Nature 2025 | 金属水解酶的计算设计
今天介绍的是发表在 Nature 的论文《金属水解酶的计算设计》。该研究围绕从头酶设计这一长期挑战展开,提出并验证了一种全新的生成式人工智能框架RFdiffusion2,用于直接从量子化学计算得到的活性位点几何结构出发设计高效金属水解酶。不同于以往需要预先指定催化残基序列位置和主链构象的方法,RFdiffusion2能够在推理过程中同时探索序列空间与构象空间,显著拓展了可采样的设计范围。
研究以锌金属水解酶为模型体系,在无需实验优化的情况下,成功获得多种催化效率达到

获取详情及资源:
0 摘要
从头酶设计旨在构建包含理想活性位点的蛋白质,使催化残基围绕并稳定目标化学反应的过渡态。生成式人工智能方法RFdiffusion能够解决这一问题,但需要为每个催化残基同时指定序列位置和主链坐标,从而限制了构象采样的范围。这里介绍RFdiffusion2,该方法消除了上述限制,并被用于从量子化学计算得到的活性位点几何结构出发设计锌金属水解酶。
在最初实验测试的96个设计中,活性最高的酶其催化效率
这四个最活跃设计的结构模型不仅彼此不同,也不同于已知蛋白结构,而其中活性最高设计的晶体结构与其设计模型高度一致,表明该设计方法具有很高的准确性。基于PLACER和Chai-1的预测,这些高活性酶具有预组织化的活性位点,能够有效定位底物,使被金属离子活化的水分子进行亲核进攻。无需实验优化即可直接通过计算生成高活性酶的能力,有望推动新一代高效人工催化剂的出现。
1 结果
金属水解酶通过其结合的金属离子活化一分子水,并将其定位在待断裂的底物键附近,从而催化生物体系中一些最具挑战性的水解反应。由于人类产生的环境污染物出现时间较短,尚不足以在自然进化中产生高效的水解酶,因此工程化构建新的金属水解酶在当前具有重要意义。蛋白质工程已经拓展了金属水解酶可作用的底物范围,但通常依赖于最初存在的催化多功能性。从头酶设计曾被用于生成新的金属水解酶,但其活性和效率普遍较低,且需要大量定向进化才能接近天然酶的水平。
在给定理想金属水解酶活性位点的前提下,从头酶设计的目标是识别或生成一种蛋白质骨架,以高精度将催化残基、金属离子和底物定位在最优的催化几何构型中。RFdiffusion已被成功用于活性位点的支架化设计,但其搜索受到限制,因为必须事先指定催化残基的序列位置和构象。研究者由此提出,如果一种生成式人工智能设计方法仅需指定围绕反应过渡态的侧链官能团位置,并能够在所有可能的序列位置和构象之间进行采样,将更容易满足复杂的催化约束。基于这一思路,研究开发了一种新方法,并利用其从包含金属辅因子的量子化学活性位点描述出发设计新的金属水解酶。
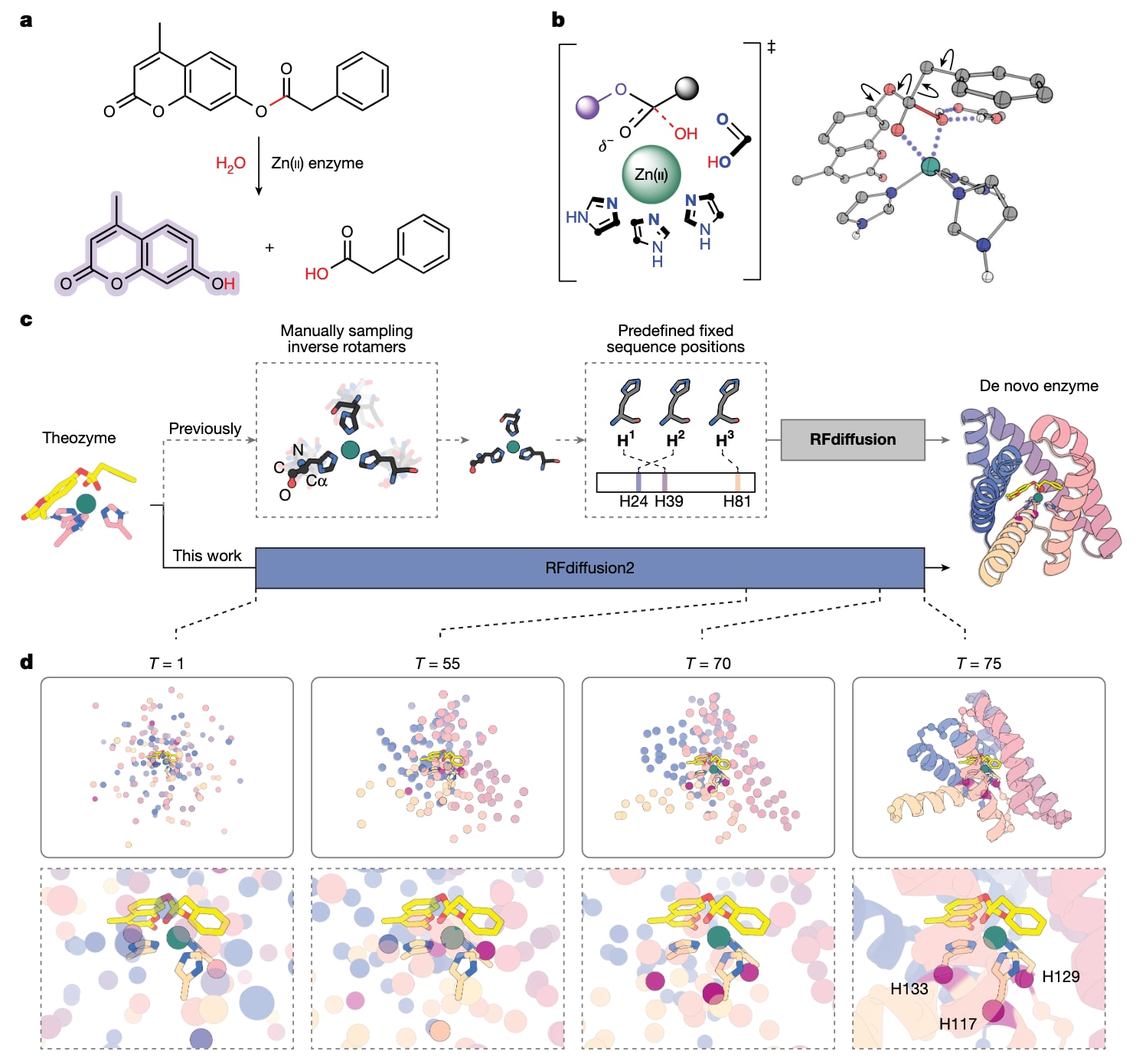
图1|RFdiffusion2设计方法。 a,4MU-PA的水解反应生成苯乙酸和具有荧光的香豆素产物。b,Zn(II)-氢氧根对4MU-PA酯键进行亲核进攻的theozyme示例,左为二维示意图,右为基于DFT的三维模型,三维模型中的箭头表示所采样的构象柔性。c,使用以往以主链为中心的RFdiffusion(上排)与以相互作用官能团为中心的RFdiffusion2,围绕输入theozyme生成蛋白支架的对比。RFdiffusion需要事先显式采样侧链构象和残基序列位置,而RFdiffusion2仅需过渡态复合物和催化侧链官能团,并在推理过程中隐式采样序列空间和旋转异构体空间。d,模型XT在RFdiffusion2推理轨迹中全局结构和活性位点的若干快照。在推理过程中,输入的过渡态复合物和催化官能团坐标保持固定,而主链结构、序列位置以及催化侧链中未指定的原子由RFdiffusion2进行采样。轨迹结束时承载催化组氨酸的
为实现对序列位置和侧链旋转异构体不敏感的酶设计,研究者开发了基于流匹配的生成式人工智能模型RFdiffusion2。与RFdiffusion相比,RFdiffusion2在两个关键方面扩展了活性位点支架化能力。首先,它支持原子级子结构支架化,不再局限于主链层面的基序,而是可以处理任意氨基酸重原子的子集,从而只需指定与反应过渡态直接相互作用的关键官能团位置。其次,RFdiffusion2实现了与序列位置无关的支架化,无需预先给定基序残基在一级序列中的位置。该模型通过在训练过程中同时提供随机选择的原子坐标和部分加噪且带有序列标注的原子坐标,在推理阶段能够在整个搜索空间内解析这些先验未知的自由度。
借助这些改进,RFdiffusion2可以直接从由官能团位置和底物坐标组成的催化构型出发生成多样化的蛋白质。让模型在推理过程中自行解析催化残基的序列位置和侧链构象,相比在推理前随机采样这些自由度更为高效,因为这一空间过于庞大,几乎无法穷举。关于RFdiffusion2训练过程及其在多种活性位点支架化任务中的系统评估,已有详细报道。
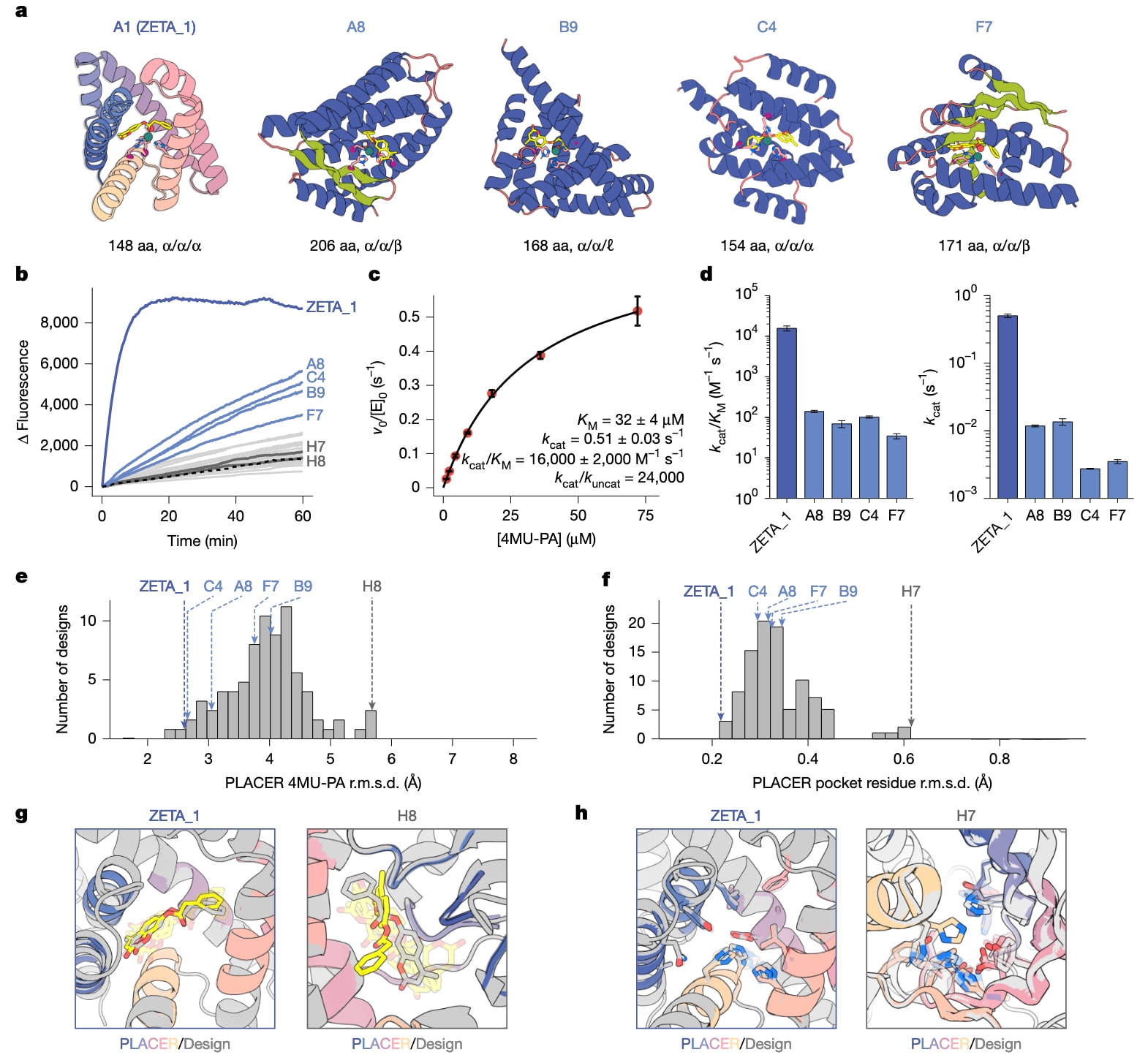
图2|活性表征与PLACER预组织化评估。 a,最活跃设计的结构模型,下方标注了序列长度(以氨基酸数计)以及各催化组氨酸所在的二级结构类型。b,反应进程曲线,黑色虚线表示缓冲液背景。c,A1(ZETA_1)的米氏动力学表征,纵轴为初始速率
作为RFdiffusion2的初步验证,研究选择了4-甲基伞形酮苯乙酸酯作为目标反应,设计一种用于其水解的锌金属水解酶。首先通过密度泛函理论确定速率决定步骤中Zn(II)-OH对底物酯键进行亲核进攻的过渡态几何结构。基于四面体中间体的立体化学以及氧阴离子孔的不同特征,共考虑了四种催化构型。这些计算提供了三个Zn(II)结合的咪唑环、金属离子以及过渡态的空间坐标。此前的RFdiffusion方法需要输入主链坐标和残基位置,这意味着必须预先对每个组氨酸的侧链构象和序列位置进行采样,而这一组合空间即便在粗略采样下也高达约
利用RFdiffusion2的推理轨迹,研究者构建了承载由DFT生成的最小活性位点构型的蛋白质骨架,即theozyme。在推理过程中,残基的
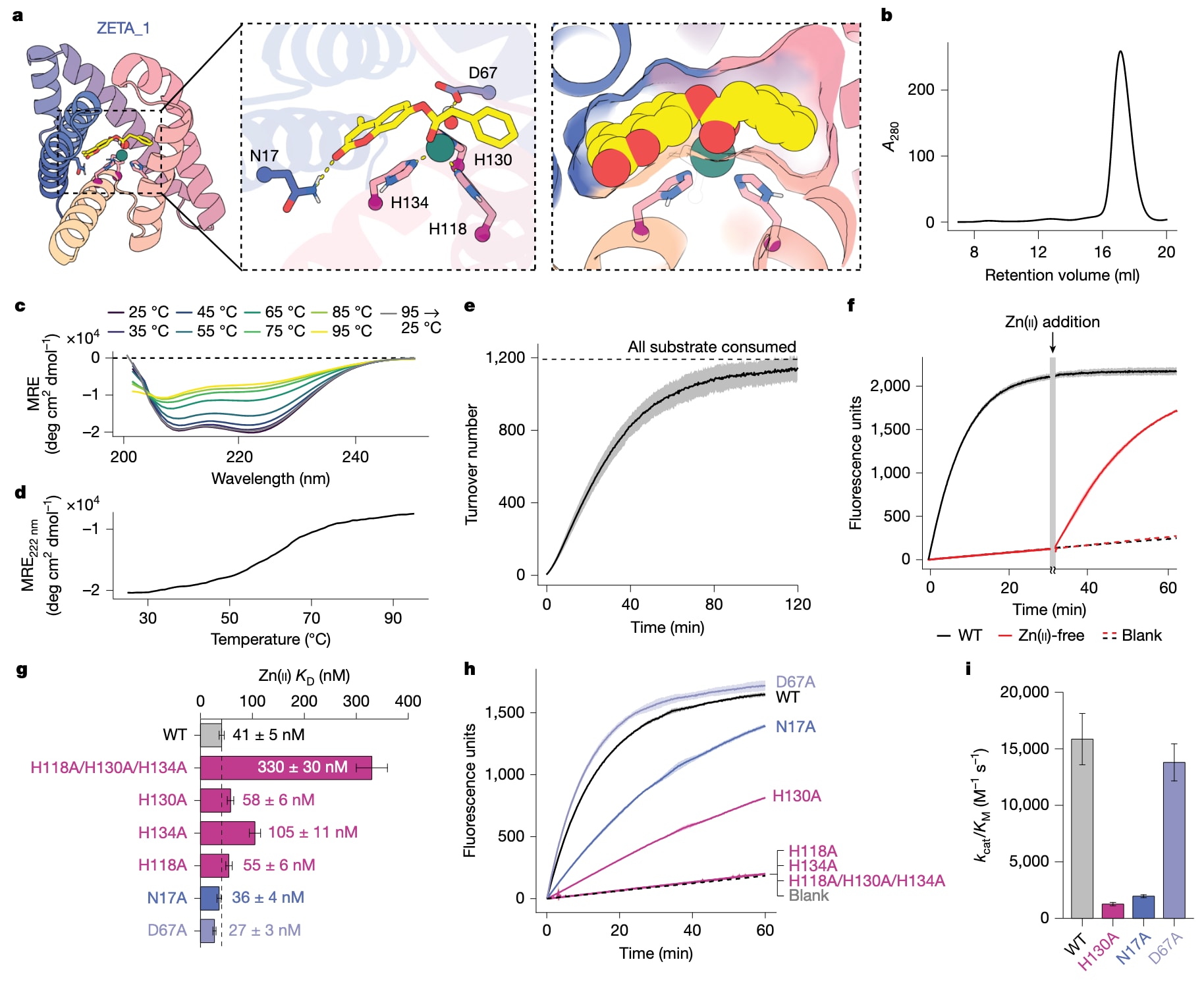
图3|ZETA_1活性的表征。 a,ZETA_1设计模型(左),中间为显示催化残基的活性位点放大图,右为设计口袋的表面视图,显示其与底物具有高度形状互补性。b,ZETA_1的体积排阻色谱图,显示单一峰,对应单体蛋白。c,ZETA_1的圆二色谱,在25 ℃至95 ℃范围内每隔10 ℃测量一次(viridis颜色梯度),并在重新冷却至25 ℃后再次测量(灰色)。结果表明ZETA_1具有α螺旋二级结构,并且在加热和部分解折叠后能够复性。MRE表示平均残基椭圆度。d,在222 nm处的圆二色信号,每隔1 ℃测量一次并随温度变化作图。e,产物与酶浓度比值的反应进程曲线显示,ZETA_1每个酶分子可水解超过1000个4MU-PA分子。背景反应已从光谱中扣除,因此每一次转化均可归因于酶催化。f,含Zn(II)的全酶态与无锌的apo态ZETA_1的反应进程曲线,显示活性依赖于锌。在30 min后向apo态样品中加入过量Zn(II)可恢复活性,表明锌对ZETA_1的催化机制至关重要。WT表示野生型。g,野生型及突变体ZETA_1对Zn(II)的亲和力,以解离常数
在满足特定设计标准后,研究者选取了96个设计进行实验表征。这些设计被克隆表达于大肠杆菌中并纯化,其中绝大多数蛋白可溶且成功表达。补充硫酸锌后,通过荧光法监测底物水解活性,发现5个设计表现出显著高于背景的催化活性。进一步纯化并进行米氏动力学分析后,最活跃的设计A1的催化效率
ZETA_1不仅在计算筛选中排名最高,其实验结构也被预测与设计模型高度一致。PLACER分析表明其活性位点高度预组织化,催化侧链构象稳定,底物被牢固定位在设计位置。相比之下,无活性的设计在PLACER轨迹中表现出底物显著波动。进一步的实验验证显示,在设计筛选阶段被PLACER指标淘汰的同源骨架确实几乎不具活性,凸显了该方法在设计筛选中的价值。
ZETA_1的活性位点主要由疏水口袋构成,三个组氨酸通过其
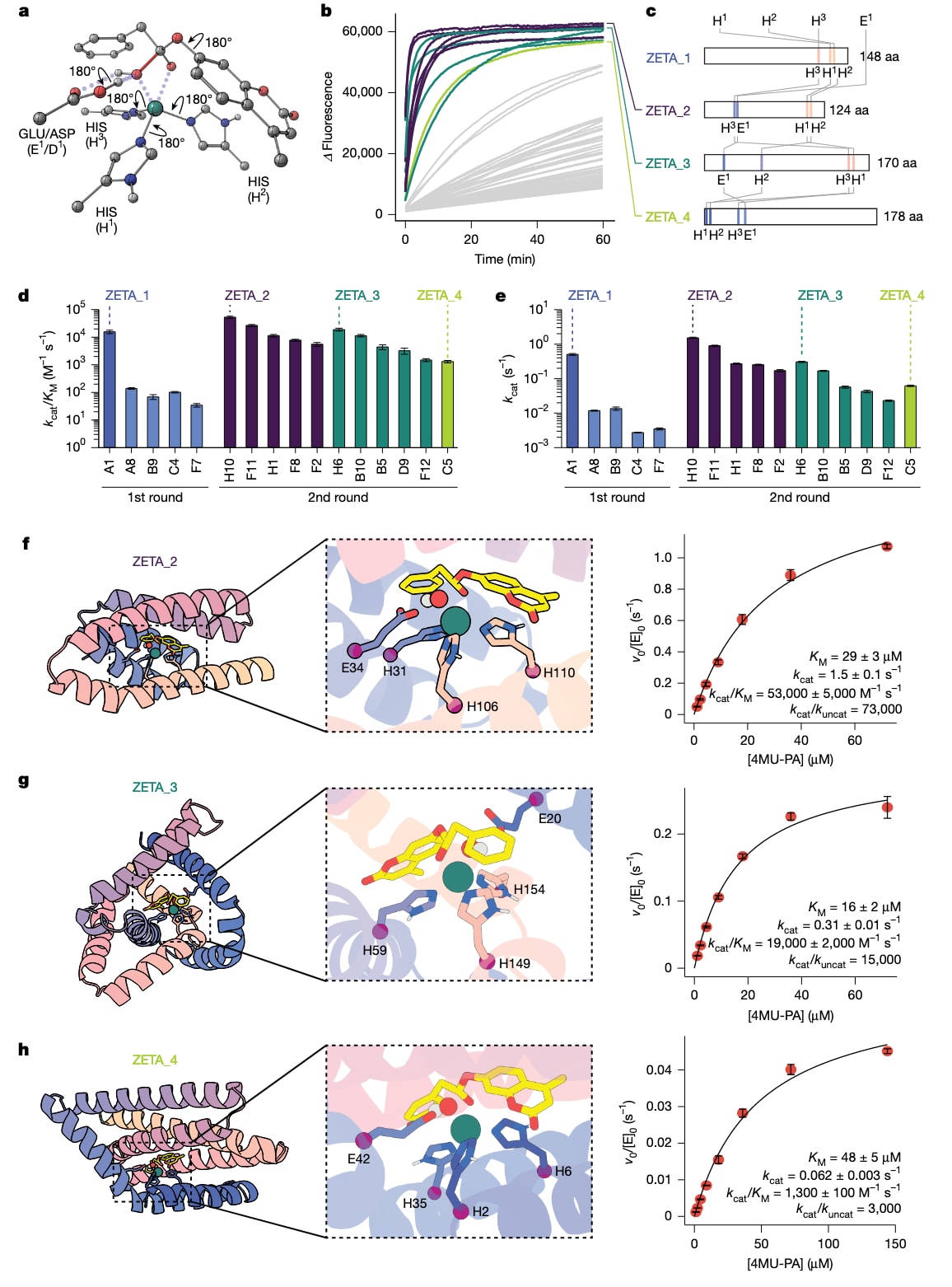
图4|第二轮设计的表征。 a,包含催化碱的第二轮DFT theozyme。在这些theozyme中,Zn(II)由组氨酸的
通过定点突变分析,研究进一步探讨了催化残基对Zn(II)结合和催化的贡献。结果显示,部分组氨酸和天冬氨酸之间可能存在竞争性的锌结合模式,这也得到了Chai-1预测的支持。基于这些发现,研究者在新一轮设计中显式引入催化碱,并使用在更大数据集上从随机权重训练的RFdiffusion2版本生成新的蛋白结构。通过Chai-1和PLACER筛选后,又有96个设计进入实验测试,其中11个表现出显著的锌依赖水解活性,成功率明显高于首轮设计。
第二轮设计中,最活跃的酶ZETA_2的
通过X射线晶体学,研究解析了ZETA_2的无底物结构和结合Zn(II)后的结构。实验结构与设计模型在主链层面高度一致,催化残基保持设计构型,活性口袋与过渡态形状互补,进一步验证了该计算设计方法在原子层面上的准确性和可靠性。
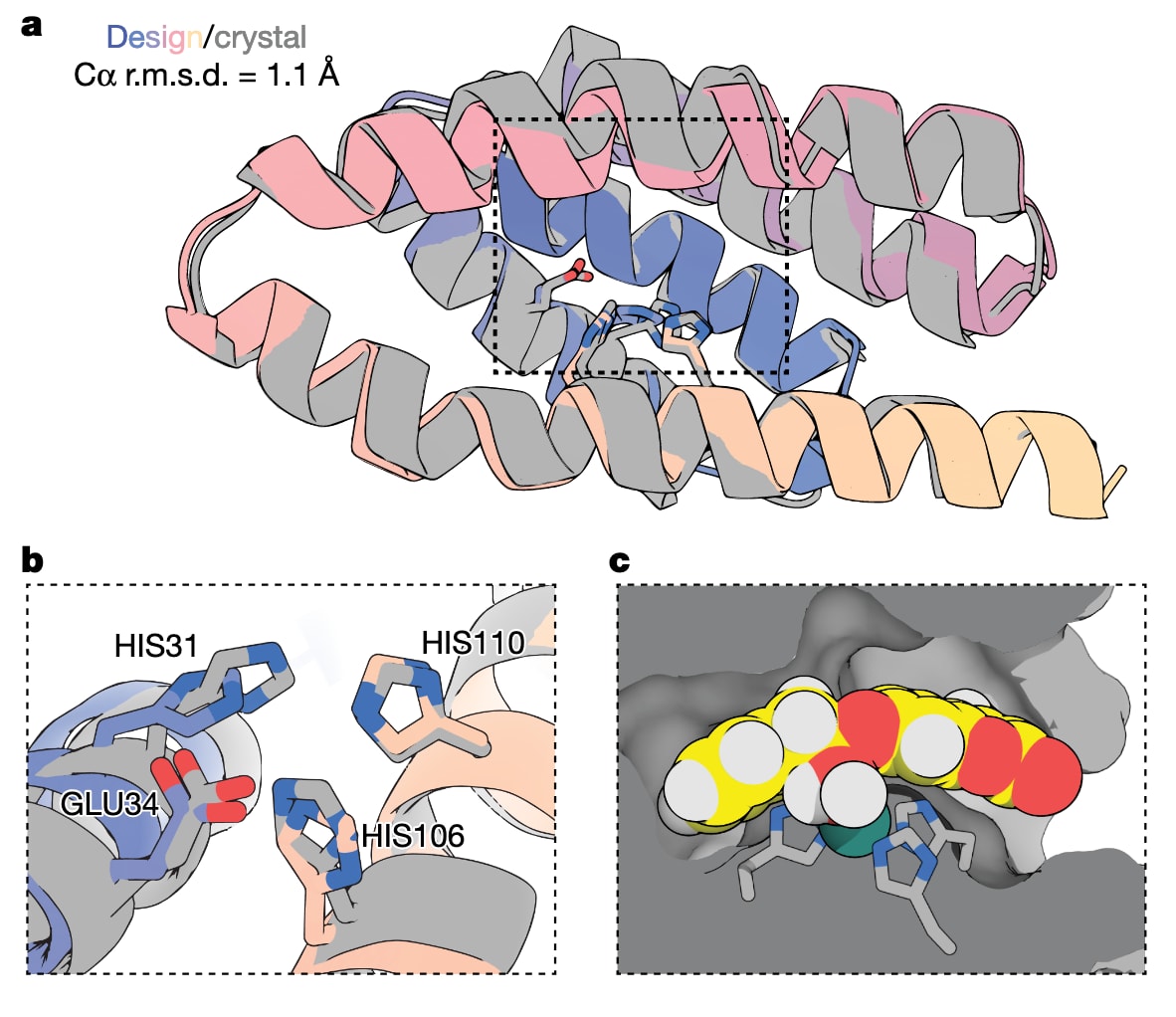
图5|ZETA_2的晶体结构与设计模型高度一致。 a,ZETA_2在apo态下的设计模型与X射线晶体结构(PDB:9PYJ)的
2 结论
研究表明,RFdiffusion2能够直接从量子化学计算得到的活性位点构型出发,生成具有高活性的金属水解酶。在完全不进行实验优化的情况下,零样本设计获得的酶ZETA_1其
ZETA_1至ZETA_3的催化效率已处于具有相似底物的天然金属水解酶常见范围内,即
未来的金属水解酶设计若能够在以下方面进一步改进,有望将活性提升至最活跃天然金属水解酶的水平:一是将一般碱定位在不易重新构型并与金属离子相互作用的位置,如ZETA_1中所示;二是更好地预组织金属结合残基;三是进一步引入侧链对氧阴离子的稳定作用。RFdiffusion2与PLACER相结合的设计策略相较以往从头酶设计方法具有多方面优势,并且有望广泛适用于为多种化学反应生成高效催化剂。通过直接对侧链官能团进行支架化,而非像RFdiffusion1所要求的那样指定主链
利用PLACER和Chai-1评估活性位点预组织化程度以及底物-过渡态定位,在筛选最活跃设计方面表现出极高的有效性。这一结果结合此前在从头设计的丝氨酸水解酶和逆羟醛裂解酶中的类似观察,表明PLACER及相关的深度学习方法将在设计排序中具有广泛用途。在实验室实践中,RFdiffusion2–PLACER设计流程已成功用于生成多种断键和成键反应的生物催化剂,这些工具已向社区免费开放,并有望推动更广泛的设计应用与创新。