NC 2025 | AI生成技术助力的双功能PKMYT1靶向PROTAC的发现
今天介绍的是发表在《Nature Communications》上的一项工作,聚焦于利用生成式AI推动靶向蛋白降解剂(PROTAC)的创新研发。该研究围绕在精准肿瘤治疗中日益突出的靶点PKMYT1展开。由于PKMYT1与CCNE1扩增、FBXW7或PPP2R1A致损突变之间存在合成致死关系,靶向该激酶有望在特定癌种中实现高选择性杀伤。然而,现有的小分子PKMYT1抑制剂普遍面临结构多样性不足与选择性欠佳等挑战。
该研究依托多模态AI平台Chemistry42生成了全新的PKMYT1抑制剂化学类型,并进一步将其与优化的CRBN配体连接,构建出双功能的PKMYT1降解剂D16-M1P2。该分子不仅能够有效结合并降解PKMYT1,还具备直接抑制其激酶活性的能力,在细胞与动物模型中均展现出强效而持久的药理作用。更重要的是,D16-M1P2对携带关键细胞周期调控突变的肿瘤表现出显著选择性,并在药代动力学方面具备良好的口服暴露与跨物种一致性,为其进一步向临床开发提供了坚实基础。

获取详情及资源:
0 摘要
PKMYT1因与多种致癌改变存在合成致死关系而逐渐成为精准癌症治疗中极具潜力的靶点,这些改变包括CCNE1扩增以及FBXW7和PPP2R1A中的突变。现有的小分子PKMYT1抑制剂仍存在分子多样性不足与选择性欠佳等局限。此项研究借助生成式AI平台,通过将全新的PKMYT1抑制剂与优化后的cereblon(CRBN)配体连接,设计出一种双功能的PKMYT1降解剂。其代表性分子D16-M1P2具备PKMYT1降解与抑制的双重机制,并凭借高度选择性展现出显著的抗增殖活性。同时,该分子具有良好的口服生物利用度、较单独使用PKMYT1抑制剂更强的药效表现,并在异种移植模型中以单药形式产生稳健的抗肿瘤响应。该PROTAC不仅为研究PKMYT1的生物学机制提供了精确的化学探针,也为进一步开发癌症治疗策略提供了有前景的先导化合物。
1 引言
细胞周期调控在癌症中常常遭到破坏,导致细胞无限增殖、基因组不稳定并最终推动肿瘤进展,同时也产生了癌症特有的脆弱性。靶向合成致死的遗传相互作用为间接作用于以往被视为“不可成药”的蛋白提供了潜在治疗途径,例如结构高度相似的细胞周期调控蛋白,并有望在降低对正常细胞影响的同时减少副作用。PKMYT1是一种定位于胞质膜的丝氨酸/苏氨酸蛋白激酶,负责编码CDK1及G2/M检查点的调控。近年来,PKMYT1因在携带CCNE1扩增以及FBXW7和PPP2R1A突变的癌症中呈现合成致死性而受到高度关注。CCNE1的扩增或过表达会增加复制压力,并使细胞依赖PKMYT1介导的对CDK1的失活性磷酸化,从而避免在基因组复制尚未完成前提前进入有丝分裂并引发灾难性的基因组不稳定。作为CCNE1直接激活的下游因子,CDK2的选择性抑制剂因为选择性不足及潜在耐药问题而面临挑战。与之相对,PKMYT1的抑制能够从正交角度干扰G2/M检查点,并在CCNE1扩增或过表达背景下诱导合成致死,从而有望规避CDK2靶向治疗所伴随的毒性和耐药风险。
此外,FBXW7的功能缺失突变(该基因编码介导cyclin E降解的E3泛素连接酶)以及PPP2R1A的功能缺失突变(该基因编码参与细胞周期调控的PP2A磷酸酶亚基)会使细胞依赖CDK1抑制作用,因此成为PKMYT1抑制的有利遗传背景。首个同类PKMYT1抑制剂RP-63068及其衍生结构通常通过内环或外环酰胺与PKMYT1的hinge区域结合。然而这些抑制剂仍面临剂量限制性毒性、临床获益有限等问题,并存在获得性耐药突变的担忧。更进一步的研究显示,PKMYT1可与β-catenin发生物理互作,并以非催化方式阻断其被E3连接酶SCFβ-TrCP识别,从而稳定β-catenin并持续激活Wnt信号通路,进而促进肿瘤形成,这提示同时靶向PKMYT1的催化与非催化功能可能提升抗肿瘤疗效。
PROTAC技术代表了一种可能规避上述问题的蛋白失活策略。PROTAC分子由两个化学基团构成,其中一个结合靶蛋白(POI),另一个结合细胞内的E3泛素连接酶亚基(如cereblon, CRBN)。PROTAC通过诱导靶蛋白与E3连接酶的近距离接触,使靶蛋白经泛素/蛋白酶体系统被降解。POI与E3连接酶之间可塑而独特的相互作用界面以及E3连接酶的组织选择性,使PROTAC在选择性方面有望优于传统的小分子配体。基于事件驱动的药理机制,PROTAC无需长时间结合或维持高饱和浓度即可发挥作用。
在该研究中,作者使用多模态AI驱动的Chemistry42平台生成了一类全新的靶蛋白结合化学类型,具有对PKMYT1的强效抑制能力。在完成CRBN配体筛选及连接臂优化后,研究者将该新型PKMYT1抑制剂构建入PKMYT1降解剂D16-M1P2中。该分子展现出强劲且高度特异的PKMYT1降解活性,其效应与CCNE1、FBXW7、PPP2R1A等遗传改变呈协同关系。D16-M1P2具备包括PKMYT1降解与抑制在内的双重作用机制,并较作为设计起点的POI抑制剂表现出更强且更持久的药效。此项成果为探索PKMYT1生物学提供了有力工具,也为针对PKMYT1活性的癌症治疗提供了具有潜力的新药物类别。
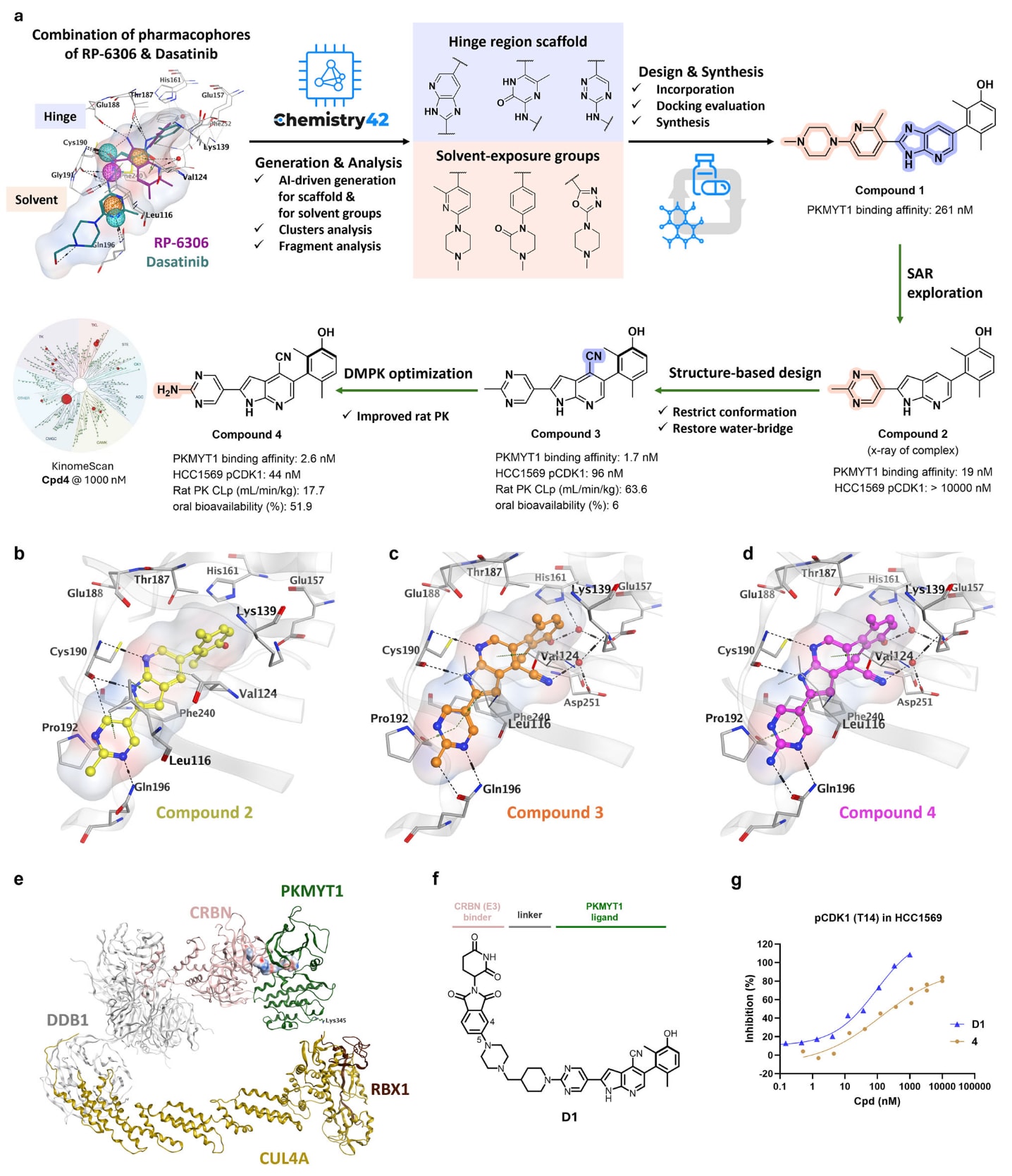
图1|基于Chemistry42平台的AI生成流程及新型PKMYT1抑制剂与PROTAC的开发过程。 a 通过AI生成与优化获得化合物4。生化结合亲和力(HTRF实验)与细胞中Thr14位点pCDK1水平(in-cell western实验)对应IC50数值。KinomeScan图中最大的红点表示在1000 nM下检测到的PKMYT1。所有数据均为至少两次重复的平均值。CLp表示血浆清除率。b 化合物2与PKMYT1的X射线共晶结构。c 化合物3的预测结合构象。d 化合物4的预测结合构象。e 建模得到的PKMYT1–CRL4CRBN复合物结构,绿色示意PKMYT1,预测的泛素化位点Lys345以绿色棒状表示。f 化合物D1的结构。g 在HCC1569细胞中比较PROTAC D1与其PKMYT1结合抑制剂组分化合物4的pCDK1抑制活性。数据以平均值表示(bioactivity实验n=2,PK实验n=3)。原始数据见Source Data文件。
2 结果
2.1 基于Chemistry42的AI生成发现新型PKMYT1抑制剂
PKMYT1抑制剂RP-6306在PKMYT1的ATP结合位点深部口袋中含有5个氢键供体,这使其作为POI配体并不理想。RP-6306与PKMYT1的浸泡共晶结构(PDB: 8D6E)显示,其伯氨基分别与gatekeeper位点Thr187和hinge位点Glu188形成氢键,酰胺基与hinge区的Cys190和Gly191形成氢键,苯酚基的羟基通过水分子介导与Glu157、Phe252和His161形成氢键,而吡啶氮则通过水分子介导与Lys139侧链形成氢键。为了进一步评估这些相互作用的动力学与稳定性,进行了200 ns分子动力学模拟,结果揭示了高度持续且稳定的氢键作用。伯氨基与Thr187的氢键占据率达88.4%,与Glu188的相互作用维持在22.2%。酰胺基分别与Cys190(80.5%)和Gly191(97.4%)形成强稳定的氢键,强化了其作为关键结构特征在结合稳定性中的作用。模拟还揭示了补充氢键网络的疏水与范德华相互作用,如与Val124和Phe240的接触,这些次级相互作用很可能进一步稳定RP-6306在PKMYT1结合口袋中的位置。整体来看,200 ns模拟过程中这些相互作用的一致性凸显了RP-6306结合模式的稳健性,并为设计可再现关键结合特征的新型抑制剂提供了重要基础。
与RP-6306不同,已获批准的广谱激酶抑制剂dasatinib(PKMYT1 IC50为63 nM)延伸进入溶剂暴露区,并与Gln196形成氢键。尽管dasatinib缺乏苯酚羟基,但其后口袋中的结构水依旧可与周围残基形成稳定氢键,提示PKMYT1抑制剂可从hinge区两个保守水分子相互作用出发,向溶剂区域延伸。因此设计中结合了RP-6306与dasatinib的结构特征。以RP-6306与PKMYT1的共晶结构(PDB: 8D6E)为起点,将其位于后口袋的2,4-二甲基-3-苯酚基团固定,同时整合RP-6306在hinge区与dasatinib在溶剂区的药效团,构建了新的药效团模型,并将其作为输入在Chemistry42平台上进行AI结构生成。策略包括在hinge区进行片段替换以及在溶剂区域进行定向片段生长,采用40余种AI生成模型。对生成结构按照蛋白-配体相互作用、三维形状与口袋匹配度、口袋体积占据以及分子新颖性等内部参数进行筛选。最终共生成2023个分子,并对其进行聚类、重新对接与可视化检查,以评估其蛋白-配体相互作用。
随后将这些重新对接的分子拆分为片段,重点分析hinge区支架及溶剂暴露基团。在此前分析基础上识别出具有良好结合特性的全新片段,并将其对应的有利官能团系统性引入新分子中,再次进行对接与评估。最终基于预测相互作用、综合潜力、可合成性及类药性,选择化合物1进行合成。之后利用Chemistry42平台的Golden Cubes工具预测潜在离靶效应,结果显示,相比dasatinib和RP-6306,化合物1对ABL1、SRC、LCK和JAKs等靶点的离靶可能性显著降低。化合物1对PKMYT1表现出亚微摩尔级结合活性,而其二甲基苯酚结构对活性至关重要,在替换为2,4-二甲基-3-吡啶基后活性完全丧失。总体而言,尽管化合物1活性相对较低,但仍被视为可进一步开展构效关系研究的新化学类型起始结构。
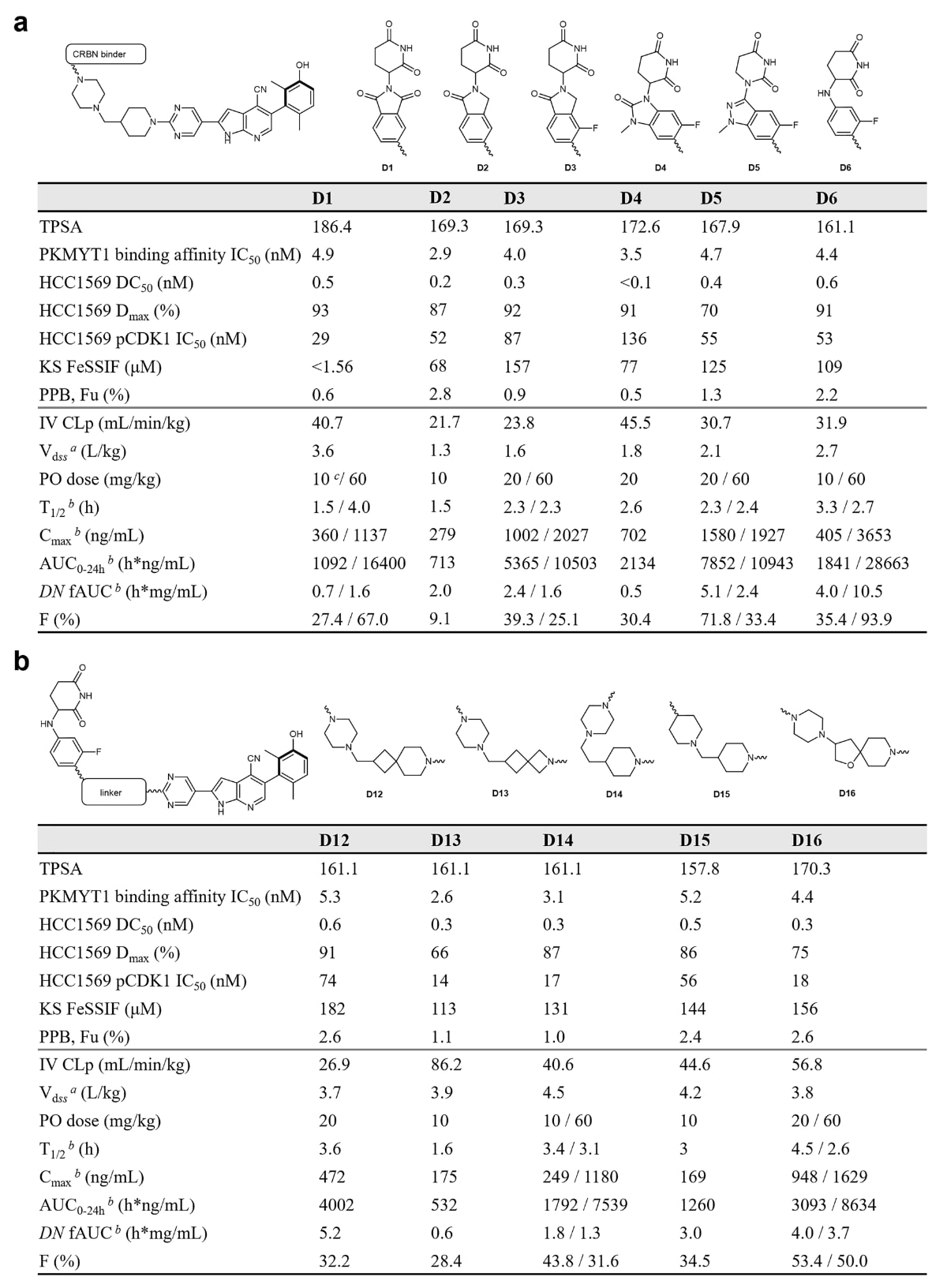
图2|PROTAC的构效关系探索。a CRBN配体的筛选。b 连接臂的筛选。 PPB Fu表示小鼠血浆蛋白结合实验中的游离比例。小鼠药代动力学实验使用CD-1品系雌性小鼠(除特别说明外),静脉给药剂量为1 mg/kg。波浪线表示成键位点。CLp为血浆清除率;PO为口服给药;Vdss为稳态分布容积;AUC0–24h为0至24小时的暴露量;DN fAUC为剂量归一化的游离暴露;F为口服生物利用度。生化结合亲和力(HTRF实验)和细胞中Thr14位点pCDK1水平(in-cell western实验)对应IC50值。细胞中PKMYT1降解(Jess实验)对应DC50及Dmax。结果以三次重复的平均值呈现,标准差为简洁起见未显示。a 静脉给药组。b 口服给药组。c 禁食小鼠组。所有数据均为至少两次生物学重复的平均值;KS、PPB实验n=2,PK实验n=3。原始数据见Source Data文件。
2.2 基于结构设计与DMPK优化获得性能提升的PKMYT1抑制剂
去除溶剂暴露区域的甲基哌嗪并对核心环进行生物电子等排替换后,获得了化合物2,其PKMYT1的IC50改善至19 nM。然而在具有CCNE1扩增特征的HCC1569乳腺癌细胞中,该化合物未能有效抑制Thr14位点的CDK1磷酸化,IC50超过10 μM。进一步的理化性质分析显示,低溶解度可能是其细胞活性不足的原因。考虑到化合物2的结构与已报道的PKMYT1抑制剂差别显著,研究者解析了其与PKMYT1的X射线共晶结构,分辨率为2.4 Å。结构显示,其嘧啶氮可在溶剂区域与Gln196形成氢键,而新的pyrrolo[2,3-b]pyridine支架在hinge区域形成两项关键氢键,从而提升酶活性抑制及细胞效应。
在进行手性拆分后,获得的对映纯化合物3表现出更高的通透性(由顶向底通量为
ADMET预测提示,嘧啶环上的甲基容易发生氧化代谢。因此,将该溶剂区域甲基替换为氨基后得到化合物4,其具有良好的通透性、水溶性、较高的构象旋转稳定性以及改善的代谢稳定性(CLp为17.7 mL/min/kg),并在大鼠中展现出51.9%的口服生物利用度。同时,化合物4在468种激酶筛选中展现出优异的选择性。此外,其末端伯氨基可与Gln196形成额外氢键,并提供了一个易于进一步连接的位点,使其适合用于构建PROTAC分子。
2.3 PROTAC D1的设计与发现
基于CRBN在组织中的广泛表达以及已有临床PROTAC成功招募CRBN的经验,研究者选择以CRBN作为E3连接酶适配体开展PROTAC的设计。首先利用MOE软件中的“Method 4”进行蛋白–蛋白对接,以构建PKMYT1–PROTAC–CRBN的虚拟三元复合物模型。该过程将PKMYT1与其结合配体(化合物4)的二元复合物结构与CRBN/CRBN配体(lenalidomide)的二元复合物结构进行对接。对接前设置输出100种构象,以便全面分析。随后选取能使PKMYT1与CRBN呈合理接近的构象。进一步地,将CRBN、DDB1、CUL4及RBX1组成的3D冷冻电镜复合物叠合到模型化三元复合物上,并以CRBN为比对参考。期望PKMYT1表面的赖氨酸可向E2延伸以促进泛素化。在多种对接构象中,作者选择了Lys345可向E2延伸的构象作为最终候选。对接结构中PKMYT1配体与CRBN配体的距离约为8 Å,对应约8至10个原子的连接臂长度。研究者用全碳骨架连接臂生成虚拟3D PROTAC模型,并以此作为迭代优化的起点。
鉴于大多数具口服可及性的PROTAC通常含有半刚性连接臂,研究者并未合成含线性碳链连接臂的PROTAC,而仅将其作为后续连接臂生成与设计的起点。E3配体方面选择lenalidomide和thalidomide分别用于连接臂设计,将E3配体与PKMYT1配体加以固定后,借助Chemistry42平台的生成式AI进行结构设计并生成连接臂片段。分析生成结果后,作者选定其中的哌啶甲撑哌嗪连接臂构建出化合物D1,并优先进行合成与检测。
化合物D1表现出对PKMYT1的高亲和力(IC50为4.9 nM)以及对CRBN的良好结合(0.37 μM),并在HCC1569细胞中实现强效PKMYT1降解(DC50为1.0 nM)。降解作用迅速且明显快于PKMYT1的自然降解速率。D1可抑制pCDK1,IC50为29 nM,是单纯抑制剂化合物4效力的近三倍,突显出靶向降解在调控信号通路方面的优势。D1在1 μM内未降解GSPT1、IKZF1/3或CK1α,显示其对CRBN已知的新生底物具有高度选择性。尽管D1在小鼠中展现适中清除率(CLp为40.7 mL/min/kg)及可接受的口服生物利用度(60 mg/kg时为67%),但其溶解度较差(FeSSIF <1.56 μM)且血浆蛋白结合率较高(PPB Fu为0.6%),因此仍需进一步结构优化。
2.4 优化类药性获得先导PROTAC D16
研究者进一步筛选了以临床阶段PROTAC中常用的不同CRBN配体为基础、并以D1为母体的多个变体,以评估其体外活性、理化性质与药代动力学特征。化合物D1至D6在PKMYT1亲和力、降解效力、清除率及pCDK1抑制活性方面表现大致相近,但其理化性质如溶解度(FeSSIF)、血浆蛋白结合比例(PPB)、口服吸收与生物利用度等则因分子量、可旋转键数量以及拓扑极性表面积(TPSA)等差异而不同。
在D1的CRBN配体上去除一个羰基后得到D2,其溶解度(FeSSIF 68 μM)更佳,小鼠体内清除率降低(CLp 21.7 mL/min/kg),但口服吸收不佳(F 9.1%)。在此基础上引入氟原子的D3在口服暴露量与生物利用度方面明显优于D2。含环状脲的D4并未显著改善pCDK1抑制活性及PK表现。将环状脲替换为吡唑的D5在低剂量时展现良好的PK,但在高剂量口服时呈现非线性暴露。D6作为苯胺类似物,其分子量与极性表面积最低,细胞效力与D1相当,并表现出109 μM的高溶解度(FeSSIF)、最高的PPB游离比例及最优的口服暴露,在60 mg/kg时实现93.9%的超线性口服生物利用度。然而D6的通透性低(0.31×10^-6 cm/s),外排比高(12.4),提示其可能是P-gp底物,这也可能解释其高剂量下暴露量的超线性提升。
将D6中CRBN配体的氟从meta移至ortho导致降解活性完全丧失(D7),在另一meta位引入第二氟原子则在1 μM出现明显hook效应(D8)。其他CRBN配体如苯甲酰基哌啶-2,6-二酮(D9)、苯基二氢尿嘧啶(D10)及三环结构(D11)在pCDK1抑制上均表现欠佳,可能由于三元复合物亲和力失衡影响降解效率。总体来看,D6凭借最高的剂量归一化游离暴露值,被选作后续连接臂优化的基础。
基于构建的PKMYT1–CRL4CRBN模型,研究者对PROTAC的连接臂进行长度与空间取向的优化。在D6的连接臂中引入环丁烷得到D12,其体外活性保持良好,并因脂溶性增加在小鼠中呈现更大分布容积(3.7 L/kg)。将4,6-螺环收缩为4,4-螺环的D13则降低了降解效率,增加清除率,并削弱口服暴露。将D6的连接臂增加一个亚甲基得到D14,恢复了降解能力并增强pCDK1抑制,但PPB游离比例下降。将连接臂中哌嗪替换为哌啶以提升碱性(D15),可提高溶解度并增加分布容积(4.2 L/kg)。采用5,6-螺环以增加连接臂刚性并减少可旋转键后,得到的D16在细胞效力方面显著提升(pCDK1 IC50为18 nM),并在溶解度与PPB游离比例等理化性质上获得改善。
在小鼠中,D16展现良好的PK表现,包括适中清除率(56.8 mL/min/kg)、较大分布容积(3.8 L/kg)、优异且剂量成比例的口服暴露(AUC0–24h分别为3093/8634 h·ng/mL,剂量为10/60 mg/kg),以及令人满意的口服生物利用度(53.4/50.0%)。将D16及D14的连接臂与其他CRBN配体组合后所得PROTAC在细胞活性方面均逊于原型。
随后对D16与PKMYT1及CRBN的结构结合模式进行了建模。D16的CRBN配体部分可与Trp380、His378及Glu377形成氢键;其PKMYT1配体部分与化合物4类似,可与保守水分子及hinge区的Cys190形成氢键,并与Leu116、Ser193形成H-π作用,与Phe240形成π-π作用。此外,连接臂中的质子化氮可与PKMYT1的Leu116形成氢键。
不同细胞与组织环境对PROTAC效应影响显著。D16在多种CCNE1扩增的细胞系中表现出稳定效力,包括HCC1599、MDA-MB-157,以及FBXW7突变的HuCC-T1,与其在HCC1569细胞中的效应一致。D16的CRBN配体在酸性条件(pH 1–2)下暴露24小时后仍保持97%的稳定性,并未发生消旋。在模拟胃液条件(SGF)下,其结构在12小时后仍保持87%,可满足口服给药要求。
2.5 一种新型PROTAC D16-M1P2实现对PKMYT1的结合与降解
通过手性超临界流体色谱(SFC)对D16进行拆分后获得四种非对映异构体。为筛选最佳立体异构体,研究者采用无细胞竞争型HTRF实验评估其与重组PKMYT1的结合能力。四种异构体的PKMYT1结合IC50介于4.1至5.0 nM之间,与化合物4(IC50 2.6 nM)及RP-6306(IC50 2.0 nM)的活性相近。对四种异构体的PKMYT1–PROTAC–CRBN三元复合物预测结合模式与其实测的PKMYT1及CRBN亲和力分析均显示差异极小。所有异构体均可在HCC1569细胞中实现剂量依赖性的PKMYT1与pCDK1(T14)下降。然而,D16-M1P2展现出最优的PKMYT1降解效力,DC50为0.7 nM,Dmax为90%,并同时展现最强的pCDK1抑制活性(IC50为9.0 nM),因此被确定为最佳先导物。
研究者观察到,在D16-M1P2的最大降解浓度前,PKMYT1降解程度与pCDK1(T14)抑制之间呈正相关,这与PKMYT1可在Thr14位点磷酸化CDK1的机制一致。然而,即便达到80–90%的PKMYT1降解,pCDK1(T14)的抑制仅为50–80%,提示部分PKMYT1残留即可维持CDK1的磷酸化,完全抑制该通路需要更彻底的PKMYT1去除。随着D16-M1P2浓度继续升高,出现典型的“hook效应”:PKMYT1降解反而下降,但pCDK1(T14)抑制继续增强。这表明在高浓度下,PKMYT1主要与D16-M1P2形成二元复合物,从而导致强效的激酶抑制,即使PKMYT1的进一步降解未发生。
在更细致的动力学分析中,100 nM D16-M1P2可在4小时内使PKMYT1下降超过80%,并在24小时达到最大降解,远快于其天然半衰期约12.5小时。同时,pCDK1(T14)的抑制也具有时间依赖性,在100 nM下4小时即可超过50%的抑制,并至少维持24小时。然而在低剂量时,PKMYT1降解与pCDK1抑制出现明显脱节。例如1 nM处理8或24小时虽可使PKMYT1下降约60%,但未导致明显的pCDK1抑制,这表明残余的PKMYT1仍足以维持CDK1的磷酸化。
相比之下,100 nM短时(2小时)处理同样可实现约60%的PKMYT1下降,却能显著更强地抑制pCDK1(T14)。这是因为高浓度下的D16-M1P2除了促进PKMYT1降解外,还可直接抑制其激酶活性。因此,该分子在高浓度时通过“降解+直接抑制”的双重机制实现更强的通路抑制。
以上结果表明,单纯的部分PKMYT1降解不足以完全抑制pCDK1(T14),而D16-M1P2实现稳健的信号通路抑制需要充分的PKMYT1耗竭与高浓度下的直接激酶抑制共同作用。
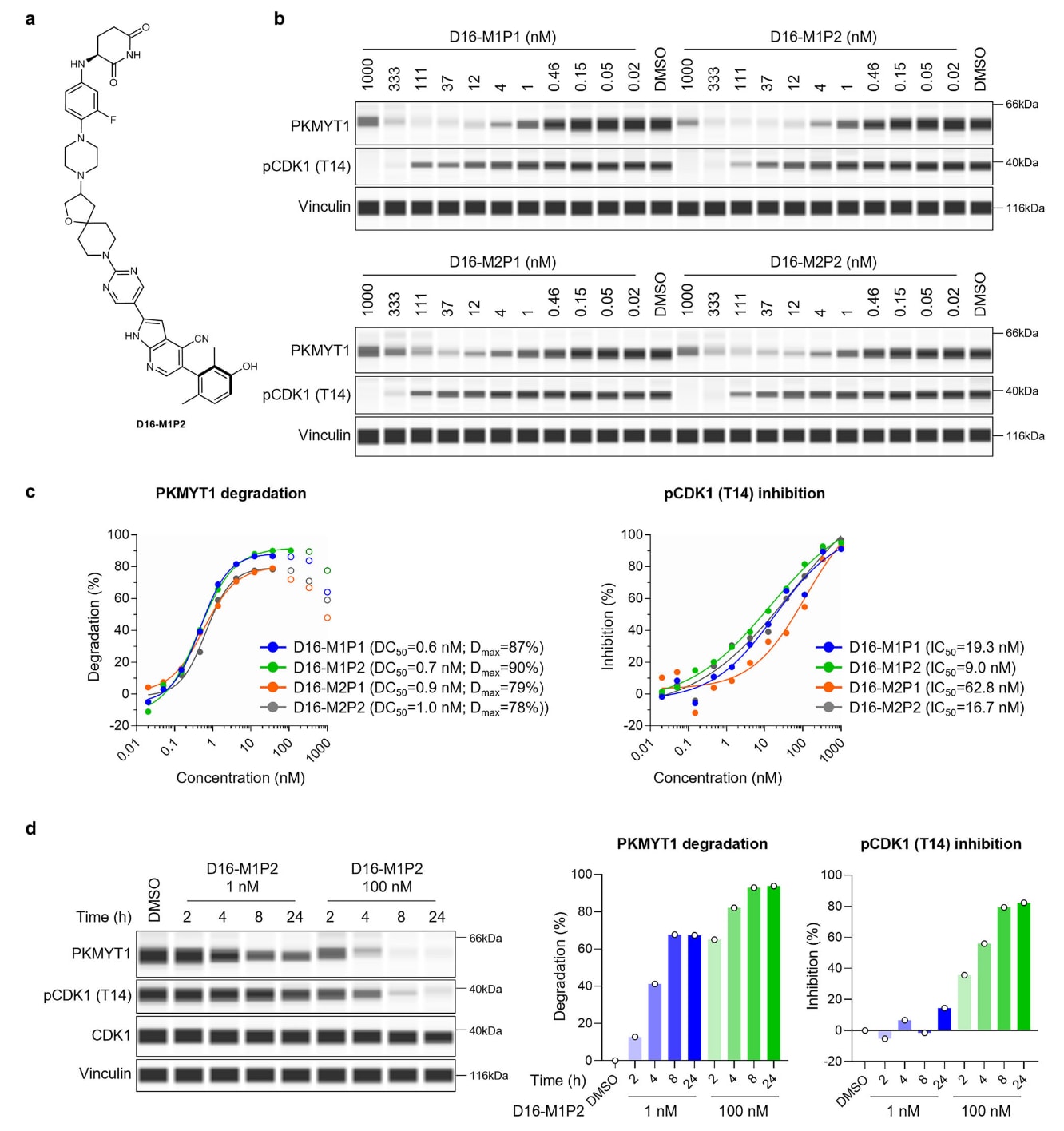
图3|新型PROTAC D16-M1P2与PKMYT1的结合及降解。 a D16-M1P2的结构(单一非对映异构体,连接臂立体化学未定义)。b、c 在HCC1569细胞中,四种D16非对映异构体处理24小时后的剂量依赖性PKMYT1降解与pCDK1(T14)抑制,分别通过JESS进行可视化(b)与定量(c)。数据来自两个独立实验的代表性结果。d HCC1569细胞在不同时间处理D16-M1P2后的PKMYT1与pCDK1(T14)蛋白水平变化,并通过JESS定量。数据为两个独立实验的代表性结果。原始数据见Source Data文件。
2.6 D16-M1P2展现出降解与抑制的双重机制
研究者首先通过联合处理实验验证了PKMYT1降解依赖于泛素-蛋白酶体系统(UPS)。当D16-M1P2与蛋白酶体抑制剂MG-132或NEDD8化抑制剂MLN-4924共同处理时,PKMYT1的降解完全被阻断;当加入CRBN配体pomalidomide或PKMYT1配体化合物4时,也同样阻断降解。这些结果表明,依赖CRBN与PKMYT1的NEDD8化与蛋白水解过程对于UPS介导的PKMYT1降解至关重要。
PROTAC具有催化式机制与事件驱动药理特征,因此在不需要持续高药物浓度的情况下也能维持抑制作用。对HCC1569细胞以100 nM D16-M1P2处理24小时后洗脱,PKMYT1的降解与pCDK1(T14)的抑制能够至少持续24小时。而RP-6306不会诱导PKMYT1降解,其对pCDK1(T14)的抑制作用在洗脱后2小时内迅速恢复,显示D16-M1P2可通过靶标降解实现更持久的通路抑制。
研究者推测,在达到最大PKMYT1降解后,进一步增强的pCDK1抑制可能来自化合物中POI-binder所带来的直接激酶抑制。为区分两种机制的贡献,作者将不与CRBN相互作用的类似物D16-NMe-M1P2与D16-M1P2进行对比。两者在24小时处理后均保持相似的PKMYT1生化抑制能力(IC50分别为8.5 nM与7.6 nM)及细胞内靶标占领(EC50分别为8.0 nM与12.0 nM),但D16-NMe-M1P2的pCDK1(T14)抑制效力仅为D16-M1P2的八分之一。与此同时,相比单纯PKMYT1抑制剂4与RP-6306,D16-M1P2虽然在生化酶抑制与细胞靶标占领方面更弱,但在HCC1569细胞中对pCDK1(T14)的抑制更为显著。
这些结果表明,PKMYT1降解是D16-M1P2长期抑制通路的主要且更有效的机制。然而在高浓度(如1000 nM)下,当PKMYT1降解因hook效应而下降时,D16-M1P2对pCDK1(T14)的抑制可增强至与D16-NMe-M1P2相当,说明在降解效率不足的情况下,其直接PKMYT1抑制作用将进一步巩固对pCDK1(T14)的抑制。这也解释了在这些浓度下PKMYT1降解程度与pCDK1抑制效果不一致的原因。

图4|D16-M1P2通过PKMYT1降解与抑制展现双重作用机制。 a 在HCC1569细胞中,分别加入或不加入蛋白酶体抑制剂MG132和/或NEDD8化抑制剂MLN-4924时,D16-M1P2对PKMYT1蛋白水平的调控,通过免疫印迹检测。DMSO与D16-M1P2(0.1 μM)组的数据以平均值(n=2)呈现。数据来自两个独立实验的代表性结果。b 在HCC1569细胞中,分别加入或不加入PKMYT1抑制剂化合物4和/或CRBN配体pomalidomide时,D16-M1P2对PKMYT1蛋白水平的调控。c 在HCC1569细胞中,用JESS可视化(上)并定量(下)D16-M1P2或RP-6306洗脱(更换为无药培养基)后的PKMYT1与pCDK1(T14)水平。数据为两个独立实验的代表性结果。d 通过ADP-Glo激酶实验测定PKMYT1酶活的剂量-反应曲线,数据以平均值(n=2)呈现。e 在NanoBRET实验中检测细胞内PKMYT1靶标占领情况,数据以平均值(n=2)呈现。f 经24小时处理后的剂量依赖性PKMYT1降解与pCDK1(T14)抑制。原始数据见Source Data文件。
2.7 D16-M1P2展现出对PKMYT1的高度特异性
研究者进一步评估了D16-M1P2的靶点特异性。在403个受检激酶中,仅有4个激酶(包含PKMYT1)在1000 nM浓度下(约为其酶学IC50的130倍)表现出超过65%的抑制。这与化合物4及RP-6306形成鲜明对比,后者分别可结合11个与46个离靶激酶,显示出D16-M1P2显著改善的激酶选择性。进一步验证三个离靶激酶BRAF、RAF1与SRC的结果显示,D16-M1P2对PKMYT1的选择性超过50倍(PKMYT1 IC50为7.6 nM,而其对BRAF、RAF1、SRC的IC50分别为485.3、373.1与398.5 nM)。相比之下,RP-6306对PKMYT1的选择性不足10倍,这可能与其在临床测试中出现剂量限制性毒性有关。
由于PROTAC分子可能在中等亲和力(≥1–500 nM)范围内降解非靶蛋白,研究者对HCC1569细胞进行8小时D16-M1P2处理(10 nM与100 nM),并进行整体蛋白质组分析。该研究共定量132,593条独特肽段,对应8,177个蛋白。结果显示,在两种浓度下,只有PKMYT1表现出显著下降,这一点也通过JESS实验得到验证。其他差异蛋白(统计学阈值P<0.001,log2倍数变化≤−1或≥1)大多在其最丰富的两至三个前体离子检测上呈现不一致性,表现为轻微变化或在某些组别中无法检测,提示这些差异多属由掉谱(dropout)导致的假阳性。因此,整体蛋白质组进一步支持D16-M1P2在细胞中对PKMYT1具有高度特异性。
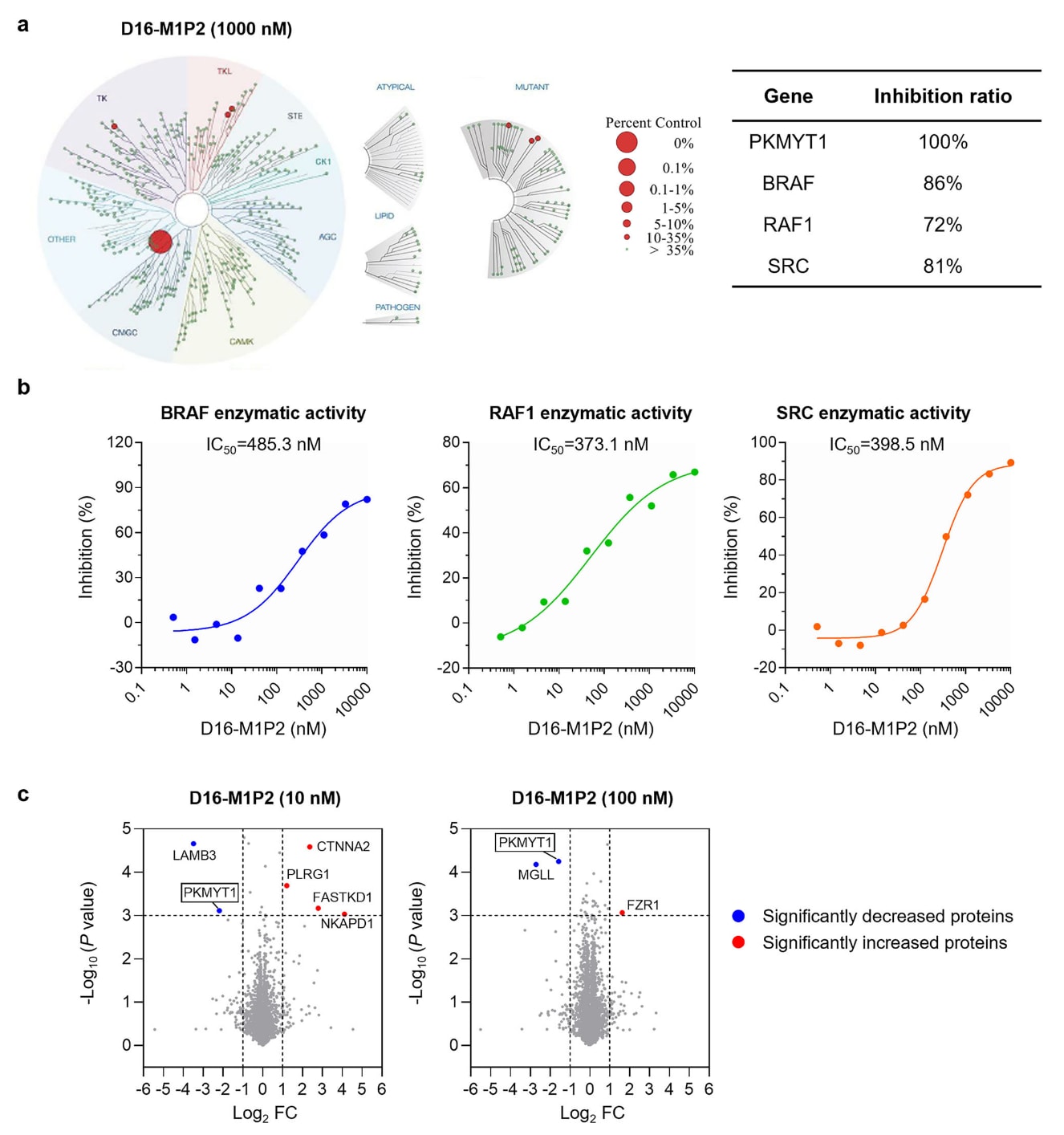
图5|D16-M1P2展现出卓越选择性。 a D16-M1P2在1000 nM下的KinomeScan结果。TREEspot图(左)中的红色圆点表示与D16-M1P2结合并产生超过65%抑制的激酶,并在右侧表格中列出。b 通过ADP-Glo激酶实验测定D16-M1P2对BRAF、RAF1及SRC的酶活抑制剂量-反应曲线,数据以平均值(n=2)呈现。c HCC1569细胞经10 nM或100 nM D16-M1P2处理(n=3)后,与DMSO对照组相比的蛋白丰度变化。统计分析采用双侧、非配对Student’s t检验,显著性阈值设定为p<0.001。原始数据见Source Data文件。
2.8 D16-M1P2抑制携带CCNE1扩增或FBXW7/PPP2R1A致损突变的肿瘤生长
研究者在包含11种肿瘤细胞系的面板中评估了D16-M1P2与RP-6306的细胞生长抑制效果,这些细胞系分别携带CCNE1扩增、FBXW7或PPP2R1A的致损突变,或均不携带相关改变。D16-M1P2在CCNE1扩增、FBXW7突变或PPP2R1A缺失的细胞中表现出平均12倍的选择性(IC50为177 nM),显著优于其在野生型细胞中的活性(IC50为2121 nM)。相比之下,RP-6306在生物标志物阳性细胞中呈现相似效力(IC50为186 nM),但对这些遗传背景的选择性仅为6倍。
值得注意的是,尽管化合物4与RP-6306具有相近的PKMYT1酶活抑制能力(分别为IC50 2.0 nM与1.5 nM),RP-6306在生物标志物阳性与阴性细胞中均表现出显著强于化合物4的抗肿瘤活性,这与其激酶选择性较低的结果一致,提示其增强效力部分可能来自离靶作用。然而,化合物4的整体效力较弱,其6倍的细胞选择性也未优于RP-6306。
与此对比,D16-M1P2在生物标志物阳性细胞中表现出强于化合物4的抗肿瘤活性,而在生物标志物阴性细胞中并无优势。尽管其酶活抑制弱于化合物4与RP-6306,但PKMYT1降解能够弥补其固有抑制活性的不足,成为其主要的抗增殖机制。
随后,研究者将D16-M1P2与RP-6306的体内疗效进行比较。在携带CCNE1扩增的HCC1569皮下移植瘤模型中,给予NOD/SCID小鼠口服D16-M1P2或RP-6306。D16-M1P2呈剂量依赖性抑制肿瘤生长,在40 mg/kg与120 mg/kg(每日两次,连续21天)的剂量下分别实现35.1%与66.4%的抑瘤率。RP-6306在20 mg/kg剂量下达到71.1%的抑瘤率,与既往报道一致。
药代与药效分析显示,D16-M1P2可诱导剂量依赖的PKMYT1降解与pCDK1(T14)抑制,并具备良好的血药暴露。然而,当比较相似程度的pCDK1抑制时(D16-M1P2的40 mg/kg比RP-6306的20 mg/kg),D16-M1P2的抑瘤效果较弱。只有在更高剂量(120 mg/kg)下达到更深层且持续的pCDK1抑制后,其效力才与RP-6306相当。这表明RP-6306的更强抗肿瘤效果部分来自其离靶机制,而不是单纯的PKMYT1通路抑制。
与此一致,在达到相似抗肿瘤效果的剂量水平下,RP-6306导致的小鼠体重下降更为明显,提示其耐受性较差、治疗窗更窄,而D16-M1P2表现出更有利的安全性特征。
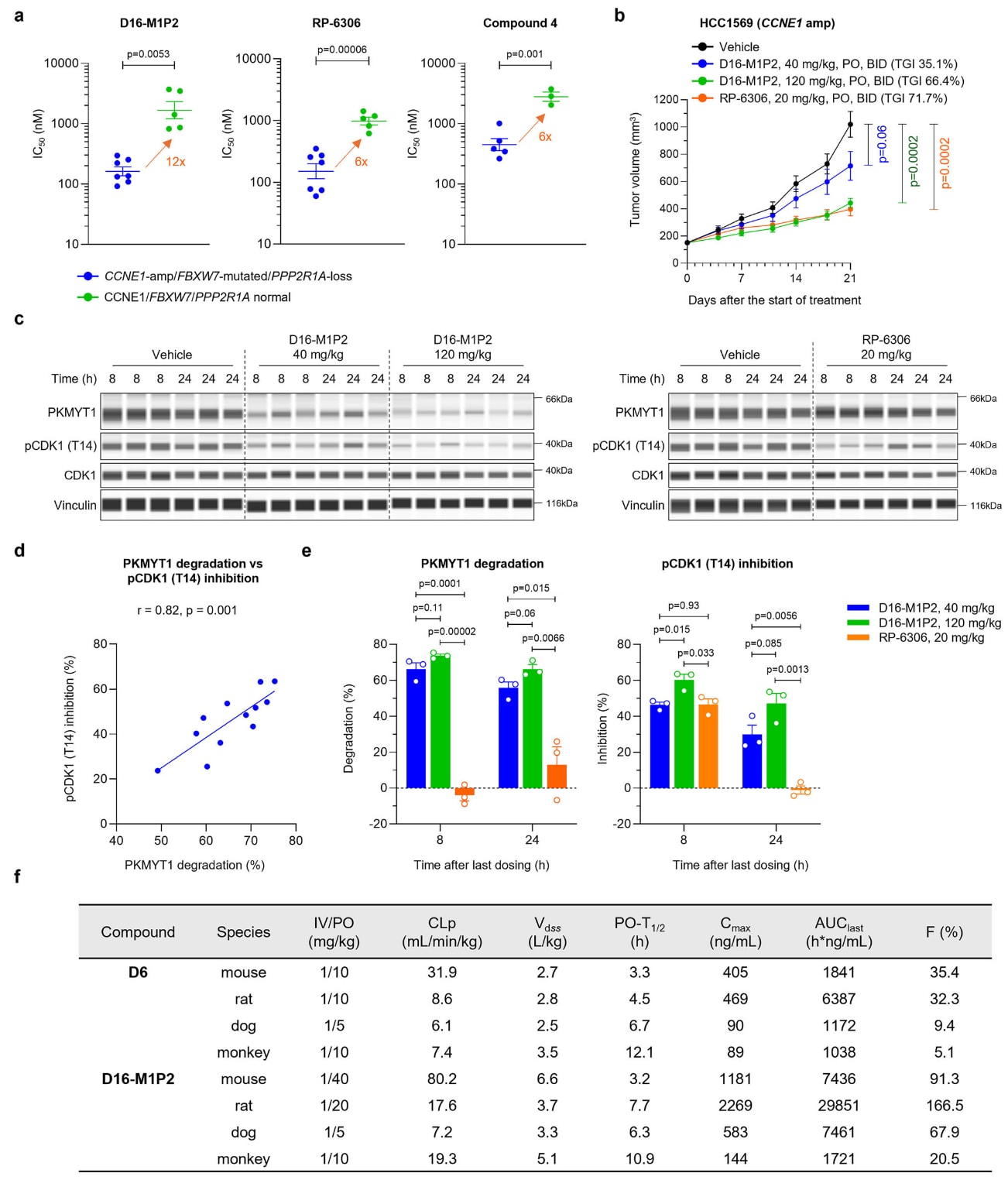
图6|D16-M1P2抑制携带CCNE1扩增或FBXW7/PPP2R1A致损突变的肿瘤生长及其PK特征。 a 对携带或不携带CCNE1扩增、FBXW7或PPP2R1A致损突变的癌细胞系进行D16-M1P2、RP-6306或化合物4的7天处理后,肿瘤生长抑制IC50的比较。每个细胞系以一个点及其对应IC50标示。数据以均值±SEM呈现。D16-M1P2与RP-6306组中,n=7(异常)与n=5(正常);化合物4组中,n=5(异常)与n=3(正常)。统计显著性采用双侧、非配对Student’s t检验。b 在NOD/SCID小鼠HCC1569异种移植模型中,每日两次口服D16-M1P2或RP-6306的体内肿瘤生长抑制效果。数据以均值±SEM呈现(n=6只小鼠/组)。终点显著性采用双侧、非配对Student’s t检验,与载体组比较。c–e HCC1569异种移植模型中,载体或每日两次口服D16-M1P2或RP-6306后的PKMYT1降解及pCDK1(T14)抑制。肿瘤组织在末次给药后8小时或24小时取样。PKMYT1降解与pCDK1(T14)抑制分别以JESS可视化(c)与定量(d、e)。在d中,利用Pearson相关系数评估PKMYT1降解与pCDK1抑制的相关性,并报告双侧p值。在e中,数据以均值±SEM呈现(n=3只小鼠/组/时间点),组间差异采用双侧、非配对Student’s t检验。f 化合物D6与D16-M1P2在小鼠(n=3)、大鼠(n=3)、犬(n=2)及猴(n=2)中的PK特征,图例同图2。原始数据见Source Data文件。
2.9 D16-M1P2在多物种中呈现良好PK特性
为评估D16-M1P2的类药性,研究者测定了其在多种临床前物种中的PK参数。在静脉给药条件下,D16-M1P2在大鼠、犬与猴中均表现出低清除率。口服给药后,D16-M1P2呈现良好的暴露量,生物利用度达到中高水平。相比具有中等刚性的D6,带有刚性螺环连接臂的D16-M1P2展现出更优越的口服吸收与生物利用度,这强调了减少可旋转键与在连接臂中引入醚环对于提升PROTAC分子生物物理性质的重要性。
3 讨论
研究结果强调了AI平台在新型激酶抑制剂及PROTAC设计中的重要潜力。与已报道的选择性PKMYT1抑制剂不同,这些抑制剂均以内环或外环酰胺为hinge区配体,而化合物4采用了完全不同的含氮杂环作为hinge结合基团。在此基础上进一步发展出的双功能降解剂D16-M1P2成功在强效抑制与卓越选择性之间取得平衡,展现出作为化学探针用于探索PKMYT1生物学的潜力。其在低纳摩尔水平即可快速降低90%的PKMYT1蛋白,并在洗脱后持续超过24小时,凸显出其作为化学敲低工具的应用潜力。
D16-M1P2同时具备降解与抑制的双重作用机制,其中降解是主要且更为有效的驱动因素。与单纯的PKMYT1配体相比,该PROTAC在细胞中展现出更强效力与更持久的药效作用。D16-M1P2在多种肿瘤细胞系中抑制细胞增殖,并对携带CCNE1扩增、FBXW7突变及PPP2R1A缺失的细胞表现出更佳选择性,这反映了PKMYT1抑制与细胞周期调控异常之间的合成致死关系。其在异种移植模型中展现出抗肿瘤活性,并伴随良好的PK特性与持续的pCDK1抑制作用。
此外,D16-M1P2在四种临床前物种中均呈现合理的清除率、良好的分布容积以及中至高的口服生物利用度,进一步强化了其作为候选药物的潜力。然而,研究仍存在局限性:尽管D16-M1P2在机制层面优于RP-6306,如更充分且持久的下游信号抑制、更佳的选择性与耐受性,但其整体抗增殖能力与体内抗肿瘤效果与RP-6306相近。这一缺口可能源于PROTAC常见的hook效应,该效应限制了高浓度时的最大降解程度,从而削弱了其治疗潜能。此外,RP-6306的离靶活性可能贡献一定抗肿瘤效果,缩小了两者间的有效性差距。
然而,已知多激酶抑制剂如RP-6306的非PKMYT1靶点(BRAF、SRC等)常导致复杂且剂量限制性的毒性,使其临床应用受限。因此,当前PKMYT1抑制剂在疗效与毒性之间的平衡,为开发更安全且具优势的靶向策略提供机会。D16-M1P2展现出更高的激酶选择性,有望减少副作用、提升安全性。
尽管如此,hook效应仍是未来开发中需重点解决的挑战。该研究提示,后续工作应在提高选择性的同时进一步优化降解特性。此外,合理的联合策略可能在维持可耐受性的前提下进一步提升抗肿瘤效果,值得更深入的探索。
总体而言,该研究为进一步开发PKMYT1靶向治疗奠定了基础,并展示了AI驱动策略在新结构发现及加速药物研发中的优势。该研究的方法具有可拓展性,也可能为其他多类靶点的发现与优化提供启发。