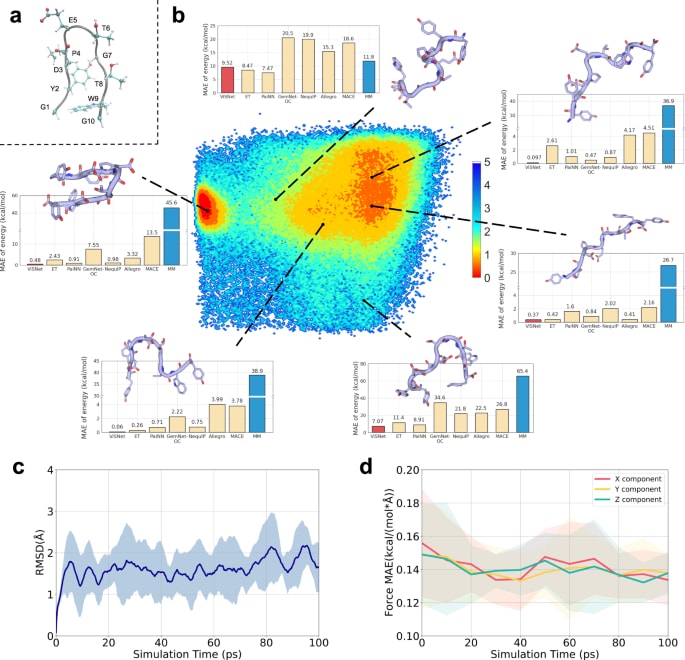
图 4|ViSNet在Chignolin构象空间评估与分子动力学模拟中的应用。 a Chignolin结构的可视化。主链以灰色表示,各残基的侧链以球棍形式突出显示。b 通过REMD采样得到的Chignolin能量景观。能量景观的x轴为D3主链羰基氧与G7主链氮之间的距离,y轴为E5主链羰基氧与T8主链氮之间的距离。随后选取六种构象进行可视化,每个构象以卡通形式呈现,残基以棍状表示。直方图展示了在这些构象上,各类机器学习力场(包括ViSNet、ET、PaiNN、GemNet-OC、NequIP、Allegro与MACE)以及分子力学计算的能量差与DFT基准的绝对误差。c 由ViSNet驱动模拟的10条Chignolin轨迹,其平均均方根偏差(RMSD)。阴影区域表示标准差范围。d 模拟过程中原子力各分量的平均绝对误差(MAE),模拟由ViSNet驱动。基准能量与受力由Gaussian 16计算得到。阴影区域表示标准差范围。源数据在附带的Source Data文件中提供。
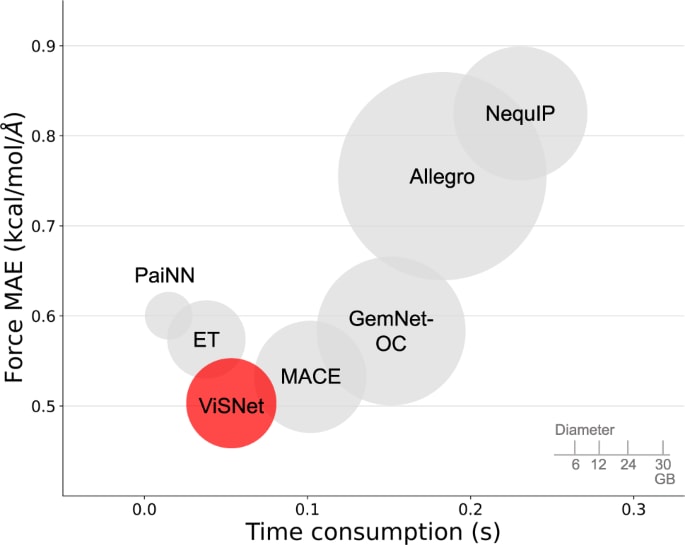
图 5|在Chignolin任务中,ViSNet(红色)与其他算法(灰色)在模型性能(纵轴)、训练时间消耗(横轴)以及训练内存消耗(体积)上的比较在这一比较中所包含的算法包括PaiNN、ET、MACE、GemNet-OC、Allegro与NequIP。PaiNN与ET的训练速度更快、模型规模更小,这是因为ViSNet进一步纳入了二面角计算。得益于运行时几何计算,ViSNet相较GemNet-OC表现更优,将显式二面角提取的复杂度从
2.5 ViSNet在分子结构上的可解释性
为了展示ViSNet捕获几何信息的能力,对其在分子结构上的可解释性进行了分析。该分析基于MD17数据集中的七种分子以及MD22中的两个分子体系,以验证ViSNet是否能够从分子结构中学习到与化学直觉一致的几何表征。
分析首先通过计算模型预测能量对原子坐标的梯度,得到原子级的敏感性评分。对MD17数据集中的七种小分子进行敏感性分析后可以观察到,敏感性评分与分子中典型的结构特征呈现高度一致性。例如,在甲醛与甲醇等分子中,碳氧双键以及羟基等具有强相互作用的化学键在敏感性图中表现为更高的梯度强度,这说明模型在能量预测中高度依赖这些关键的局部结构特征。更重要的是,ViSNet中的方向单元向量
在MD22中的较大体系Ac-Ala3-NHMe和Uracil-Water中,类似的分析同样显示出模型对关键几何结构的敏感性。例如,在肽段体系中,与肽键扭转相关的二面角区域在模型的敏感性图中具有更高的响应;在Uracil-Water体系中,涉及氢键相互作用的结构单元因其在体系能量中占据核心地位,也表现出更高的敏感性。这些现象说明,ViSNet不仅能够有效捕获局部结构中的简单角度与距离特征,也能够识别更复杂的高阶几何关系。
为了更加系统地解释ViSNet对几何表征的学习方式,还将方向单元的内积、向量拒斥量以及高阶张量扩展的标量输出与传统化学几何量进行对比。结果表明,这些内部量与真实的角度分布、二面角分布以及不正确扭转角分布之间均具有明显相关性,如在数据集中不同温度与构象区域中均能看到一致的趋势。由此可见,ViSNet通过RGC与向量–标量交互机制,确实在内部构建了与传统分子力场中使用的几何特征近似一致的表征结构。
总体而言,ViSNet在学习分子几何表征方面展现出较高的可解释性,使其在能量预测与分子动力学模拟任务中不仅具有精度优势,也具备良好的透明度与可信度。
3 总结
该研究提出了ViSNet,这是一种用于分子动力学模拟的几何深度学习势能模型。基于群表示理论的方法与基于方向信息的方法是实现SE(3)等变性的两类主要几何深度学习势能模型。ViSNet在设计中融合了两者的优势,通过RGC策略与ViS-MP机制实现高效且充分的几何信息利用。一方面,RGC策略能够以轻量级计算方式显式提取并利用方向几何信息,使模型训练与推理更为迅速;另一方面,ViS-MP包含一系列高效的向量–标量交互操作,能够更充分地发挥几何信息的作用。
此外,根据多体展开理论,一个体系的总势能可视为每个原子的单体势能与从两体到多体的能量修正项之和。以往研究通常通过堆叠
ViSNet的普适性或完备性目前尚未通过几何Weisfeiler–Leman测试验证,这是由于模型采用了内积操作,该操作具有计算效率,但无法区分某些具有相同角度信息的原子反射结构。若要通过相关反例或通过GWL测试,未来模型需在更高阶球谐函数的基础上引入Clebsch–Gordan积。
除了在能量、力以及化学性质上实现高精度预测外,以接近从头算精度执行分子动力学模拟,同时保持经验力场级别的计算成本,也是一个重要挑战。ViSNet在真实场景的从头算分子动力学模拟中表现出可行性,并能扩展至蛋白等大型分子体系。未来的研究方向将致力于进一步拓展ViSNet以支持更大规模且更复杂的分子体系。