JACS Au 2024 | 基于结构的“首尾相连”大环PROTACs设计
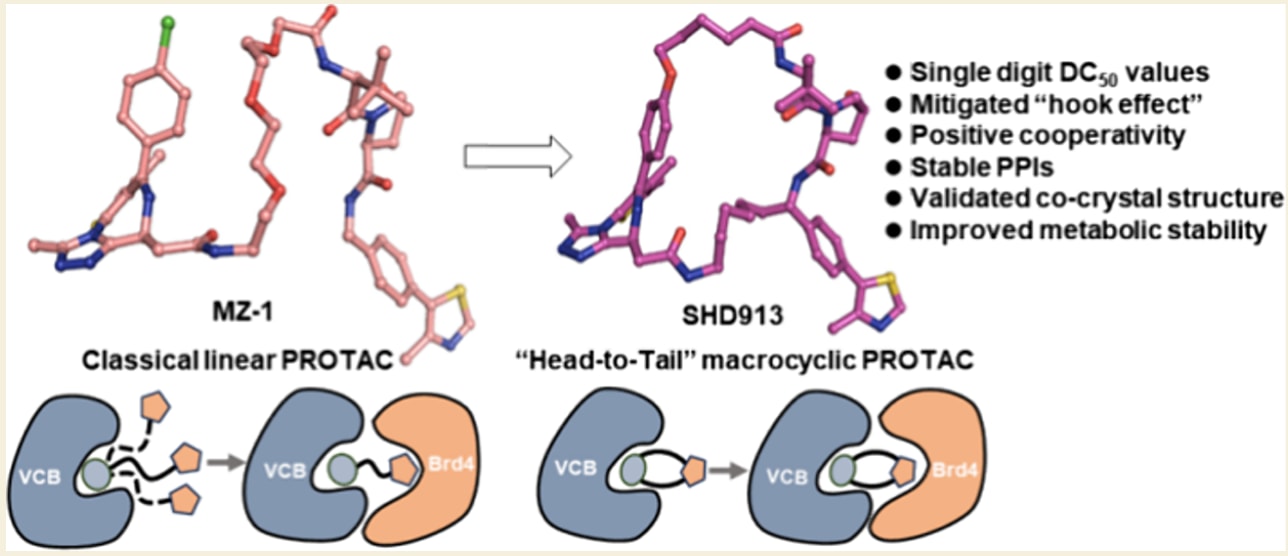
今天介绍的是发表在 JACS Au 的一项研究,主题为基于结构的“首尾相连”大环PROTACs设计。该工作围绕靶向蛋白降解领域中长期存在的关键挑战展开,即线性PROTAC由于构象高度柔性,容易引发不理想的药代性质以及典型的“钩状效应”。研究者以Brd4-VHL体系为模型,在已有三元复合物结构信息的基础上,提出并实现了一种全新的“首尾相连”大环化设计思路,通过构象受限策略稳定有利于三元复合物形成的空间排布。代表性化合物SHD913在多种癌细胞中表现出低nM水平的高效Brd4降解活性,同时在远高于有效浓度的条件下仍未出现“钩状效应”。结合生物物理实验、细胞NanoBRET分析、共晶结构解析以及分子动力学模拟,该研究系统阐明了大环化在增强正协同性、诱导新的蛋白-蛋白相互作用以及改善代谢稳定性方面的独特优势,为新一代PROTAC的理性设计提供了重要范式。

获取详情及资源:
0 摘要
大环化是传统药物设计中一种极具吸引力的策略,可用于提升生物活性、靶标特异性以及代谢稳定性,但由于PROTACs作用机制及结构复杂性,这一策略在PROTAC设计中很少被采用。该研究报道了首个“首尾相连”大环PROTACs系列的理性设计。其中代表性化合物SHD913在多种癌细胞系中表现出显著的Brd4蛋白降解能力,其DC50达到低nM水平,同时几乎完全消除了PROTACs中普遍存在且备受关注的“钩状效应”。进一步的生物学研究表明,该化合物具有正协同性,并在生物物理和细胞NanoBRET实验中诱导了新的蛋白-蛋白相互作用,其细胞活性优于已报道的首个大环Brd4 PROTAC macroPROTAC-1。体外肝微粒体稳定性评估显示,与线性对照分子相比,SHD913在不同物种中均具有更高的代谢稳定性。Brd4BD2:SHD913:VCB(VHL、Elongin C和Elongin B)三元复合物的共晶结构解析结合分子动力学模拟,揭示了化学诱导蛋白-蛋白相互作用的结构细节,并突出了SHD913构象受限特性在三元复合物形成中的关键作用。上述结果共同表明,大环化是一种具有吸引力且可行的全新PROTAC设计策略。
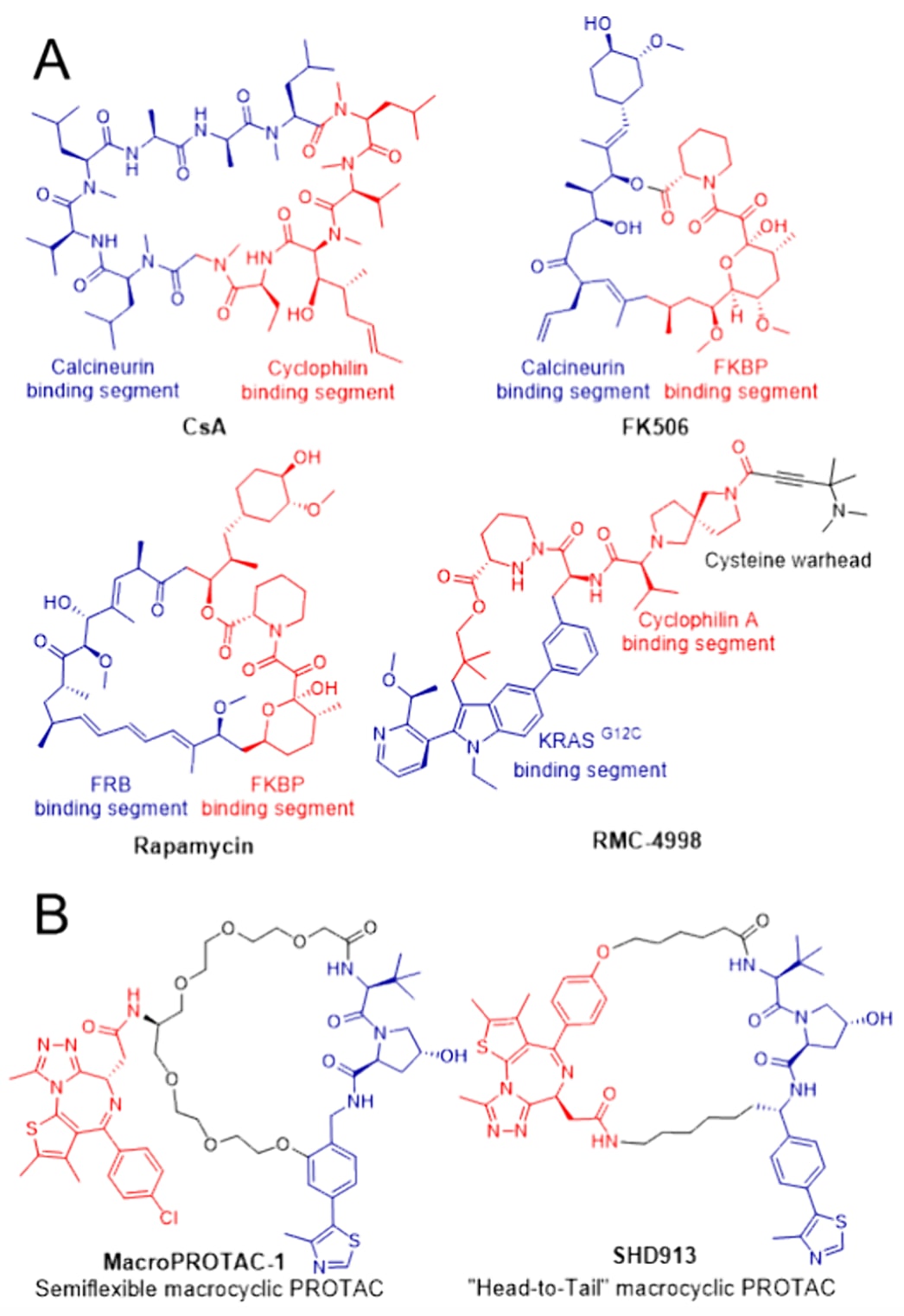
图1 (A)能够诱导和/或稳定蛋白-蛋白相互作用的大环分子实例。(B)macroPROTAC-1和SHD913的结构示意图。
1 引言
靶向蛋白降解(Targeted protein degradation,TPD)因其事件驱动的作用模式,以及在功能层面上高度模拟基因敲除,正在成为药物发现和化学生物学研究中极具吸引力的策略。目前已有30余种蛋白降解靶向嵌合体(PROTACs)进入不同阶段的临床开发。PROTACs是一类双功能分子,通过将靶蛋白与E3泛素连接酶拉近至空间邻近位置,触发靶蛋白的蛋白酶体降解。与传统小分子药物相比,PROTACs具有催化或亚化学计量级的药理效应、更持久的作用时间,并能够作用于传统方法难以靶向的蛋白。然而,由于其作用依赖于E3:PROTAC:靶蛋白三元复合物的形成,理论上的“钩状效应”(钟形剂量-效应关系)成为PROTAC治疗中普遍关注的问题。
典型的PROTAC通常呈“哑铃状”结构,通过柔性或刚性的线性连接臂促进三元复合物的形成。但线性PROTAC所具有的构象柔性往往会带来不理想的药代动力学性质,例如溶解度差和代谢不稳定性,这也是PROTAC研发中的另一项关键挑战。大环化是传统药物设计中常用且有效的策略,通过限制分子构象来提升化合物的效力和类药性,同时大环骨架也被认为是调控具有大而平坦、动态界面的蛋白-蛋白相互作用(PPIs)的优势结构单元。例如,环孢素A、他克莫司和雷帕霉素等天然大环产物,早在20世纪90年代就因其能够诱导并稳定PPIs而被视为最早的“分子胶”。近期,一种合成的大环分子RMC-4998也被报道可诱导环孢素A与KRASG12C之间的蛋白相互作用,并在非小细胞肺癌模型中展现出良好的治疗潜力。
鉴于PROTAC通常与靶蛋白和E3连接酶的表面口袋结合,且在配体和E3结合基团上均存在多个潜在的连接位点,可以设想通过设计大环衍生物来降低分子熵,将PROTAC锁定在有利于三元复合物形成的构象中。值得注意的是,近期报道的一种针对BET蛋白Brd4的半柔性大环PROTAC(macroPROTAC-1)表现出强效的靶蛋白降解活性,其与VHL和Brd4BD2形成的三元复合物共晶结构也验证了该设计思路,为大环化用于PROTAC设计提供了首个概念验证。不过,该分子也可被视为一种携带大环VHL配体的线性PROTAC,因为其靶蛋白结合基序位于大环体系之外。
以VHL-Brd4体系为模型,基于VHL、Brd4第二溴结构域(Brd4BD2)与线性PEG连接臂PROTAC分子MZ1形成的三元复合物的先导结构信息,开展了“首尾相连”大环PROTACs的基于结构的设计。**所获得的新型大环PROTAC在低nM水平DC50下即可实现快速而显著的靶蛋白降解。**尤为引人注目的是,即使在浓度高出DC50约10000倍的条件下,该化合物在多种癌细胞系中仍未观察到可检测的“钩状效应”。进一步的生物物理实验和NanoBRET分析证实,该分子在溶液和细胞环境中均表现出正协同性,并诱导Brd4BD2与VHL之间产生新的蛋白-蛋白相互作用。同时,三元复合物的共晶结构解析揭示了Brd4BD2、VHL与PROTAC之间的精细相互作用细节。据目前所知,该研究首次实现了“首尾相连”大环PROTACs的基于结构的理性设计。
2 结果与讨论
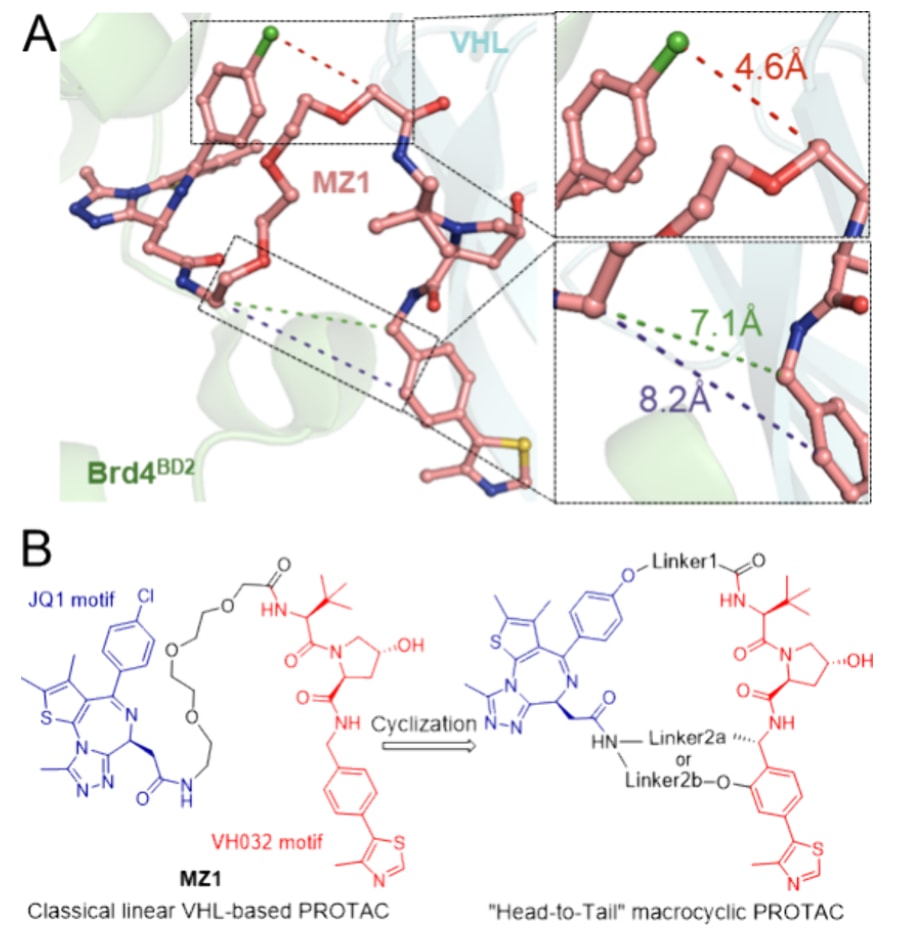
图2|基于结构的“首尾相连”大环PROTACs设计。 (A)Brd4BD2:MZ1:VHL三元复合物(PDB:5T35)的结构特征分析。MZ1以鲑红色棒状形式显示,Brd4BD2和VHL分别以绿色和青色的卡通模型表示。(B)“首尾相连”大环PROTACs的基于结构的设计示意图。
2.1 基于结构的“首尾相连”大环PROTACs设计
Alessio Ciulli及其同事此前报道了Brd4BD2:MZ1:VCB(VHL、Elongin C和Elongin B)三元复合物的晶体结构(图2A,PDB:5T35)。研究表明,线性PROTAC分子MZ1能够诱导Brd4的BD2结构域与VHL蛋白之间形成新的蛋白-蛋白相互作用,从而构建出一个“碗状”的相互作用界面,分子本身嵌入其中。在该三元复合物中,MZ1通常采取“Z”型构象,其中JQ1基序和VH032基序分别以与其原始单价配体相似的结合模式结合于Brd4BD2和VHL(图2A)。
进一步的结构特征分析显示,战头分子中的氯原子朝向蛋白-蛋白相互作用界面,并与VH032基序中的乙酰基保持约4.6 Å的距离,提示该位置可能适合作为通过短连接臂将VHL配体连接起来的新位点。X射线结构还表明,JQ1基序中的酰胺基团指向VH032基序的苄基位点和邻位苯环位置,两者之间的距离分别为7.1 Å和8.2 Å(图2A)。已有研究证实,VH032的苄基和邻位苯环位置均暴露于溶剂环境中,适合用于基于VHL的PROTAC设计。
受上述结构信息的启发,设计了一系列新的“首尾相连”大环PROTACs,其目标在于降低结合过程中的能量损失,从而将分子锁定在有利于三元复合物形成的构象中。同时,大环化策略也有望提升基于VHL的PROTACs的代谢稳定性。在结构设计上,这些大环分子包含两段较短的连接臂,其中一段连接JQ1的苯环与VH032的乙酰基(上方,连接臂1),另一段则通过VH032的苄基或邻位苯环位置将JQ1的酰胺基团与VH032连接(下方,连接臂2a和2b)(图2B)。
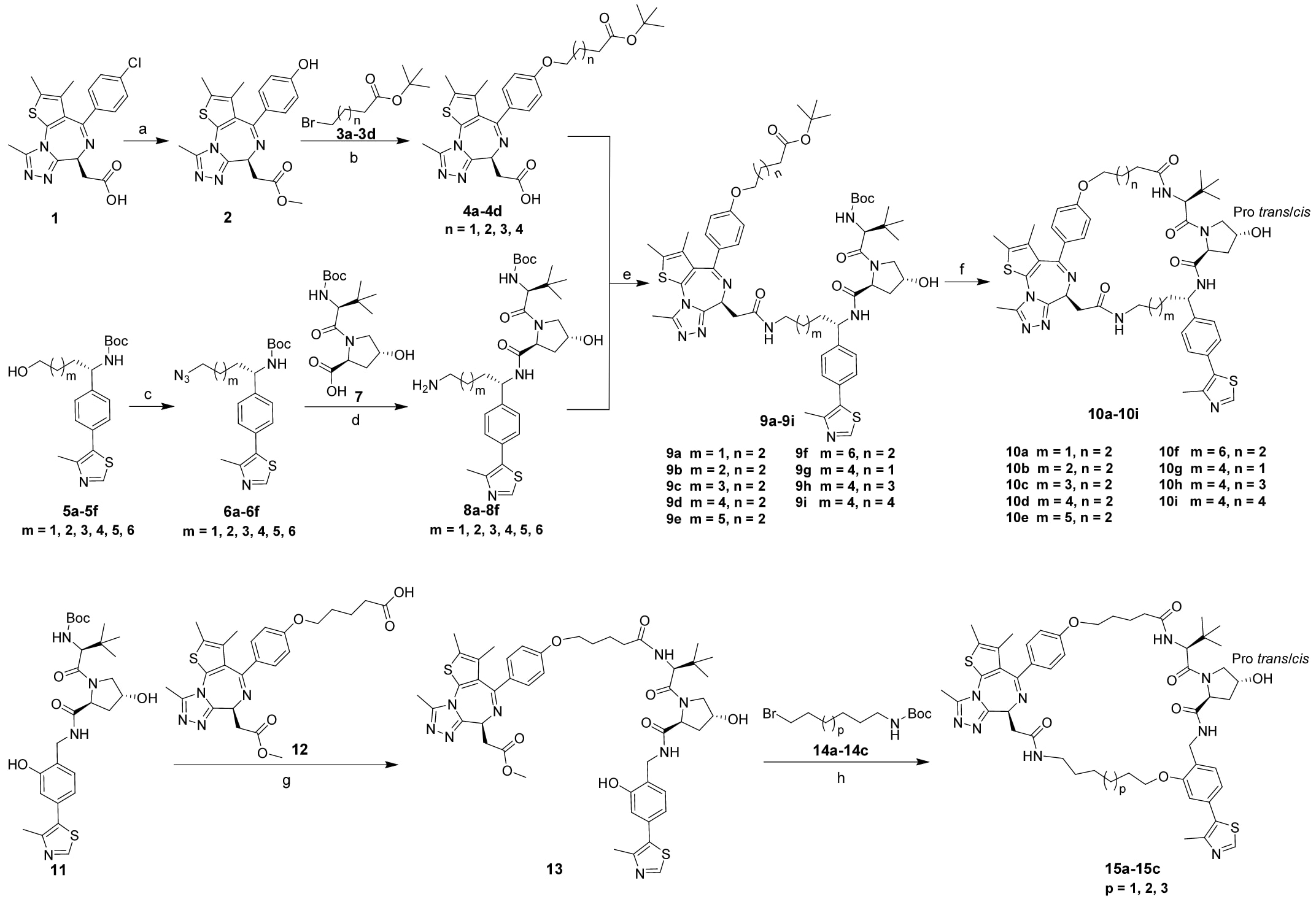
方案1|化合物10a−10i和15a−15c的合成路线示意图。
2.2 大环PROTACs的化学合成
所设计分子的合成路线如方案1所示。JQ1基序由商品化的(S)-2-(4-(4-氯苯基)-2,3,9-三甲基-6H-噻吩[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂卓-6-基)乙酸1制备得到。首先在三(二亚苄基丙酮)二钯(Pd2(dba)3)催化、t-BuBrettPhos配体以及氢氧化钾作为碱的条件下进行钯催化羟基化反应,随后对所得中间体进行酯化反应,得到化合物2。化合物2与溴取代的酯类3a−3d发生醚化反应,再经水解生成4a−4d。
VHL基序由5a至5f合成,其制备方法参考已报道的文献流程。5a−5f经Mitsunobu反应得到叠氮化合物6a−6f,随后脱保护并与化合物7反应生成相应的酰胺,其中叠氮基进一步被还原为伯胺,得到8a−8f。随后,JQ1基序4a−4d与8a−8f偶联生成9a−9i,经脱保护并进行大环化反应,最终得到目标化合物10a−10i。
化合物15a−15c以起始原料11为原料合成,该起始物按照已有报道的方法制备。首先脱去Boc保护基并进行酰胺化反应得到13,随后化合物13与溴取代连接臂14a−14c反应,并依次经过水解、Boc基团脱保护以及环化反应,获得最终产物15a−15c。
1H NMR结果表明,由于VHL结合基序中羟脯氨酸对酰胺键旋转的限制,大环化合物10c−10i和15a−15c以顺式/反式旋转异构体不可分离的形式存在。进一步的1H−1H ROESY谱图证实反式构象更加稳定,且通过加热至80 °C,这些化合物可转变为单一构象,这一过程已通过1H NMR加以确认(支持信息)。
表1|化合物10a−10i和15a−15c的蛋白降解效率。
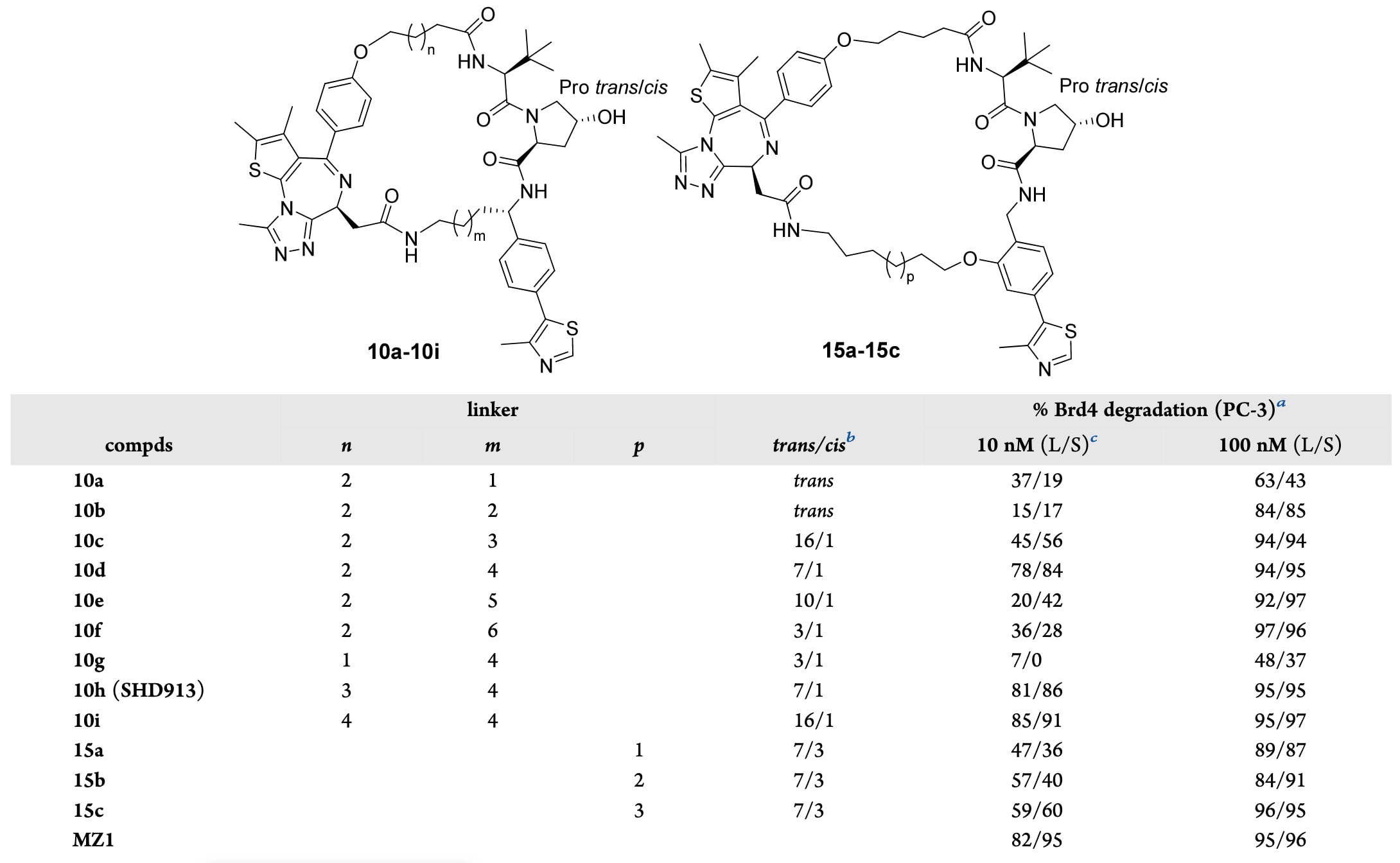
2.3 大环PROTACs的结构-降解关系
Brd4BD2:MZ1:VCB(PDB:5T35)三元复合物的结构显示,MZ1分子中氯原子与乙酰基之间的距离为4.6 Å,大致相当于三个C−C键的长度。因此,首先设计了以3个−CH2−单元(n=2)作为连接臂1的化合物,并以VH032的苄基位点作为大环化锚点,系统考察连接臂2a长度对活性的影响。如前所述,由于VHL结合基序中羟脯氨酸的存在,这些大环分子以顺式/反式旋转异构体不可分离的形式存在,其trans/cis比例通过1H NMR测定。
化合物的蛋白降解活性通过免疫印迹实验进行评估,在PC-3前列腺癌细胞中分别以10 nM和100 nM处理24 h,并以MZ1作为阳性对照(表1)。结果表明,大多数新型“首尾相连”大环分子均表现出明显的蛋白降解能力,且连接臂2a的长度对活性具有决定性影响(化合物10a−10f)。在第一组化合物中,连接臂2a为6个−CH2−单元(m=4)的化合物10d表现出最强的降解活性,在10 nM和100 nM条件下,对Brd4长、短两种亚型的降解率分别达到78%/84%和94%/95%。当连接臂2a进一步延长或缩短时,在10 nM条件下对Brd4长、短亚型的降解活性均显著降低,尽管在100 nM条件下所有化合物仍表现出较强的降解能力。例如,连接臂2a为4个−CH2−单元(m=2)的化合物10b在10 nM时对Brd4长、短亚型的降解率仅为15%和17%,而在100 nM时则分别提高至84%和85%。连接臂2a更长的化合物10e和10f在10 nM条件下同样表现出明显降低的活性,但在100 nM时其降解效率与10d几乎相当。
在确定连接臂2a的最优长度(m=4)后,进一步考察了连接臂1长度的影响。结果显示,较短的连接臂1(n=1,10g)在低浓度下几乎完全丧失蛋白降解活性,而具有更长连接臂1的化合物(n=3,10h,即SHD913;n=4,10i)在低、高浓度条件下均表现出与10d相当的活性。值得注意的是,化合物10d、SHD913和10i在实验条件下与MZ1具有相当的降解效力。
尽管在Brd4BD2:MZ1:VCB三元复合物中,VH032的邻位苯环位置(8.2 Å)相较于苄基位置(7.1 Å)与JQ1酰胺基团的距离略远,但该位点同样可作为大环PROTAC设计的连接向量。事实上,此前报道的半柔性macroPROTAC-1正是利用该位点实现环化的。因此,还基于邻位苯环位置设计并合成了一系列新的“首尾相连”大环PROTACs,并相应采用更长的连接臂2b以匹配其与JQ1酰胺基团之间更远的距离。所得化合物15a(p=1)、15b(p=2)和15c(p=3)在100 nM条件下同样能够有效降解Brd4,但在低浓度下其活性明显弱于通过苄基连接的化合物10d、SHD913和10i(表1,图S1)。由于大环化反应在该位点上存在较大的化学合成挑战,进一步的结构优化研究受到了一定限制。
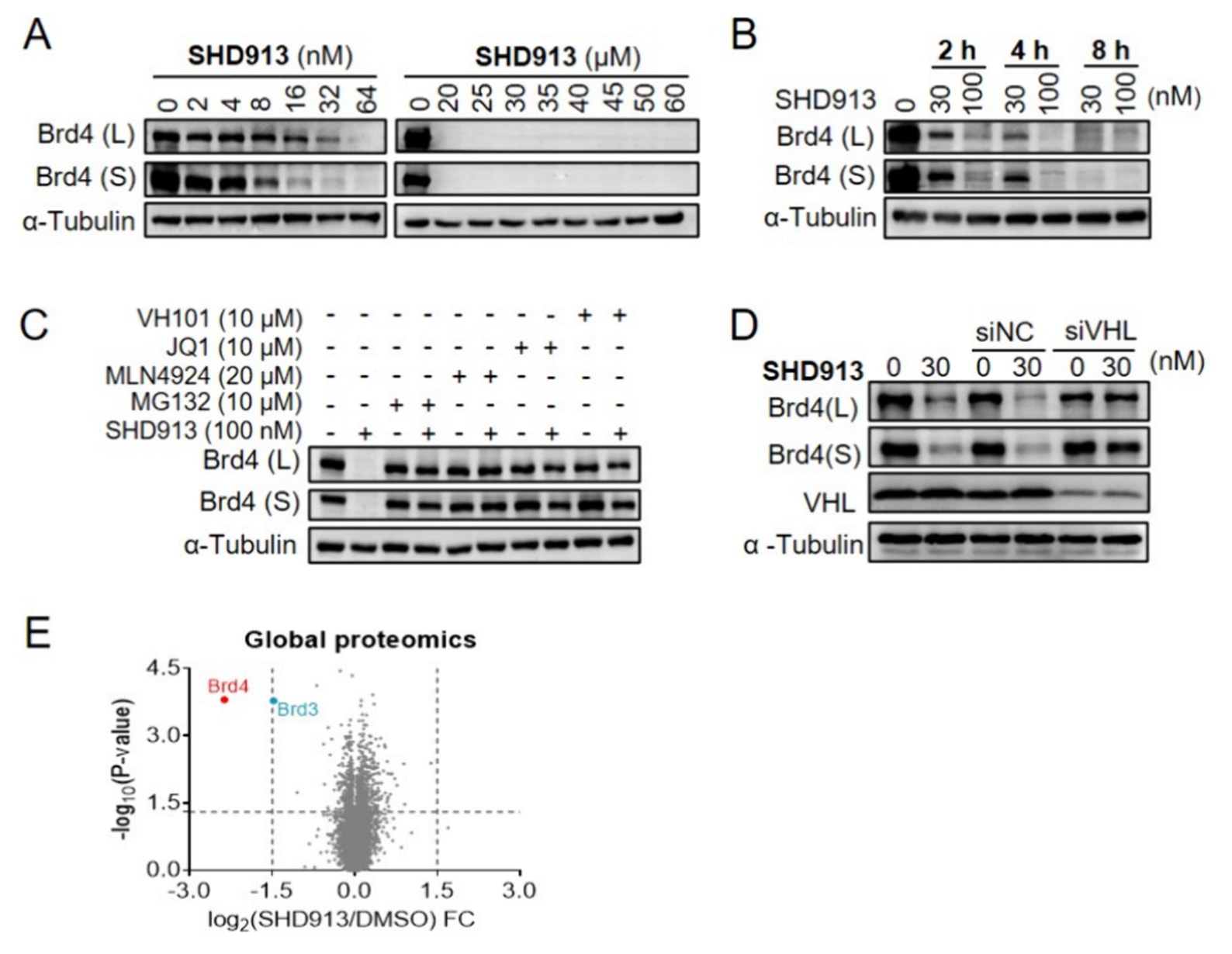
图3|SHD913通过泛素-蛋白酶体降解机制选择性降解Brd4。 (A)PC-3细胞中以不同浓度的SHD913处理24 h后Brd4水平的免疫印迹分析。(B)PC-3细胞中以30 nM或100 nM的SHD913处理不同时间后Brd4水平的免疫印迹分析。(C)PC-3细胞分别预处理MG132(10 μM)、MLN-4924(20 μM)、JQ1(10 μM)或VH101(10 μM)2 h后,再以100 nM SHD913处理2 h时Brd4水平的免疫印迹分析。(D)PC-3细胞经siRNA-VHL(50 nM)处理72 h后,再分别加入DMSO或30 nM SHD913处理4 h后Brd4水平的免疫印迹分析。(E)PC-3细胞分别以DMSO或100 nM SHD913处理2 h后蛋白质组学分析得到的蛋白水平变化情况。
2.4 化合物SHD913通过蛋白酶体降解过程选择性降解Brd4
由于兼具强效的降解活性和良好的合成可行性,SHD913被选作代表化合物开展进一步的生物学表征。实验结果表明,在PC-3细胞中,SHD913能够以剂量依赖和时间依赖的方式高效诱导Brd4长、短两种亚型的降解。该化合物在低至2−4 nM的浓度下即可显著降低Brd4蛋白水平,在64 nM处理24 h后几乎实现完全降解(>90%)。其对Brd4长、短亚型的DC50分别为7.7 nM和5.0 nM(图3A和图S2A)。此外,SHD913表现出快速起效的特征,在100 nM条件下处理2 h即可使Brd4两种亚型的降解率分别达到82%和86%(图3B)。
进一步验证了SHD913介导的泛素-蛋白酶体降解机制。预先加入蛋白酶体抑制剂MG132或NEDD8激活酶抑制剂MLN4924均可显著阻断SHD913诱导的Brd4降解(图3C)。BET溴结构域抑制剂JQ1和VHL配体VH101通过竞争性结合同样抑制了该降解过程。此外,采用siVHL预处理也能够逆转SHD913介导的Brd4降解(图3D),进一步支持该过程依赖于VHL的蛋白酶体降解机制。
为评估这一新型大环PROTAC的靶标选择性,采用TMT标记的质谱方法进行了全蛋白质组学分析。结果显示,Brd4是下调最为显著的蛋白。尽管Brd3的蛋白水平也有所下降,但幅度明显较小(图3E和图S2B、C)。在该实验中未检测到Brd2,这可能与Brd2在PC-3细胞中的丰度较低有关。
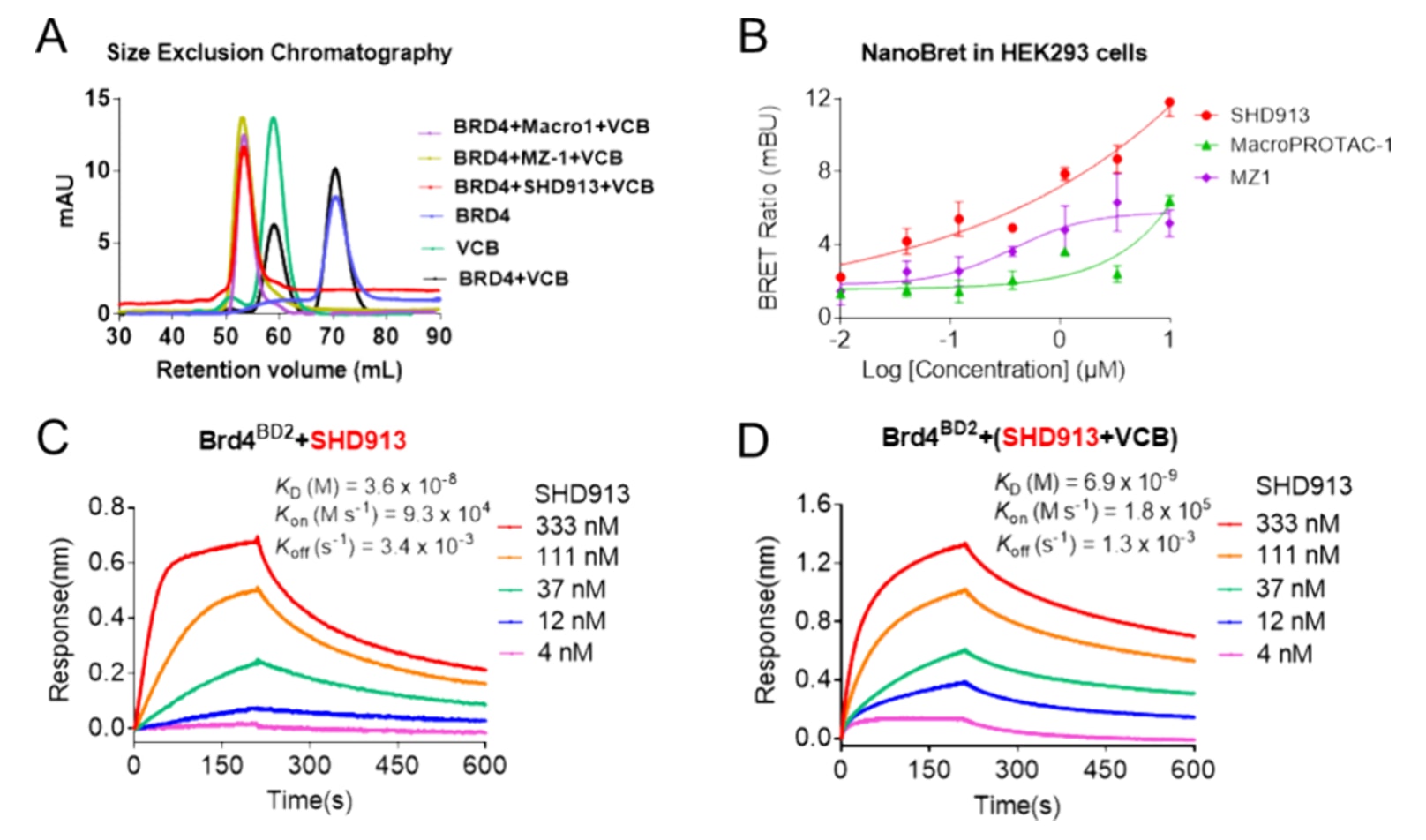
图4|SHD913诱导稳定的三元复合物形成并表现出正协同性。 (A)采用体积排阻色谱对不同组分进行分析,包括Brd4BD2(蓝色)、VCB(浅绿色)、Brd4BD2与VCB混合物(黑色),以及Brd4BD2与VCB在SHD913(红色)、MZ1(黄色)或macroPROTAC-1(紫色)存在下形成的复合物。(B)在HEK293细胞中通过NanoBRET实验验证三元复合物的形成情况,其中SHD913为红色,macroPROTAC-1为绿色,MZ1为紫色。(C)利用生物层干涉(BLI)实验测定SHD913与Brd4BD2形成二元复合物的Kd值,将生物素化Brd4BD2固定于传感器表面后,与不同浓度的SHD913孵育。(D)利用BLI实验测定SHD913:VCB:Brd4BD2三元复合物的Kd值,将生物素化Brd4BD2固定于传感器表面后,加入预先与VCB孵育的不同浓度SHD913进行测定。
2.5 SHD913在癌细胞中表现出正协同性并消除了“钩状效应”
“钩状效应”是PROTACs中常见且具有理论意义的问题,其来源于化合物与E3连接酶或靶蛋白之间的二元结合达到饱和状态。令人瞩目的是,即使在60 μM这一可应用的最高浓度下,即约为在PC-3细胞中测得DC50值的10000倍,仍未检测到SHD913的“钩状效应”(图3A)。SHD913强效且近似线性剂量依赖的蛋白降解能力在多种其他人源癌细胞中同样得到验证,包括非小细胞肺癌细胞系A549和NCI-H23、三阴性乳腺癌细胞MDA-MB-231以及胰腺癌细胞Panc 10.05(图S2C−F)。相比之下,MZ1在PC-3细胞中自30 μM起即出现明显的“钩状效应”,这一现象与此前报道一致(图S2G)。为排除SHD913未出现“钩状效应是由于溶解度或细胞摄取受限所致的可能性,进一步开展了细胞内质谱定量分析,结果表明该化合物可在细胞内呈剂量依赖性累积,且其细胞内浓度高于MZ1和macroPROTAC-1(图S2H)。
基于上述结果,进一步评估了SHD913是否能够诱导形成稳定的Brd4:SHD913:VCB三元复合物。首先通过体积排阻色谱实验发现,将SHD913与纯化的Brd4BD2和VCB蛋白混合后,洗脱体积明显向左偏移,而在缺少化合物的情况下未观察到该变化(图4A),表明稳定三元复合物的形成。SHD913在细胞中诱导三元复合物形成的能力也通过NanoBRET实验得到验证。在相同实验条件下,SHD913诱导的发光信号强度明显高于MZ1和macroPROTAC-1,且值得注意的是,MZ1在3.3 μM时即达到信号饱和,而SHD913在该实验中即使达到最高可行浓度10 μM仍未出现饱和现象(图4B)。这些结果共同表明,该大环PROTAC相较于先导化合物能够更高效地诱导三元复合物的形成。
为进一步评估SHD913诱导新的蛋白-蛋白相互作用的能力,首先开展了生物层干涉(BLI)实验,结果证实Brd4BD2与VCB之间不存在内在相互作用(图S3A)。然而,在将SHD913与不同浓度的VCB预孵育后,显著诱导了固定化生物素化Brd4BD2与VCB之间的相互作用,其Kd值为20.0 nM(图S3B)。协同性因子α(α=Kdbinary/Kdternary)用于反映化合物诱导两种不同蛋白形成三元复合物的能力。正协同性(α>1)意味着化合物与其中一种蛋白形成二元复合物后,可促进并稳定其与另一种蛋白形成三元复合物,通常源于有利的化学诱导蛋白-蛋白相互作用;负协同性(α<1)则提示化合物可能在两种蛋白之间引入不利的空间位阻,使二元结合不利于三元复合物的形成;而非协同性结合(α=1)表示双价分子体积足够大,与其中一种蛋白的结合不会影响其与另一种蛋白的相互作用。
为评估SHD913的协同性,基于BLI实验测得的Kd值计算了其α值。通过分别在有无VCB蛋白存在的条件下,将SHD913与生物素化Brd4BD2孵育,测定其Kdbinary和Kdternary。结果显示,在缺乏VCB时,SHD913与Brd4BD2结合紧密,Kdbinary为36.2 nM(图4C);而在与VCB预孵育后,其与Brd4BD2的结合显著增强,Kdternary降低至6.9 nM(图4D),对应的α值为5.2,表明其对Brd4BD2具有明显的正协同性。
此外,还通过等温滴定量热法(ITC)对SHD913、MZ1和macroPROTAC-1的协同性因子进行了验证。结果表明,SHD913在ITC实验中对Brd4BD2表现出高达117的α值(表2),在相同条件下略高于MZ1和macroPROTAC-1(图S4,表S1)。不同实验方法下α值的差异可归因于测定条件的不同。这些数据与前述生物物理实验和细胞NanoBRET结果高度一致,充分表明SHD913能够有效诱导Brd4BD2与VHL之间的相互作用,并显著缓解甚至消除“钩状效应”。
表2|通过等温滴定量热法(ITC)测定的SHD913、VHL−Elongin C−Elongin B(VCB)与Brd4BD2之间形成二元及三元复合物的热力学参数。

2.6 Brd4BD2:SHD913:VCB三元复合物的结构分析
为深入理解SHD913诱导的三元复合物结构特征,解析了Brd4BD2:SHD913:VCB的共晶结构,分辨率为2.95 Å(PDB:8YMB,表S2)。该三元复合物的整体四元构型与Brd4BD2:MZ1:VCB以及Brd4BD2:macroPROTAC-1:VCB高度相似,通过主链Cα原子比对得到的均方根偏差(RMSD)分别为1.092和1.079(图S5)。结构显示,SHD913以清晰的电子密度占据Brd4BD2与VHL之间的界面,作为桥梁将两种蛋白连接在一起,从而促进三元复合物的形成(图5A和图S6)。
在结合模式上,SHD913中的JQ1基序通过与Brd4BD2中N433形成氢键,并与WPF架构产生疏水相互作用,结合于乙酰赖氨酸识别口袋;VH032基序则通过多重氢键和堆叠相互作用结合于VHL的羟脯氨酸结合位点(图5B)。这些结合特征与此前报道的二元复合物晶体结构高度一致。此外,SHD913中的连接臂2a与Brd4BD2的ZA环第二段螺旋形成疏水相互作用,并有效填补了两种蛋白界面形成的空腔(图5B和图S7)。尤为重要的是,优化后的连接臂2a能够推动两种蛋白靠近,并通过Asp381Brd4(BD2)、Glu383Brd4(BD2)与Arg108VHL之间保守的双齿盐桥促进蛋白-蛋白相互作用的形成(图5C)。
在三种复合物结构中,仅在Brd4BD2:MZ1:VCB中观察到Arg69VHL与Glu438Brd4(BD2)之间的盐桥。相比之下,在Brd4BD2:MZ1:VCB和Brd4BD2:macroPROTAC-1:VCB中均可观察到由PEG连接臂与His437Brd4(BD2)介导的氢键,以及Arg107VHL、His110VHL与Ala384Brd4(BD2)之间的氢键,而在Brd4BD2:SHD913:VCB共晶结构的上部区域并未观察到明确的蛋白-蛋白相互作用(图5C)。因此,SHD913诱导的Brd4BD2与VHL之间的蛋白界面埋藏表面积略有降低,为603 Å2,低于MZ1(689 Å2)和macroPROTAC-1(681 Å2)。然而,SHD913中疏水性的连接臂1充当了“铰链”,将两种蛋白劫持至空间邻近位置,从而弥补了蛋白-蛋白相互作用的减少,并促进和稳定三元复合物的形成。
为考察该复合物在动态条件下的构象特征,进一步开展了分子动力学(MD)模拟。以三元复合物晶体结构为起点,构建了Brd4BD2:SHD913:VCB、Brd4BD2:SHD913以及SHD913:VCB三种体系,并分别进行了500 ns的MD模拟。结果显示,所有体系均表现出良好的稳定性,RMSD波动较小(图5D和图S8A、B)。对三元复合物的氢键分析证实了氢键网络的重要作用,与晶体结构中的观察一致。其中,JQ1三唑氮原子与Asn433Brd4(BD2)、SHD913羟脯氨酸与His110VHL、以及Arg108VHL与Asp381Brd4(BD2)之间的氢键占有率约为95%。相比之下,His110VHL与Ala384Brd4(BD2)之间的氢键占有率仅为12%,贡献明显较小(图5E)。
同时,通过分析大环PROTAC在二元复合物中的RMSD及三唑氮原子与羟脯氨酸之间的距离(dN−O),评估了其构象稳定性(图S8A、B)。结果表明,SHD913在两种二元复合物中均保持高度稳定的构象,RMSD变化幅度较小(图S8C、D)。在SHD913:Brd4BD2复合物中,dN−O距离为13.7±0.5 Å,在SHD913:VCB复合物中为15.1±1.0 Å,均与晶体结构中观察到的距离(dN−O=16.0 Å)以及三元复合物MD模拟中的结果(dN−O=16.3±0.4 Å)高度一致。这些结果表明,SHD913在二元复合物中保持了与三元复合物相似的构象,进一步支持了该大环PROTAC的构象稳定性。综合晶体结构解析与分子动力学模拟结果,为SHD913在诱导三元复合物形成方面的高效性提供了有力解释,并凸显了通过构象受限的大环化策略进行新型PROTAC设计的显著优势。
3 结论
综上所述,通过设计并合成首个“首尾相连”的大环Brd4 PROTAC,系统展示了大环化策略在新型PROTAC开发中的应用潜力。代表性大环分子SHD913在蛋白降解效力方面与此前报道的线性PROTAC MZ1相当,其DC50处于低nM水平。然而,SHD913在多种人源癌细胞系中显著缓解了PROTAC治疗中普遍存在的“钩状效应”,即使在浓度高出其DC50约10000倍的条件下,仍未检测到可测量的“钩状效应”。
生物物理实验结果表明,SHD913具有明显的正协同性,其在BLI和ITC实验中测得的协同性因子α分别为5.2和117。NanoBRET分析进一步显示,SHD913能够在细胞中强效诱导Brd4BD2与VHL之间产生新的蛋白-蛋白相互作用。通过解析Brd4BD2:SHD913:VCB三元复合物的共晶结构,揭示了化合物诱导蛋白-蛋白相互作用的具体结构基础,而分子动力学模拟结果进一步证实了SHD913构象受限特性在三元复合物形成中的关键作用。
尽管仍需开展更深入的表征研究,SHD913已清晰展现出显著减弱的“钩状效应”、正协同性以及诱导新的蛋白-蛋白相互作用的能力。此外,对SHD913进行的初步体外肝微粒体稳定性评估表明,与线性先导化合物相比,该大环分子在不同物种中均表现出更优的代谢稳定性(表S3)。上述结果共同支持了大环化策略在新型PROTAC设计中的潜在优势。