AS 2025 | ALLSites:对所有药物分子形态蛋白结合位点的精确识别
今天介绍的这项工作来自 Advanced Science。蛋白质通过与多种分子形态发生相互作用来执行其生物学功能,但长期以来,不同类型药物分子对应的蛋白结合位点缺乏系统而准确的鉴定,使得整个蛋白质组的可成药性仍然被严重低估。传统药物研发主要集中于小分子,但事实表明,小分子只能调控人类蛋白质组中不到一小部分蛋白,这促使研究者不断探索蛋白、多肽、核酸以及糖类等新型药物分子形态。在这一背景下,如何在全蛋白质组尺度上统一识别不同药物分子形态的结合位点,成为拓展“可成药蛋白”边界的关键问题。现有的蛋白结合位点预测方法大致可分为基于结构和基于序列两类。基于结构的方法在精度上具有优势,但高度依赖高质量三维结构,而大量蛋白缺乏实验解析结构,预测结构的不确定性也会显著影响性能。基于序列的方法适用范围更广,却往往难以充分刻画残基之间的相互作用,从而限制了预测准确性。此外,多数已有方法仅针对单一药物分子形态设计,难以满足跨模态、统一预测的需求。针对这些局限,该工作提出了一个统一的序列驱动框架ALLSites,用于在不依赖结构信息的情况下,准确识别多种药物分子形态的蛋白结合位点。该方法以蛋白语言模型ESM-2为基础,从序列中提取高维残基表示,并结合门控卷积网络与Transformer架构,同时学习局部上下文特征和全局序列信息,从而在序列层面有效建模残基之间的复杂相互作用。这种设计在一定程度上弥合了序列方法与结构方法之间的性能差距。系统评测结果表明,ALLSites在蛋白-蛋白、蛋白-多肽、蛋白-小分子、蛋白-糖类以及蛋白-核酸等多种结合位点预测任务中均表现出稳定而优越的性能。其整体精度不仅显著优于现有序列方法,而且在多项任务上可达到甚至接近最优结构方法的水平。同时,该方法具备较高的计算效率,能够支持全蛋白质组尺度的快速预测。总体而言,该研究提供了一种兼顾准确性与适用性的统一解决方案,为系统性描绘蛋白质组的可成药性图谱提供了重要工具,也为多种药物分子形态的发现与设计奠定了方法学基础。

获取详情及资源:
- 📄 论文: https://advanced.onlinelibrary.wiley.com/doi/full/10.1002/advs.202516530
- 💻 代码: https://github.com/idrblab/ALLSites
0 摘要
蛋白质能够与多种不同的分子形态发生相互作用,然而其结合位点尚未被全面识别,这使得蛋白质组尺度上的可成药性仍然缺乏系统认识。尽管已经开发了多种用于预测蛋白结合位点的计算方法,现有方法仍受到诸多限制,例如通常仅适用于单一药物分子形态,高度依赖高质量结构数据,或预测精度不足。为此,构建了一种统一的基于序列的框架ALLSites,用于在蛋白质组范围内识别所有药物分子形态的结合位点。该方法基于ESM-2嵌入表示,将门控卷积网络与Transformer架构相结合,同时捕获序列的全局与局部特征,并直接从序列层面有效建模残基之间的相互作用。这一设计在序列方法与结构方法之间建立了联系,使ALLSites在多种药物分子形态上均表现出优异的预测性能,涵盖蛋白、多肽、小分子、糖类、DNA和RNA等。该方法在序列方法中达到了当前最优水平,并在精度上可与最佳结构方法相当。通过实现跨所有药物分子形态的高精度、无结构依赖的结合位点预测,ALLSites有望拓展可成药蛋白质组的边界,并为药物发现提供有力的研究工具。
1 引言
蛋白质通过与多种分子形态发生相互作用在细胞过程中发挥基础性作用。然而,由于可被配体调控的蛋白数量有限以及作用机制认识不充分,蛋白的可成药性在整体蛋白质组尺度上仍然缺乏系统探索。即便小分子是目前最主要的药物分子形态,其也只能调控不到15%的人类蛋白质组,这一事实清楚地反映了现有药物开发范式的局限性。为突破这一瓶颈,研究者开始探索替代性的药物分子形态,包括蛋白类,多肽类,核酸类以及糖类治疗手段,以实现对蛋白功能的调控。因此,对所有药物分子形态对应的蛋白结合位点进行全面识别具有关键意义,因为这有助于在不同分子形态之间重新界定蛋白的可成药性,从而将某一分子形态下被认为“不可成药”的蛋白转化为另一分子形态下的“可成药”靶点。由于药物分子形态本身具有高度的多样性和复杂性,通过实验手段鉴定蛋白结合位点面临巨大挑战,这也促使研究者投入大量精力开发用于预测不同药物分子形态蛋白结合位点的计算方法。
根据输入信息的不同,现有计算方法大体可分为基于结构和基于序列两类。以蛋白-蛋白相互作用位点预测为例,DeepPPISP,DELPHI和EnsemPPIS等方法分别利用蛋白三维结构或序列信息来识别相互作用残基。PepBind和PepSite等工具则专门用于预测蛋白-多肽相互作用位点,为蛋白和多肽类药物的设计提供支持。在小分子相关研究中,已有多种方法用于预测蛋白-小分子相互作用位点,例如P2Rank通过机器学习识别潜在的小分子结合口袋,而CAPSIF:V等方法则针对蛋白-糖类相互作用位点进行了专门设计。此外,针对核酸分子,研究者也开发了多种DNA或RNA结合位点预测工具,以加深对生物学过程的理解并推动核酸类药物的设计,例如用于DNA-蛋白相互作用预测的DNAPred和DNABind,以及用于RNA-蛋白相互作用预测的NucleicNet。
尽管上述方法在各自领域取得了一定进展,但在蛋白质组尺度上准确区分和识别所有药物分子形态的结合位点仍然面临显著困难。一方面,许多方法高度依赖精确的三维结构或预测精度有限。基于结构的方法受限于高分辨率结构的稀缺性,且对结构误差高度敏感,尤其是大量被认为不可成药的蛋白缺乏实验解析结构,使用预测结构往往会进一步降低方法性能。相比之下,基于序列的方法具有更广泛的适用性,但由于难以充分刻画残基之间的相互作用信息,预测性能通常不理想。另一方面,现有方法大多针对单一药物分子形态设计,尚缺乏能够统一预测所有药物分子形态结合位点的通用方法。当前仅有少数方法支持多模态预测,例如GraphBind主要针对DNA和RNA结合位点,而PepBCL仅在多肽场景下表现良好,在DNA或RNA任务中性能较差。总体来看,蛋白质组和药物分子形态覆盖不足以及预测性能受限,严重制约了这些方法的实际应用,从而迫切需要一种能够在蛋白质组范围内准确预测所有药物分子形态结合位点的新方法。
基于不同药物分子形态结合位点的关键信息本质上蕴含于蛋白序列之中这一认识,该研究构建了一种统一的基于序列的框架ALLSites,用于识别蛋白质组尺度上所有药物分子形态的结合位点。该方法以蛋白语言模型ESM-2为序列嵌入基础,将门控卷积网络与Transformer架构相结合,同时学习全局序列特征和局部上下文模式,并直接从序列中建模复杂的残基相互作用,从而在一定程度上弥合了序列方法与结构方法之间的差距。ALLSites能够在保持广泛适用性的同时,对蛋白,多肽,小分子,糖类,DNA和RNA等多种药物分子形态实现高精度的结合位点预测。在各类任务中,该方法在序列方法中达到了当前最优水平,并在性能上可与最佳结构方法相媲美。准确性与适用性的平衡使ALLSites成为推动蛋白质组可成药性研究以及加速多种分子形态向临床应用转化的重要工具。
2 结果和讨论
2.1 用于预测所有药物分子形态结合位点的ALLSites框架
为实现对所有药物分子形态蛋白结合位点的精确识别,基于Transformer架构设计了一种统一的深度学习框架ALLSites。其整体框架如图1a所示。模型首先以蛋白序列作为输入,利用蛋白语言模型ESM-2生成残基级别的嵌入表示。随后,这些嵌入被输入至包含门控卷积网络的编码器中,其结构如图1b所示。编码器的主要作用是为每个残基提取局部上下文模式,并进一步整合形成蛋白的全局序列特征。在此基础上,原始的残基嵌入与编码器提取的全局序列特征共同输入到改进的Transformer解码器中,其结构如图1c所示。该解码器引入了跨注意力机制,并对原始Transformer中的掩码操作进行了调整,以确保模型能够在蛋白全序列范围内进行有效学习。多头跨注意力机制使ALLSites能够捕获序列中任一残基与其他残基之间的相互作用关系。最终,解码器输出的特征被送入由多层全连接层组成的分类器,用于预测每个残基在不同药物分子形态下作为结合位点的概率。ALLSites能够直接从蛋白序列中学习多样化的残基特征,从而作为一个统一框架,在蛋白质组尺度上识别包括蛋白,多肽,小分子,糖类,DNA和RNA在内的所有药物分子形态的结合位点。
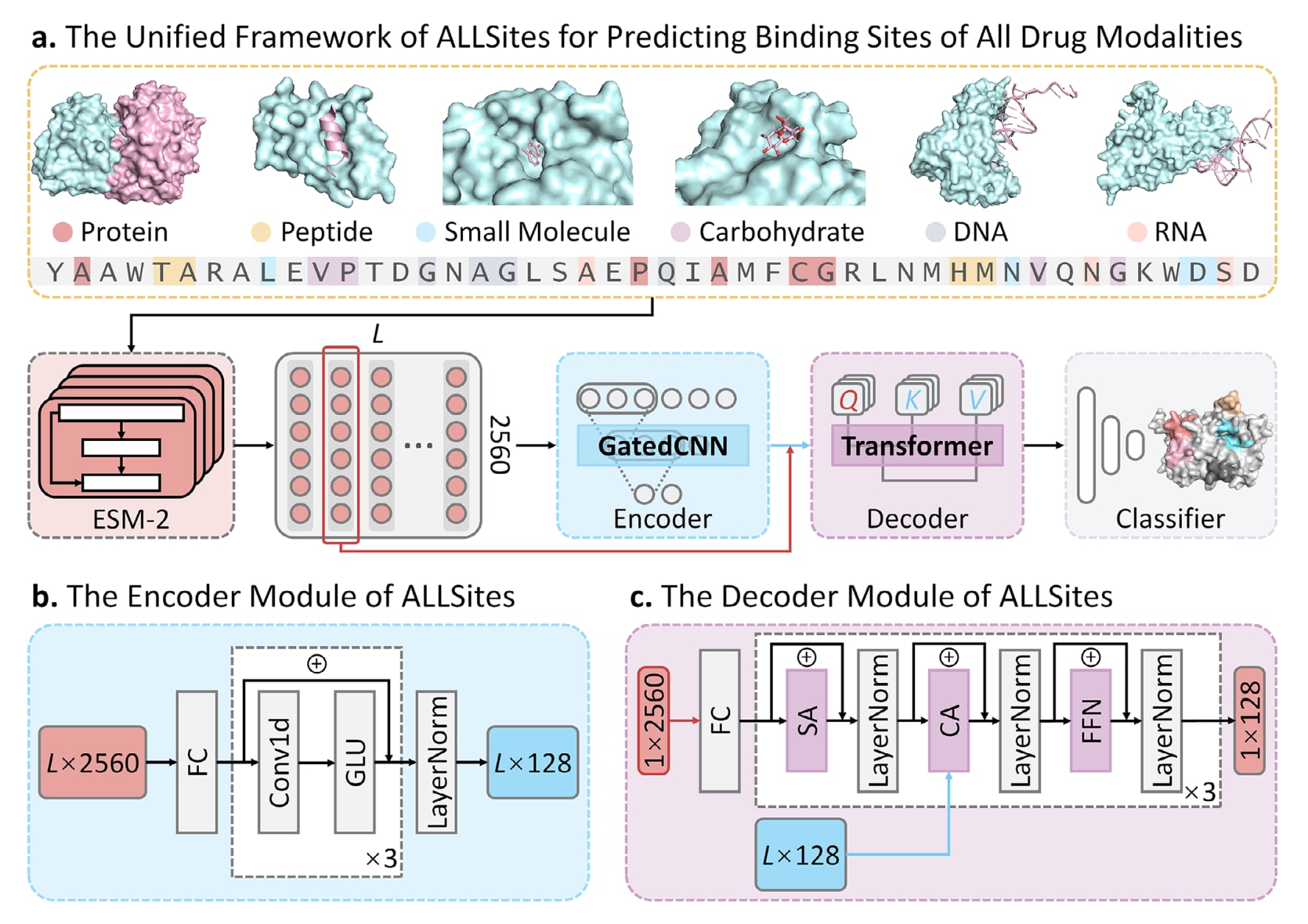
图1|用于预测所有药物分子形态蛋白结合位点的ALLSites框架。 a)ALLSites的整体框架。该方法仅以蛋白序列作为输入,即可实现对多种药物分子形态结合位点的预测,包括蛋白,多肽,小分子,糖类,DNA和RNA。模型整体由三个主要模块组成,分别为用于处理ESM-2生成表示的蛋白特征编码器,跨注意力解码器以及分类模块。b)编码器模块的结构。编码器本质上是一个门控卷积网络,用于从蛋白序列中同时提取局部特征和全局特征,其由一维卷积层和门控线性单元激活函数构成。c)解码器模块的结构。解码器以残基级表示和全局蛋白特征作为输入,通过跨注意力机制学习残基之间的相互作用信息。编码器和解码器均由三层结构组成。FC表示全连接层,SA表示自注意力机制,CA表示跨注意力机制,FFN表示位置前馈网络。
2.2 ALLSites在蛋白与多肽结合位点识别中的评估
识别蛋白-蛋白相互作用位点和蛋白-多肽相互作用位点对于蛋白类和多肽类生物药物的开发具有重要意义。为全面评估ALLSites在蛋白与多肽结合位点识别中的性能,该研究选用了四个常用的蛋白-蛋白相互作用位点基准数据集以及两个蛋白-多肽相互作用位点基准数据集进行系统测试。
2.2.1 ALLSites在蛋白-蛋白相互作用位点预测中的性能评估
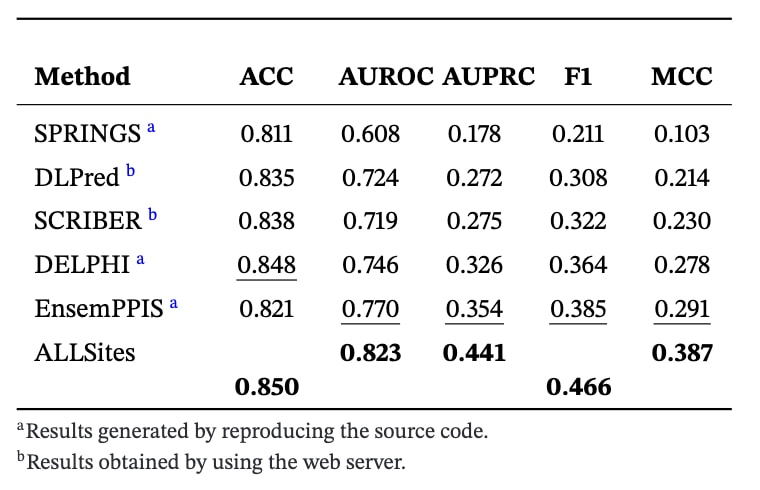
首先在PPI-Test70数据集上对ALLSites与13种对比方法的性能进行了系统比较。对比方法包括4种基于结构的方法和9种基于序列的方法。除SPPIDER,ProNA2020,SCRIBER和DLPred的结果来自其在线服务器外,其余方法的结果均通过复现源代码或直接整理自相关文献获得,以确保训练数据一致。结果如表1所示,ALLSites在AUROC,AUPRC,F1和MCC等核心指标上均取得最优表现,同时超越了所有基于序列和基于结构的方法。与性能排名第二的EnsemPPIS相比,ALLSites在AUROC和AUPRC上分别提升了5.0%和8.1%,在F1和MCC指标上也分别提高了0.034和0.042。在另一项常用的PPI位点预测任务中,采用PPI-Train9982作为训练集,PPI-Test355作为测试集,由于训练数据不包含结构信息,因此仅在基于序列的方法之间进行比较。结果如表2所示,ALLSites在所有评估指标上依然表现最优,整体性能显著优于其他序列方法。相较于EnsemPPIS,ALLSites在AUROC和AUPRC上分别提高了6.9%和24.6%,在F1和MCC指标上分别提升了0.081和0.096。
表1|ALLSites在PPI-Test70数据集上的性能评估结果。每一项评估指标中表现最优的结果以加粗标示,次优结果以下划线标示。
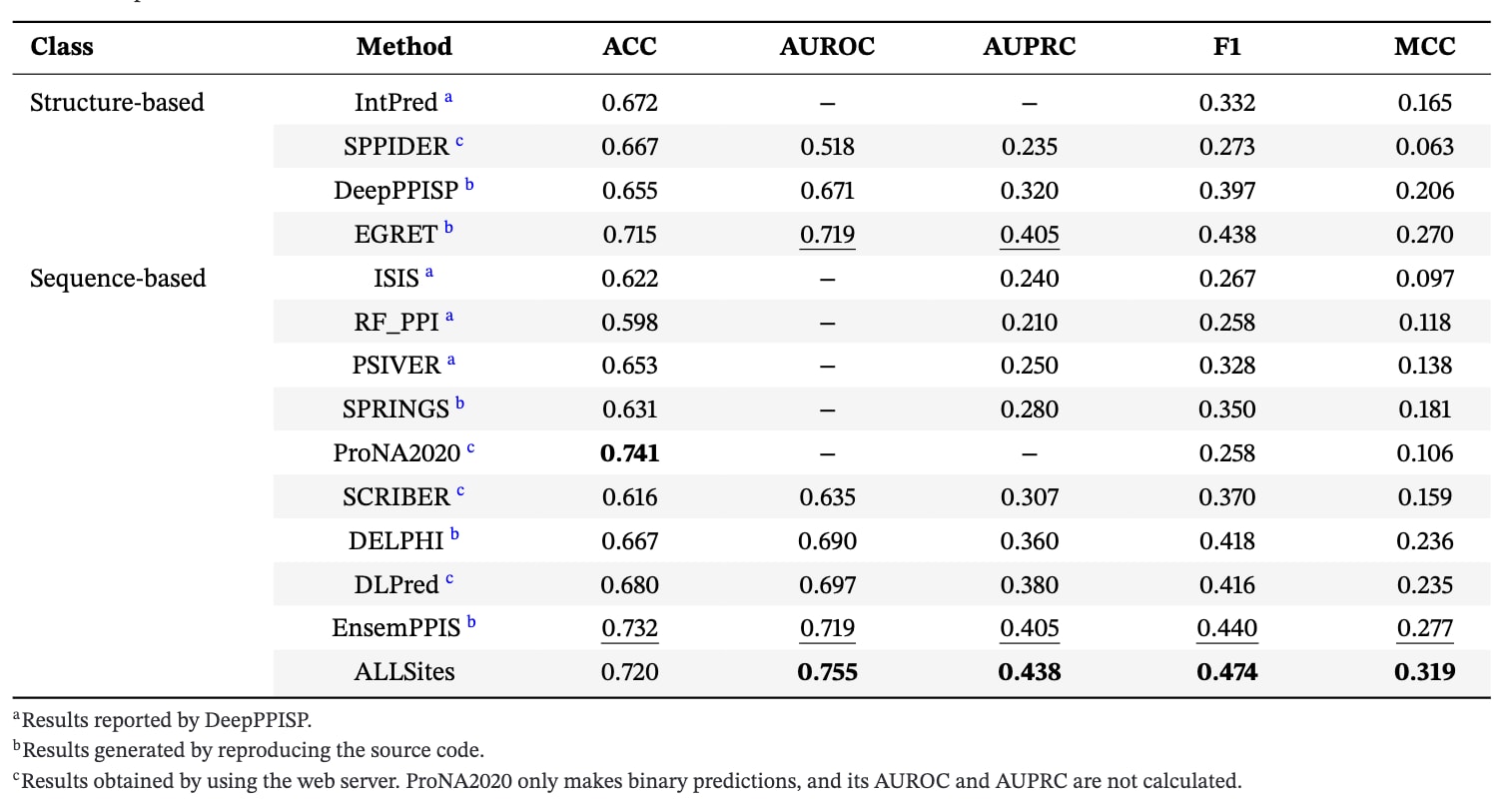
表2|ALLSites在PPI-Test355数据集上的性能评估结果。所有对比方法均仅使用蛋白序列信息进行预测。每一项评估指标中表现最优的结果以加粗标示,次优结果以下划线标示。
此外,ALLSites还在PPI-Test60和PPI-Test315数据集上进行了评估,所使用的模型均基于相同训练策略在PPI-Train335数据集上训练得到。如图2a所示,在AUROC和MCC等关键指标上,ALLSites明显优于所有基于序列的方法,并且整体性能可与最佳基于结构的方法RGN相当。相比之下,作为序列方法中表现次优的EnsemPPIS,其MCC指标始终低于GraphPPIS和RGN等结构方法。值得注意的是,在PPI-Test60数据集上,ALLSites的表现甚至略优于采用几何深度学习从蛋白表面结构中学习特征的MaSIF-site。进一步的校准分析结果表明,ALLSites在这两个测试集上的预测具有良好的可靠性,其Brier得分分别为0.153和0.163,均低于0.25这一常用阈值,说明预测概率具有较高可信度。
考虑到ALLSites的蛋白表示模块理论上可以结合更新的蛋白语言模型,研究中进一步比较了使用ESM-2和ESM-C两种表示方式的性能差异。受硬件条件限制,选用了ESM-C 600M模型进行评估,并在PPI-Test70,PPI-Test315和PPI-Test60数据集上进行测试。结果显示,在PPI-Test70数据集上,ESM-C在AUROC和AUPRC上略有提升,但在ACC,F1和MCC等指标上整体性能下降。尤其是在PPI-Test60和PPI-Test315数据集上,几乎所有评估指标均出现明显下降,MCC分别降低了0.037和0.032。这表明在PPI位点预测任务中,采用更新的ESM-C模型并未进一步提升ALLSites的整体性能,这一结论也与近期关于蛋白语言模型的大规模基准评测结果一致,从而验证了在ALLSites中选择ESM-2作为蛋白表示骨干的合理性。
在蛋白层面,ALLSites在PPI-Test315和PPI-Test60数据集上的表现同样得到了进一步分析。如图2b所示,在PPI-Test315数据集中,ALLSites在73.7%的蛋白上取得了高于EnsemPPIS的MCC值。以PDB编号为6G4JB的蛋白为例,ALLSites显著减少了假阳性和假阴性预测,其MCC达到0.834,而EnsemPPIS仅为0.425。在PPI-Test60数据集中也观察到类似趋势,ALLSites在81.7%的蛋白上优于EnsemPPIS。然而,在该数据集中仍有18.3%的蛋白上ALLSites的MCC低于EnsemPPIS。例如在PDB编号为4HLUA的蛋白中,ALLSites的MCC仅为0.095,而EnsemPPIS为0.333,其原因在于ALLSites虽然减少了假阳性预测,但真实阳性预测数量极少,从而显著拉低了MCC值。
从AUROC指标来看,ALLSites在大多数蛋白上的表现同样优于EnsemPPIS。综合残基层面与蛋白层面的评估结果可以看出,ALLSites在蛋白-蛋白相互作用位点预测任务中均达到了当前最优水平。
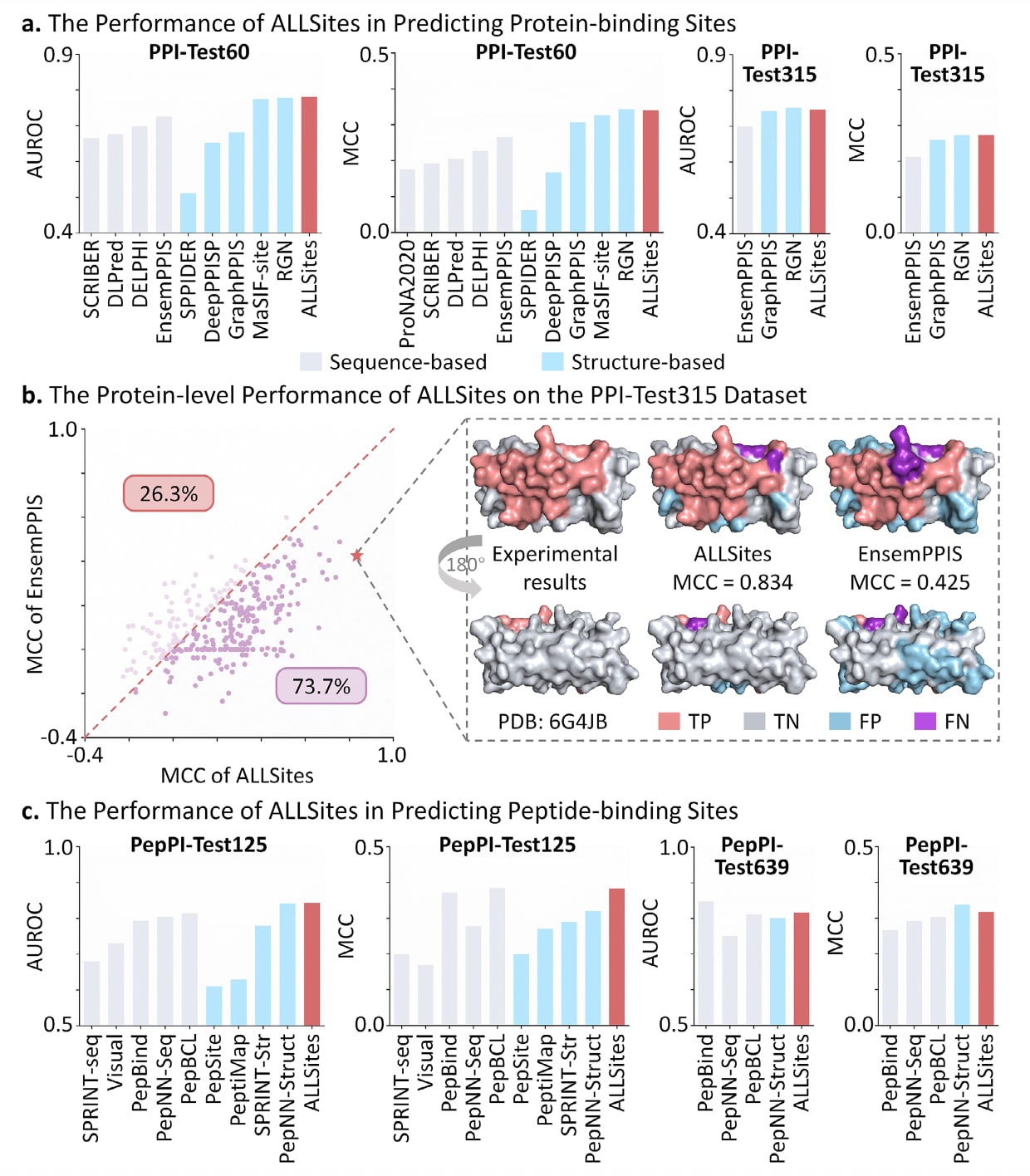
图2|ALLSites在蛋白与多肽结合位点识别中的性能表现。 a)ALLSites在蛋白结合位点预测中的整体性能,在PPI-Test60和PPI-Test315数据集上以AUROC和MCC作为评估指标进行比较,灰色柱表示基于序列的方法,蓝色柱表示基于结构的方法。b)ALLSites在PPI-Test315数据集上的蛋白层面预测性能。基于ALLSites的预测结果为每个蛋白计算MCC指标,并以一个具体蛋白为例进行展示,PDB编号为6G4JB,用于对比ALLSites与EnsemPPIS的预测结果及其对应的实验标注。c)ALLSites在多肽结合位点预测中的性能表现,在PepPI-Test125和PepPI-Test639数据集上以AUROC和MCC作为评估指标进行比较。相关原始数据见表S2至S5。
2.2.2 ALLSites在蛋白-多肽相互作用位点预测中的性能评估
针对蛋白-多肽相互作用位点预测,从两个任务对ALLSites的性能进行了系统评估。第一项任务以PepPI-Train1154作为训练集,PepPI-Test125作为测试集;第二项任务则采用PepPI-Train640进行训练,并在PepPI-Test639数据集上进行测试。在评估过程中,将ALLSites与9种现有的蛋白-多肽相互作用位点预测方法进行了比较,其中包括5种基于序列的方法和4种基于结构的方法。由于各方法采用了相同的训练数据集和一致的模型训练策略,对比方法的性能指标直接引自相关文献,从而保证了评估的公平性。
如图2c所示,ALLSites在两项任务中均展现出优异的预测性能。在PepPI-Test125数据集上,ALLSites在AUROC和MCC两个关键指标上同时优于所有基于序列和基于结构的方法。在AUROC指标方面,其表现不仅超过了所有序列方法,还略高于表现最优的结构方法PepNN-Struct;在MCC指标上,ALLSites优于全部结构方法,仅比性能最佳的PepBCL低0.002,差距极小。在PepPI-Test639数据集上,PepBind和PepNN-Struct分别在AUROC和MCC指标上取得最高分,而ALLSites在两项指标上均稳定排名第二,显示出良好的泛化能力和稳健性。综合上述结果可以看出,ALLSites能够准确识别蛋白中的多肽结合位点,在不同数据规模和任务设置下均保持较高性能,为多肽药物的发现与设计提供了一种可靠而有效的计算工具。
2.3 ALLSites在小分子与糖类结合位点识别中的评估
2.3.1 ALLSites在蛋白-小分子相互作用位点预测中的性能评估
小分子是当前已获批治疗药物中最为常见的分子形态,因此在蛋白上准确识别潜在的蛋白-小分子相互作用位点对于新型小分子药物的研发至关重要。为评估ALLSites在小分子结合位点预测中的性能,基于sc-PDB数据库构建了一个新的蛋白-小分子相互作用位点基准数据集,共包含2324条非冗余蛋白序列,且两两序列相似性较低。该数据集被随机划分为训练集SMPI-Train1628,验证集SMPI-Valid348和测试集SMPI-Test348。在SMPI-Test348数据集上,将ALLSites与一种广泛使用的基于结构的方法P2Rank进行了对比评估。按照原始研究中的设置,P2Rank的性能基于其预测得分最高的结合口袋进行计算。需要指出的是,P2Rank的预测结果直接来自其预训练模型,未在该研究数据集上重新训练,因此在训练数据来源上与ALLSites并不完全一致,这一差异可能在一定程度上影响比较的公平性。尽管如此,从实际应用角度出发,将ALLSites与预训练的P2Rank进行对比仍具有重要参考价值。如图3a左图所示,ALLSites在准确率,召回率,精确率,F1和MCC等五项指标上均优于P2Rank。尤其是在F1,MCC和召回率方面,ALLSites分别达到0.601,0.560和0.593,相较P2Rank分别提升了0.151,0.136和0.232。
进一步从蛋白层面对SMPI-Test348数据集的预测性能进行了分析。如图3b所示,在348个蛋白中,ALLSites在66.4%的情况下取得了高于P2Rank的MCC值。以两个代表性蛋白为例,在PDB编号为2QA1A和3OUMA的蛋白中,ALLSites分别获得了0.755和0.717的MCC,显著高于P2Rank的对应结果。从预测结果可以直观地看出,ALLSites识别的结合位点更接近真实的小分子结合口袋,而P2Rank则产生了较多的假阴性预测。值得强调的是,ALLSites在完全不依赖任何结构信息的情况下,整体性能仍优于成熟的结构方法P2Rank,充分体现了其仅基于序列提取小分子结合位点特征的强大能力。
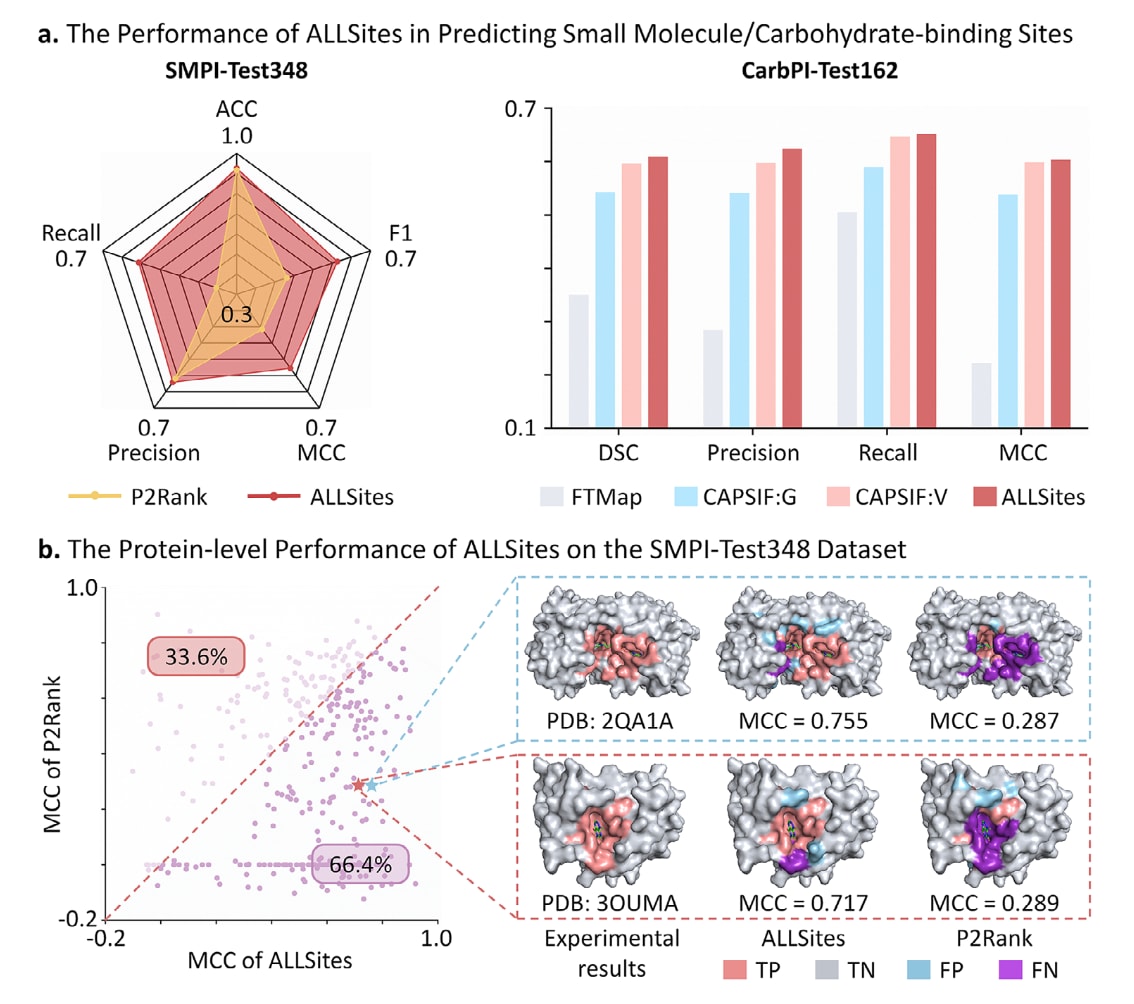
图3|ALLSites在小分子与糖类结合位点识别中的性能表现。 a)ALLSites在残基层面预测蛋白-小分子相互作用位点和蛋白-糖类相互作用位点的性能评估,分别在SMPI-Test348和CarbPI-Test162数据集上进行测试,所有对比方法均基于结构信息。相关原始数据见表S6和表S7。b)ALLSites在SMPI-Test348数据集上的蛋白层面预测性能,基于ALLSites的预测结果为每个蛋白计算MCC指标,并选取两个具体蛋白作为示例,PDB编号分别为2QA1A和3OUMA,用于展示ALLSites与P2Rank的预测结果及其对应的实验标注。
2.3.2 ALLSites在蛋白-糖类相互作用位点预测中的性能评估
关于糖类与蛋白相互作用的研究已经促成了多种获批药物的出现。尽管相当一部分糖类分子在分子尺寸上可归类为小分子,其独特的化学性质,尤其是富含羟基的结构特征,使其对应的蛋白结合位点在本质上不同于典型的小分子结合位点。因此,有必要单独评估ALLSites在蛋白-糖类相互作用位点识别中的预测能力。
用于评估ALLSites在蛋白-糖类相互作用位点预测性能的基准数据集来源于已有研究,包含训练集CarbPI-Train517,验证集CarbPI-Valid129以及测试集CarbPI-Test162。性能比较中选用了三种基于结构的方法,其中包括一种通用的小分子结合位点预测工具FTMap,以及两种针对糖类分子专门设计的结合位点预测方法CAPSIF:V30和CAPSIF:G30。由于这些方法采用了相同的训练与评估流程,对比所需的性能指标直接引自原始研究,从而保证了评估结果的可比性。评估时采用与原研究一致的指标体系,并对测试集中所有蛋白的结果取平均值。如图3a右图所示,ALLSites在平均DICE,精确率,召回率和MCC等指标上均优于所有对比方法。相较于FTMap,ALLSites的平均DICE和MCC分别提高了0.258和0.381,性能提升尤为显著。FTMap表现较差的原因在于其设计目标是通用的小分子结合口袋预测,并未针对蛋白-糖类相互作用位点进行专门优化。上述结果进一步表明,糖类结合位点在特征上与常规小分子结合位点存在显著差异。同时,ALLSites具有良好的可移植性,不仅适用于糖类分子,还可扩展用于其他小分子药物形态的结合位点预测,例如共价结合位点的识别。
2.4 ALLSites在核酸结合位点识别中的评估
2.4.1 ALLSites在DNA与RNA结合位点预测中的性能评估
核酸-蛋白相互作用在多种关键细胞过程中发挥着核心作用,包括DNA复制,转录以及翻译等。阐明这些相互作用背后的分子机制,尤其是对蛋白中DNA和RNA结合位点的刻画,有助于推动针对蛋白与核酸异常调控相关疾病的药物研发。为评估ALLSites在核酸结合位点识别方面的能力,该研究采用了两个用于DNA-蛋白相互作用位点预测的基准数据集以及一个用于RNA-蛋白相互作用位点预测的数据集。
在DNA-蛋白相互作用位点预测任务中,将ALLSites与3种基于序列的方法以及4种基于结构的方法进行了性能比较。其中,GraphBind的结果通过复现其源代码获得,其余方法的性能则通过各自的在线服务器获取。使用在DPI-Train573数据集上训练得到的单一模型,分别在DPI-Test129和DPI-Test181两个测试集上进行评估。如图4a所示,ALLSites在两个测试集上的AUROC和MCC指标均显著优于所有基于序列的方法,并超过了大多数基于结构的方法。与表现最优的序列方法相比,ALLSites在DPI-Test129和DPI-Test181上的AUROC分别提高了9.7%和12.6%,MCC分别提升了0.148和0.150。尽管在MCC指标上略低于表现最佳的结构方法GraphBind,但ALLSites在AUROC上的表现与其相当。
在RNA-蛋白相互作用位点预测的基准评测中,ALLSites与2种基于序列的方法以及5种基于结构的方法进行了比较。NucleicNet和GraphBind的结果通过复现源代码获得,其余方法的性能来自对应的在线服务器。如图4b所示,ALLSites在该任务中的表现与DNA结合位点预测结果一致,在所有序列方法中表现最优,并优于大多数结构方法。在AUROC和MCC两个指标上,ALLSites均排名第二,其中AUROC与表现最佳的结构方法GraphBind处于相同水平。这些结果表明,ALLSites仅依赖蛋白序列信息,即可对核酸结合位点实现准确识别。
2.4.2 基于预测结构的结构方法性能评估
尽管在DNA和RNA结合位点预测任务中,ALLSites的性能整体上略低于表现最优的结构方法GraphBind,但由于仅依赖蛋白序列信息,其在蛋白质组尺度上具有更广泛的适用性。这一优势尤为重要,因为结构方法受到高分辨率实验结构数量有限以及对结构误差高度敏感等因素的显著制约。实际上,仅约35%的人类蛋白拥有实验解析的晶体结构,且在许多情况下,这些结构只覆盖了完整序列的一部分。虽然近年来出现了AlphaFold2和RoseTTaFold等先进的蛋白结构预测工具,在一定程度上缓解了结构数据匮乏的问题,但预测结构与天然结构之间不可避免的偏差,往往会显著降低结构方法的预测性能。
为进一步揭示结构方法的局限性,研究中评估了使用AlphaFold2预测结构对GraphBind性能的影响,并在DPI-Test129和DPI-Test181数据集上进行了测试。如图4c所示,当使用预测结构替代实验解析结构时,GraphBind在AUROC,AUPRC和MCC三个指标上均出现了明显的性能下降。在DPI-Test129数据集上,GraphBind的AUROC和AUPRC已低于ALLSites,MCC则与ALLSites接近;在DPI-Test181数据集上,GraphBind在全部三项指标上均低于ALLSites。这些结果表明,结构方法对结构误差具有高度敏感性,即便采用当前最先进的结构预测工具,也难以完全弥补实验结构稀缺所带来的限制。所有基准评测的原始数据见表S2至S14。
在计算效率方面,ALLSites展现出极高的推理速度。在单张NVIDIA V100 GPU上,该方法可在16小时内完成对整个人类蛋白质组的筛选,涵盖来自UniProt数据库的20420条经人工审校的人类蛋白序列。平均而言,每条蛋白的预测耗时为2.81 s,每个残基仅需0.0075 s。综合来看,由于仅依赖蛋白序列且具有快速推理能力,ALLSites非常适合用于在蛋白质组尺度上绘制所有药物分子形态的结合位点分布图谱。
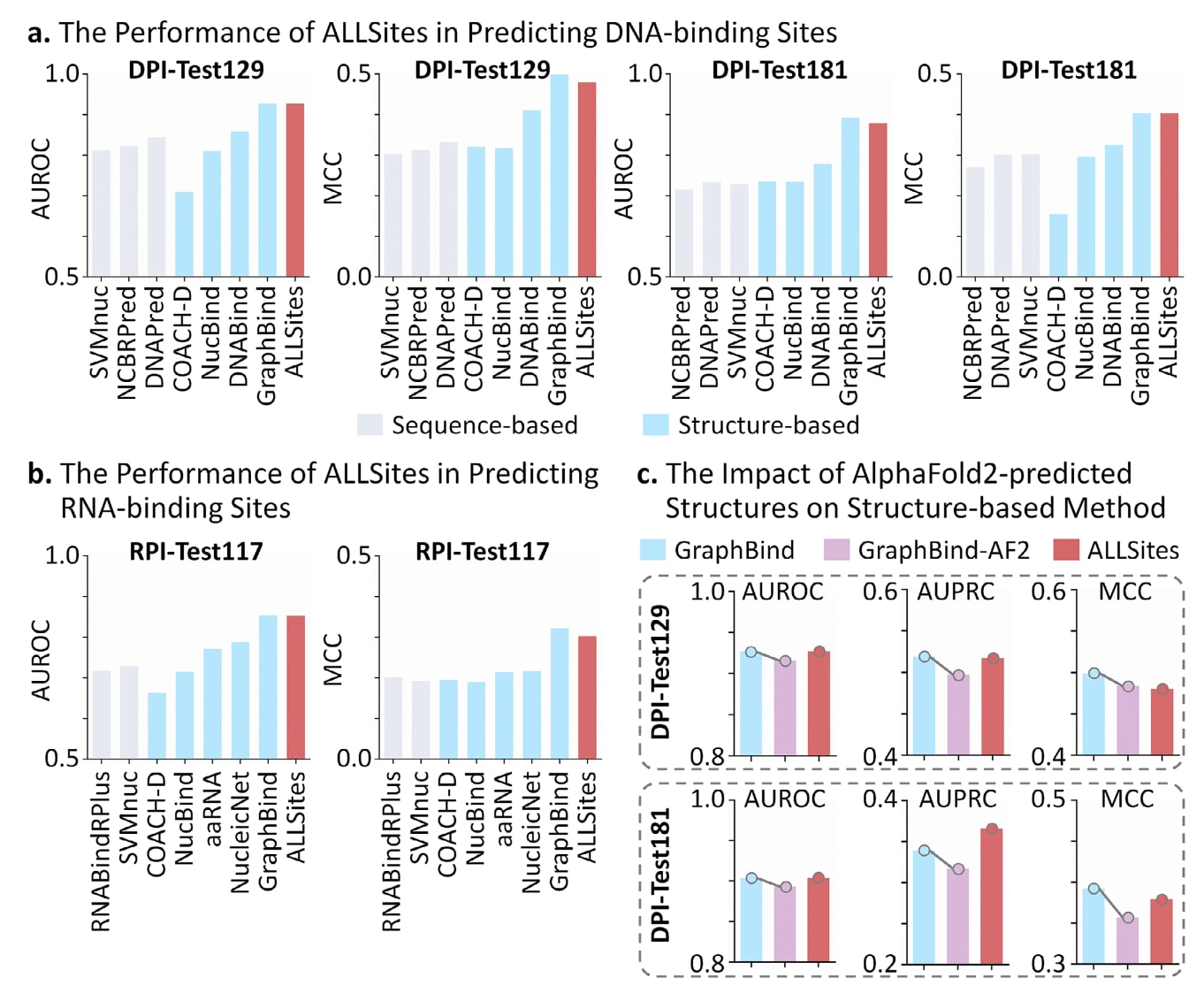
图4|ALLSites在核酸结合位点识别中的性能表现。 a)ALLSites在DNA结合位点预测中的性能,在DPI-Test129和DPI-Test181数据集上以AUROC和MCC作为评估指标,灰色柱表示基于序列的方法,蓝色柱表示基于结构的方法。b)ALLSites在RNA结合位点预测中的性能,在RPI-Test117数据集上以AUROC和MCC作为评估指标进行评估。c)使用AlphaFold2预测结构对结构方法性能的影响,在DPI-Test129和DPI-Test181数据集上评估预测结构对GraphBind性能的影响,蓝色柱表示使用实验解析结构的结果,紫色柱表示使用预测结构的结果。相关原始数据见表S8至表S14。