Angew. Chem. Int. Ed. 2025 | CrystalGAT: 一种基于人工智能的柔性晶体材料智能设计计算平台

获取详情及资源:
- 📄 论文: https://doi.org/10.1002/anie.202518543
- 💻 代码: https://github.com/ZhaoCY001/CrystalGAT-An-AI-based-intelligent-flexible-crystal-materials-design-computing-platform
- 🛜 网站: https://huggingface.co/spaces/ZZZCCCYYY/CrystalGAT
0 摘要
赋予功能晶体以柔性能够显著拓展其在柔性智能器件领域中的潜在应用。尽管在理解晶体柔性的基础结构原理方面已经投入了大量研究,但具有机械柔性的晶体发现至今仍多为偶然。近年来,机器学习已成为材料科学中的一种变革性研究范式。基于此,提出了一种创新方法并开发了一个用于预测分子晶体机械性能的平台CrystalGAT。CrystalGAT是一种基于注意力机制的图神经网络模型,通过数据增强策略构建了稳健的最优模型,其预测性能相较于已有模型表现出显著优势。在验证集上,该模型实现了90%的高预测准确率,同时展现出可扩展至多组分体系的良好泛化能力。更为重要的是,利用CrystalGAT识别了影响分子晶体机械性能的关键结构片段,成功实现了将脆性晶体转变为柔性光响应晶体的突破。此外,该平台还能够快速筛选具有塑性的多组分药物晶体,从而提升压片性能。CrystalGAT为柔性分子晶体的设计提供了一种高效方法,展示了其在材料发现与药物分子改性方面的应用潜力。为方便使用者,已建立专用网站:。
1 引言
在晶体自适应技术这一宏观框架下,分子晶体的动态行为通常由多种环境刺激所触发,包括机械作用、热处理、光照、湿度变化以及磁场作用等,从而产生丰富的宏观动态效应。这类动态晶体在执行器、偏振旋转器、固态激光器、有机电子器件、机械生物光子学、软体机器人、光波导以及药物制剂等多个领域中,其机械形变或运动被认为具有广阔的应用前景。然而,大多数有机晶体在外力作用下表现出明显的脆性,这一特性严重限制了其在柔性智能材料中的应用,并且与金属、高分子和凝胶等材料所固有的柔性形成了鲜明对比。自Reddy和Ghosh首次报道弹性晶体以来,柔性有机晶体受到了广泛关注。随着研究重心逐渐转向其力学性能,越来越多的研究尝试总结柔性晶体的共性结构特征,尽管此类材料的发现往往仍具有偶然性。例如,Desiraju等人以及Reddy等人提出了获得机械柔性分子晶体的设计策略,引入了由弱范德华作用或卤素-卤素相互作用形成的滑移面概念。然而,部分可发生机械形变的材料并不严格符合这些设计原则,使得柔性晶体的设计仍然带有较强的经验性。此类试错式方法不仅耗时耗力,而且单晶生长过程本身具有不可预测性和挑战性,进一步加剧了材料发现的难度。
近年来,机器学习技术被广泛应用于材料性能预测,其核心目标在于学习分子材料与特定性能之间的映射关系。已有研究通过逻辑回归模型预测溶剂化物形成概率,利用基于化学式的描述符预测振动自由能与熵,以及结合主成分分析实现包合物形成的多用途预测。这些工作主要依赖描述符提取分子的平均或关键特征,以实现性能判别预测。然而,这种特征提取过程往往依赖高精度的DFT计算,计算成本高且易引入模型内部偏差。同时,基于描述符的模型在预测路径上是单向的,只能从分子结构到描述符再到性能,无法反向推断能够产生特定性能的分子结构,因此难以建立材料性能与化学结构之间的可逆关系。基于此,亟需发展一种能够直接关联结构与性能的模型,以实现柔性分子晶体的高效筛选与预测。
在深度学习技术快速发展的推动下,图神经网络凭借其端到端学习分子化学结构图表示的能力,逐步取代了繁琐的人工特征工程,在捕捉原子成键信息和预测复杂化学性质方面展现出显著优势。例如,基于原子指纹的深度学习增量理论为可迁移且可解释的性能预测提供了新范式,而引入不确定性感知的图神经网络框架则为概率优化驱动的可靠分子设计奠定了基础。这些研究表明,材料设计模型不仅需要具备高预测精度,还应能够解释其决策依据并量化预测的不确定性。
在此背景下,基于图神经网络的上述优势,该研究提出了一种用于预测晶体材料力学性能的方法Crystal Graph Attention Networks,即CrystalGAT。通过系统整理文献数据,构建了一个同时包含单组分和多组分晶体的数据集,共涵盖368种具有弹性、塑性和脆性特征的分子。分子结构通过原子特征矩阵和邻接矩阵进行表示,其中原子特征矩阵描述分子中各原子的属性,邻接矩阵用于模拟原子与化学键之间的拓扑关系。在模型初步训练过程中发现,由于数据规模有限,模型容易出现过拟合现象。为此,引入了基于SMILES分子表示的多样性数据增强策略。同时,在模型中融合注意力机制,以捕捉原子局部邻域中的关键信息,并对不同原子环境向量进行加权聚合,从而更准确地反映不同原子对晶体材料性能的差异化贡献。
研究采用多种评价指标对模型性能进行了系统评估,并将CrystalGAT与多种基线模型在预测能力上进行了对比。模型验证通过两个具体案例展开,一是基于模型识别的关键原子对分子进行修饰,获得兼具多种功能的柔性分子;二是发现能够提升压片性能的多组分药物晶体。研究结果表明,该方法在预测精度和泛化能力方面均表现优异,充分展示了基于深度学习的方法在加速柔性晶体材料设计与发现过程中的巨大潜力。最后,还将建立一个便于用户使用的在线平台,以实现相关模型与功能的开放共享。
方法学
CrystalGAT用于预测有机分子力学性能及其分子修饰的整体流程如图2所示。研究从117篇文献中收集了单组分晶体225条数据和多组分晶体143条数据作为样本,这些数据来源于该领域数十年来大量研究工作的积累。为表征分子结构,首先将分子的SMILES字符串转换为图结构,在该过程中显式编码原子属性以及化学键特征。针对数据量有限且模型易发生过拟合的问题,进一步引入了基于SMILES的 数据增强策略,该方法能够生成在化学上合理的分子变体,从而有效扩充训练数据集并提升模型的泛化能力。收集到的数据中有10%被划分为验证集,其余数据在增强后按照80/20的比例随机划分为训练集和测试集,相关数据划分见Excel-S1。
在模型选择方面,以CrystalGAT作为主要研究模型,并将其预测性能与多种基线模型进行了系统比较,包括图卷积神经网络GCN、Transformer编码器模型、多层感知机MLP、支持向量机SVM、随机森林RF以及极端梯度提升模型XGBoost。对于CrystalGAT、GCN和Transformer编码器模型,原子特征矩阵与邻接矩阵被作为模型输入。而在MLP、SVM、RF和XGBoost模型中,则将弹性晶体分子MolEs、塑性晶体分子MolPs以及脆性晶体分子MolBs的SMILES表示转换为独热编码形式用于模型训练。用于训练各基线模型的数据集均选取自与CrystalGAT取得最佳训练效果时相对应的数据集,以保证不同模型之间比较的公平性与一致性。
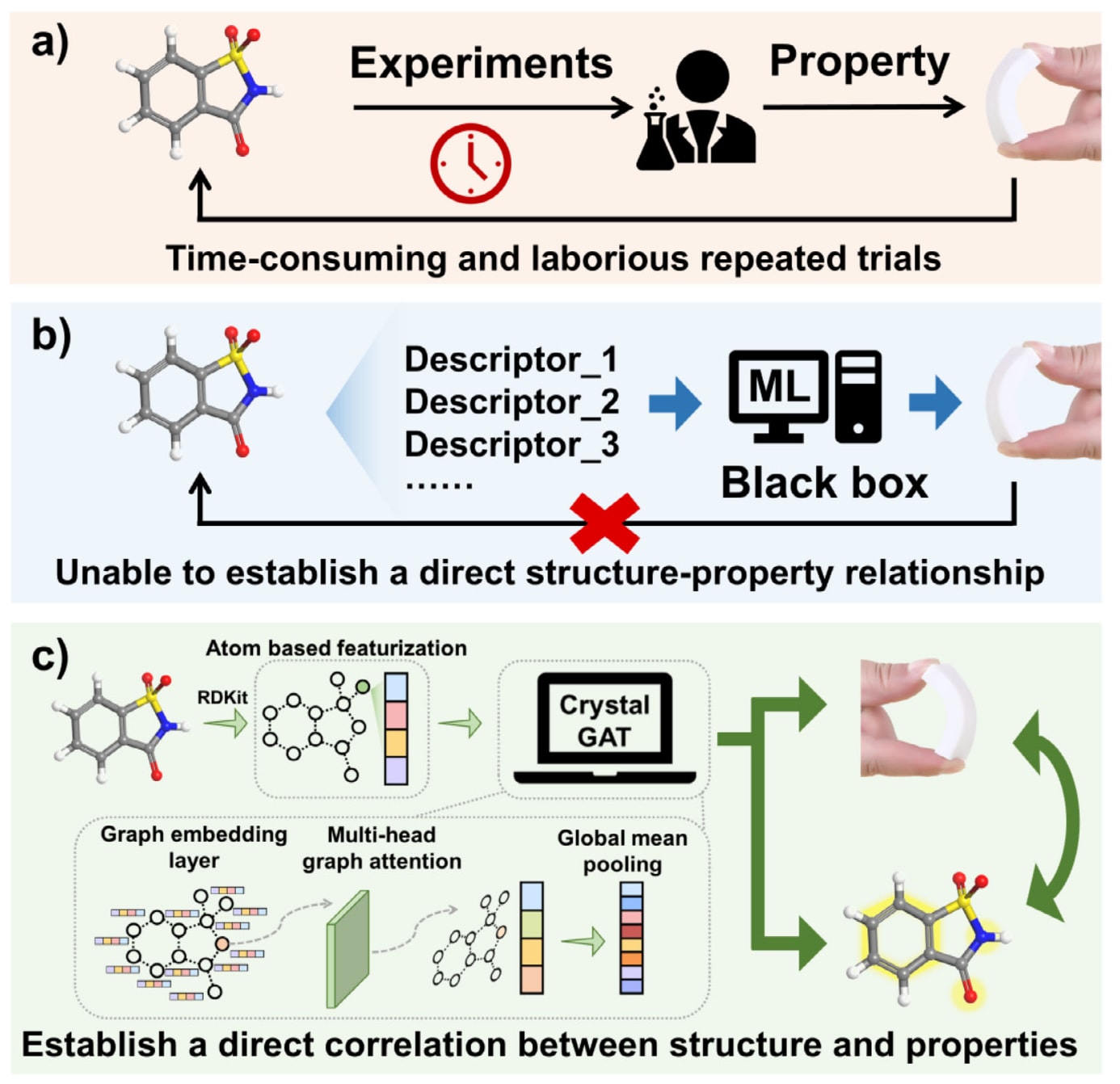
图1|不同柔性分子晶体制备方法的示意图。(b)中缩写“ML”表示机器学习。
2 结果与讨论
2.1 模型结果展示
首先,利用RDKit提取所收集分子的SMILES字符串并将其转换为分子图结构,相关示意见图S1。通过数据增强策略,由348条SMILES生成了超过6000条规范化SMILES数据集,用于模型训练。增强后的数据在12种原子特征的分布上与原始数据相比呈现出显著差异,其分布范围更加宽广,表明模型学习到了更加全面的信息,相关结果见图S3至S5。图3a展示了随着增强数据量增加,用于MolEs预测模型的多种评价指标,包括准确率、精确率、召回率、F1值以及MCC的变化趋势。当增强数据量达到500时,模型的准确率,包括测试集和验证集准确率,达到峰值,因此在该节点保留了鲁棒性最优的模型。对于MolPs和MolBs的训练过程,评价指标整体呈现先上升后趋于稳定的趋势,分别如图S6a和图S7a所示。在综合平衡测试集准确率与验证集准确率最大值后,最终选取使用5000条和6000条增强数据训练的模型,分别作为MolPs和MolBs的最优模型。
在模型选择过程中,主要依据验证集准确率来判断最优模型。当随着数据集规模增加验证集准确率趋于稳定时,再参考测试集准确率以确定最终模型。如图3b至3d、图S6、图S7以及表S10至S12所示,CrystalGAT在整体性能上显著优于其他基线模型。具体而言,其在验证集上的准确率稳定保持在80%至90%之间,体现了其对未见样本的优异判别能力。此外,在MolEs和MolPs的预测任务中,SVM在召回率方面与CrystalGAT表现相近,说明其能够较好地识别正类样本。然而,在数据分布不均衡的情况下,F1值相较于准确率更能全面反映模型的整体性能。结果表明,CrystalGAT在各类任务中均持续获得显著高于其他基线模型的F1值。
同时,研究以受试者工作特征曲线下面积即ROC-AUC作为核心评价指标,并辅以分类准确率进行综合评估。ROC-AUC通过刻画不同分类阈值下真阳性率与假阳性率之间的权衡关系,量化分类器的整体判别能力,其数值越接近1,表示模型区分正负样本的能力越强。与其他模型相比,GCN和Transformer模型在AUC水平上与CrystalGAT较为接近。其中,在MolPs和MolBs数据上训练的Transformer模型在ROC曲线形态上优于CrystalGAT,但其验证集准确率低于CrystalGAT,表明其测试集准确率和AUC值可能受到过拟合的影响而被高估。需要指出的是,GCN和Transformer模型与CrystalGAT采用相同的输入形式,均基于分子图结构。CrystalGAT通过引入注意力机制,能够更有效地捕捉分子中原子之间的长程依赖关系。总体而言,在该研究所涉及的特定任务和小规模数据条件下,CrystalGAT所具备的归纳偏置更加契合问题本身的结构特征,从而在实际预测性能上表现出明显优势。
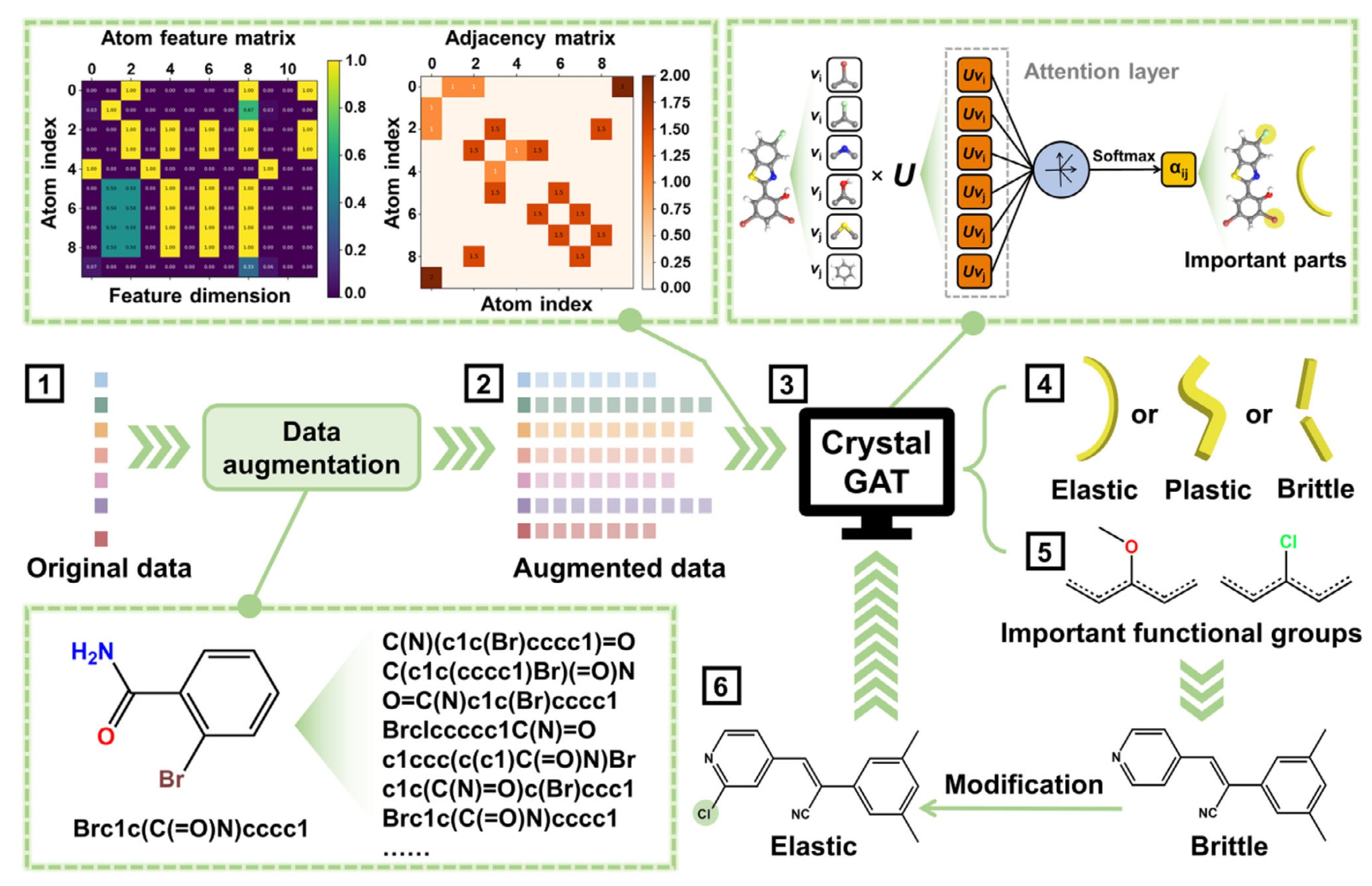
图2|CrystalGAT用于分子力学性能预测及特定性能分子修饰的整体流程示意图。
2.2 原子重要性分析的可视化
机器学习模型常被视为“黑箱”,其内部运行机制和决策过程对使用者而言并不直观。尽管这类模型能够给出准确的预测结果,但其透明性不足会给研究者基于模型决策结果进一步分析实验带来困难。为缓解这一问题,引入注意力机制对原子重要性进行分析。原子重要性得分用于量化各个原子对最终预测结果的贡献。图4a展示了MolPs验证集中的20个样本,其中对勾表示模型预测正确,叉号表示预测错误。每个分子中的颜色条表明,颜色越深的蓝色对应的原子在模型决策中越重要。MolEs和MolBs在验证集中的对应结果分别见图S7和图S8。
以MolPs验证集中的分子1,3,5-三氟-2,4,6-三碘苯(CFI)为例进行说明,如图4b所示。其原子重要性得分表明,苯环对塑性预测的影响最大,其次是碘原子,而氟原子的贡献相对较小。重要性数值为0到1之间的归一化相对值。图4c所示的能量框架表明,强相互作用主要集中在苯环的π-π堆积上,而图4d显示的氢键则主要存在于I···I之间。由π-π堆积形成的层内相互作用明显强于由I···I作用形成的层间相互作用,使得分子在外力作用下能够沿滑移面发生滑动。这一结构特征决定了CFI晶体表现出宏观塑性行为,与模型的分类结果以及原子重要性分析高度一致。
上述结果表明,该模型能够有效捕捉分子结构特征如何影响晶体力学性能的关键信息,为理解预测结果提供了可靠依据。
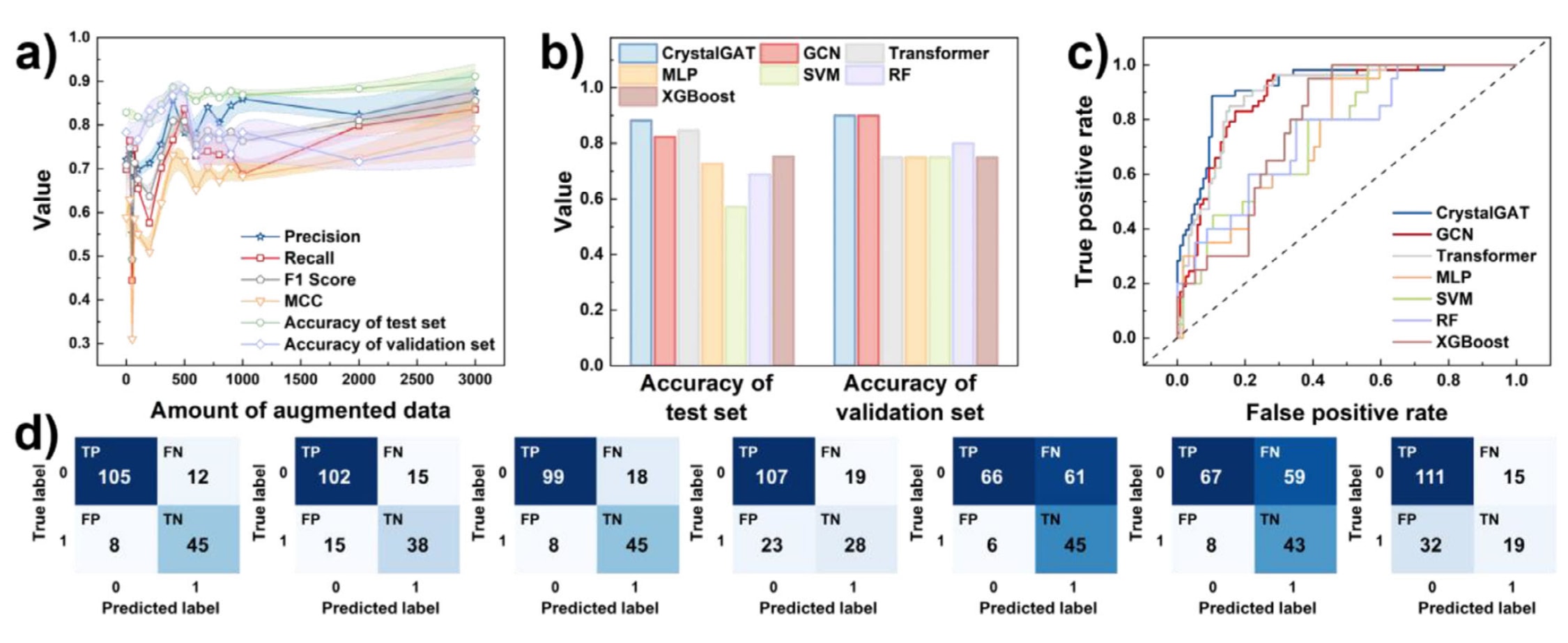
图3| a) 在引入增强数据条件下,CrystalGAT对MolEs预测的准确率、精确率、召回率、F1值和MCC。 b) 不同模型在MolEs任务中测试集与验证集的准确率对比。 c) 不同模型在MolEs任务中的ROC曲线。 d) CrystalGAT、GCN、Transformer、MLP、SVM、RF和XGBoost模型在MolEs测试集上的混淆矩阵。
2.3 案例研究与验证(案例1)
动态分子晶体在受到机械力、热和光等外界刺激时,能够将分子尺度的运动显著放大为宏观机械运动。 在众多动态分子体系中,氰基苯乙烯衍生物由于合成简便、结构可调以及功能多样,在智能材料的开发与应用中展现出巨大潜力。 尤其是弹性分子晶体,其同时具备晶态特征和可逆弯曲性能,在机械执行器、光学器件、有机电子学等领域具有重要应用前景。 基于此,研究计划对实验室已合成但表现出宏观脆性断裂行为的PAPA分子进行修饰,以期赋予其力学柔性和光响应特性。
首先进行原子重要性计算。 每个原子的综合重要性由基于注意力的贡献和基于特征的贡献加权得到,其中注意力贡献占60%,特征贡献占40%。 注意力贡献通过对第一层GAT中与该原子相连的所有边的归一化注意力权重求和获得,特征贡献则依据预设权重对原子特征进行加权求和。 最终得到的综合重要性被归一化到0到1之间。 随后提取关键原子环境,对每个分子中重要性最高的前三个原子,提取其在2键半径范围内的局部子结构即功能基团。 在此基础上进行基团统计与聚类分析,将所有提取的功能基团汇总,并按中心原子的原子重要性对其出现频率进行加权。 为筛选具有代表性的功能基团,计算半径为2的Morgan指纹,并采用基于密度的空间聚类算法DBSCAN进行聚类,其中邻域半径设为0.6,最小簇大小为1。 每个簇中出现频率最高的功能基团被选作代表基团。 最终筛选得到的关键功能基团通过RDKit进行可视化,为分子弹性设计提供直观指导。 图S10展示了对弹性结果影响显著的前30种功能基团。
为实现分子弹性的定向优化,构建了一种基于功能基团替换的分子修饰引擎。 其工作原理如下:首先,在分子中的环状结构包括芳香环和非芳香环以及非环状链结构上,识别所有氢原子以及可替换基团如甲基、乙基等且原子数不超过5的基团,以避免破坏分子核心骨架,将其作为潜在修饰位点。 通过去除一个氢原子或可替换基团并连接新的功能基团对这些位点进行修饰。 其次,允许在同一分子上同时进行1到2个位点的修饰。 修饰位点类型包括环状和链状结构以及功能基团组合通过笛卡尔积方式生成所有可能组合。 具体而言,对于修饰深度k(k=1或2),枚举所有k位点组合,并从对应的功能基团库中为每个位点独立选择功能基团。 最后,基于标准SMILES去除重复结构,并进行相似性筛选,即保留与原始分子基于半径为2的Morgan指纹计算得到的Tanimoto相似度大于等于0.4的分子。 利用预训练的CrystalGAT模型预测分子的弹性概率,并保留预测概率大于等于0.5的分子,如图5a所示。 该策略实现了对分子化学空间的定向探索,在结构多样性与功能优化之间取得平衡,为弹性晶体设计提供了一种高效工具。
综合实验工作量与计算成本的考虑,最终选取排名前五的功能基团用于分子修饰,如图5b所示。 对PAPA分子进行一次修饰共得到49个候选分子(图S11a),并根据合成可行性筛选出5个分子用于实验验证。 随后对PAOA分子进行二次修饰得到14个候选分子(图S11b),同样选择其中1个分子进行后续合成与测试。 最终获得的CAPA、BPPA、PAFA、PAOA、PABA和DCOA均成功合成,并得到了适用于结构分析和性能测试的高质量单晶(图6,表S13)。 上述所有分子均通过一步Knoevenagel缩合反应制备,并通过1H NMR进行表征,相关数据见图S9和图S10。
在365 nm紫外光照射下,所有晶体均表现出明亮荧光,其发射峰位于350至550 nm之间(图7a),对应的荧光颜色在CIE色坐标图中给出(图7b)。 它们的UV-vis吸收光谱均呈现相似的宽吸收带,表明具有较强的可见光捕获能力(图7c)。 所研究的七种化合物在紫外光激发下均表现出显著荧光,其发射波长与HOMO-LUMO能隙相关。 其中,DCOA具有较小的能隙3.56 eV,因此发射波长更长的荧光(图7f)。 在紫外光照射下,DCOA、BPPA和CAPA表现出明显的光响应行为,具体表现为DCOA的光致开裂以及BPPA和CAPA的光致弯曲(图S14)。
以CAPA为例,其“烯烃对”的几何参数满足Schmidt判据,如表S14和图S15所示。 具体而言,潜在反应双键中心之间的距离d为3.842 Å,一条双键相对于另一条的旋转角θ1为0°,由双键中四个碳原子形成的平行四边形的钝角θ2为104.05°,该平行四边形与同一双键碳原子上所连两个取代基构成平面的二面角θ3为78.81°,如图7d所示。 这些参数表明CAPA可能发生光诱导的[2+2]环加成反应,DCOA和BPPA的情况亦类似。 同时,当在(0 0 1)晶面上施加外力时,CAPA和BPPA均表现出显著的柔性,在不发生明显断裂的情况下分别可承受高达3.3%和3.1%的弯曲应变(图S13和图S14)。
为进一步探究其弹性特征,对CAPA晶体的分子堆积方式进行了分析(图7e)。 沿a轴方向观察,CAPA的堆积模式通过两类氢键相互连接,分别为中等强度的C-H···N作用(D、d、θ分别为3.663 Å、2.774 Å、151.14°)和C-H···Cl作用(3.853 Å、2.950 Å、159.11°)。 分子还通过C-H···N(3.477 Å、2.577 Å、158.14°)、C-H···N(3.664 Å、2.795 Å、152.60°)以及C-H···Cl(3.551 Å、3.044 Å、114.94°)等相互作用,在垂直于(1 0 0)晶面的方向形成氢键网络,并进一步通过π-π相互作用沿a轴方向堆积,其相互作用能为−46.3 kJ mol−1(图S18)。 这种分子堆积方式能够在弹性有机晶体弯曲过程中同时容纳内弧的压缩和外弧的伸展,BPPA同样遵循这一机制。 因此,可以提出如图7e所示的CAPA晶体弹性弯曲模型。
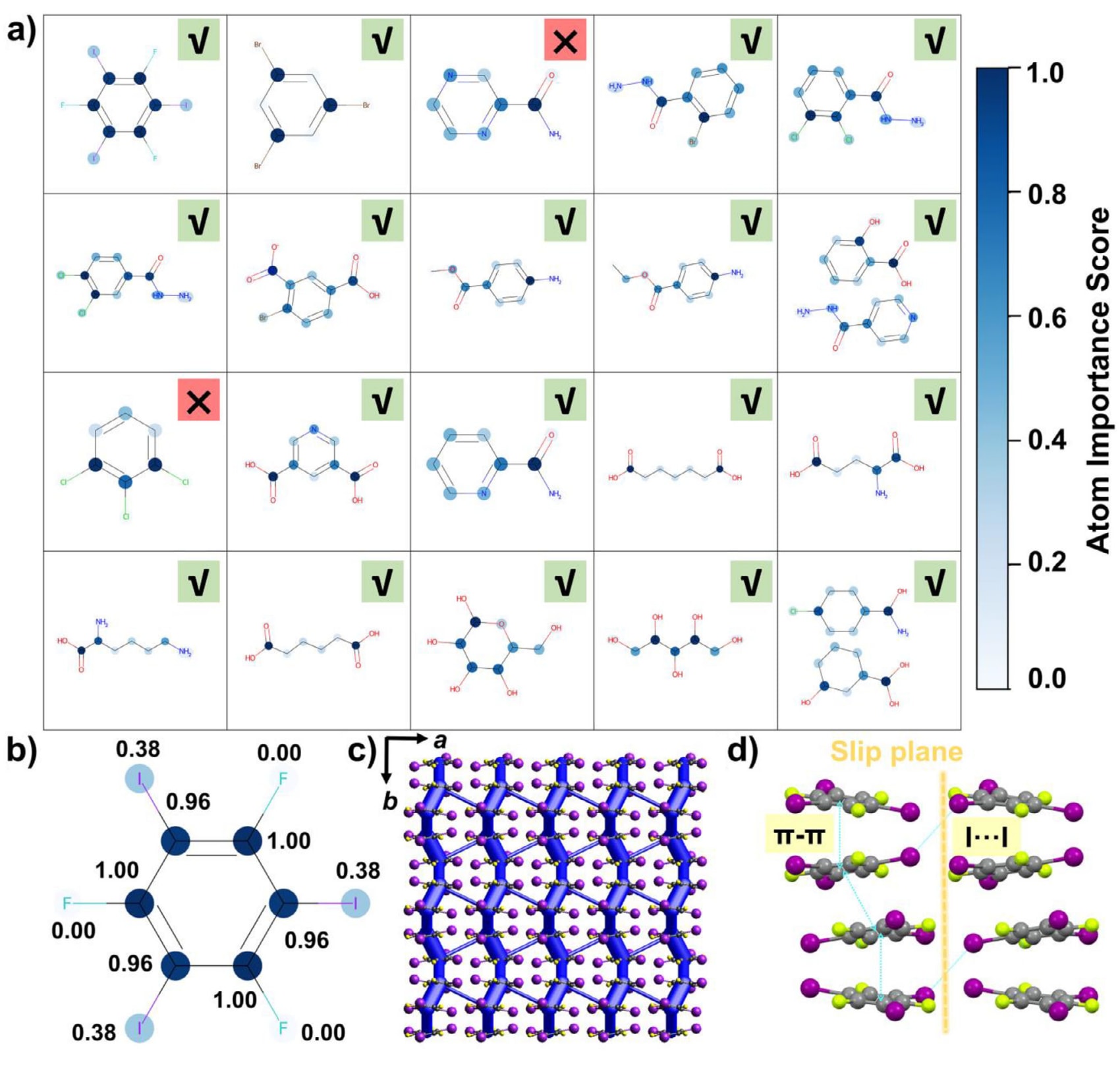
图4|a) MolPs验证集中分子的模型预测结果及原子重要性分析,✓表示预测结果与实际结果一致,×表示预测结果与实际结果不一致。 b) CFI的原子重要性评分。 c) CFI的能量框架。 d) CFI晶体中的分子间相互作用模式。
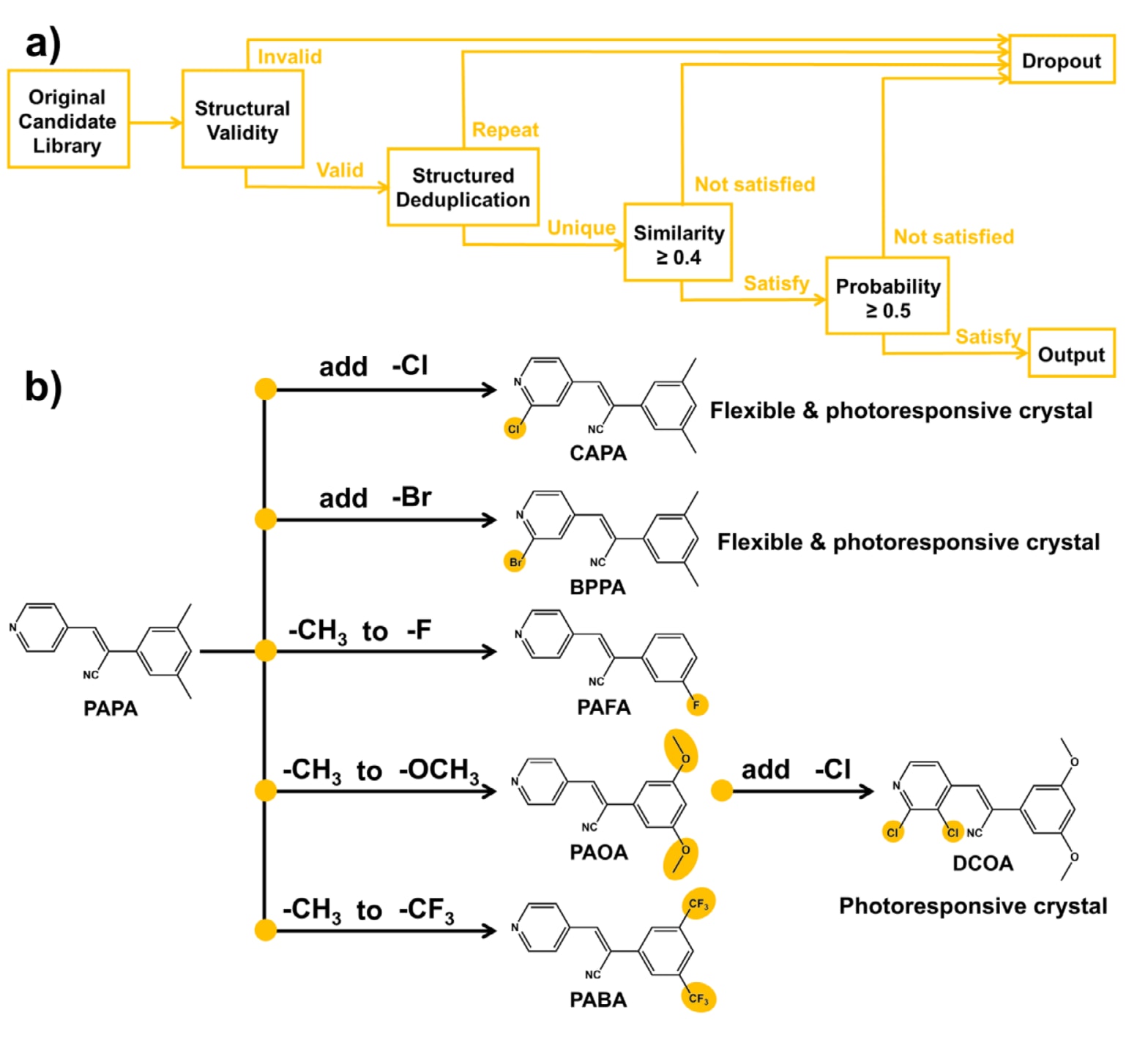
图5|a) 分子修饰流程图。 b) 基于CrystalGAT所提供的重要功能基团进行分子修饰的示意图。
2.4 案例研究与验证(案例2)
在多种药物剂型中,片剂因其优良的理化稳定性和较低的生产成本而成为最常见的商业化剂型。已有研究表明,片剂的可压缩性与固体材料的力学性能密切相关,是决定片剂产品可制造性的关键因素之一。加巴喷丁(GBP)是一种用于治疗癫痫发作和周围神经病变的药物,但其片剂拉伸强度较低。该研究旨在通过预测模型筛选能够有效改善GBP压片性能的共晶体系。
首先,利用CrystalGAT对GBP的八种共晶的力学性能进行了预测,结果如图8所示。预测结果表明,除GBP-RS-MA(RS-MA表示外消旋扁桃酸)外,其余共晶均表现出塑性。基于此,进一步制备了这八种共晶并进行了三点弯曲实验。其中,GBP-S-MA(S-MA表示S-扁桃酸)、GBP-R-MA(R-MA表示R-扁桃酸)以及GBP-CA(CA表示樟脑酸)的晶体结构为该研究首次解析。实验结果显示,GBP-3HA(3HA表示3-羟基苯甲酸)、GBP-4HA(4HA表示4-羟基苯甲酸)、GBP-SA(SA表示水杨酸)以及GBP-1H2NA(1H2NA表示1-羟基-2-萘甲酸)为脆性晶体,与模型预测的脆性结果一致。尽管模型对这四种体系预测出两种力学性质(塑性和脆性),但这并不直接表明CrystalGAT的判断存在错误,也可能源于这些分子中存在其他晶型。
GBP-S-MA和GBP-R-MA晶体表现出明显的塑性弯曲行为,其实验结果与模型预测高度一致。此外,在以扁桃酸三种形式作为共晶形成剂与GBP形成共晶时,模型预测结果与实验结果之间存在一定偏差。这是由于CrystalGAT当前采用的原子层面特征中缺乏足够的“手性标签”,不足以全面表征外消旋体与对映体之间复杂的三维空间构型差异,从而导致性能预测出现误差。在上述八种共晶体系中,仅GBP-RS-MA和GBP-CA在模型预测与实验结果之间存在不一致。这一结果充分表明,尽管训练集中多组分晶体样本数量有限,该模型在多组分晶体预测方面仍具有较强的泛化能力。未来,通过在大规模数据集上进行预训练,该类模型有望发展为适用于分子晶体的通用基础模型,并通过迁移学习进一步降低对领域特定数据的依赖,实现更广泛且更精确的性能预测。
通过PXRD表征可以确认GBP及其多组分晶体均已成功制备且具有良好的相纯度(图S19)。以GBP-RS-MA为例分析其塑性行为与片剂压缩性能之间的关系。GBP-RS-MA结晶于P1̄空间群,Z=2(表S15)。GBP与RS-MA分子沿b轴方向交替排列,并通过N-H···O范德华相互作用(2.812 Å、1.921 Å、168.49°)相连接。GBP-RS-MA的塑性弯曲源于其晶体中存在明显的滑移面。滑移面两侧通过较强的氢键O─H···O(2.574 Å、1.797 Å、163.43°)、N···O(3.062 Å)以及O···O(3.009 Å)相互连接。从能量框架图可以看出,层内相互作用显著强于层间相互作用(图9a)。由于滑移面内分子之间的连接较弱,仅需较小的外力即可使一个分子层相对于另一分子层发生滑动。GBP-S-MA和GBP-R-MA的塑性结构亦可采用类似方式进行分析(图S18和图S19)。
共晶的形成能够调控固体药物的压实性能,从而影响其可制造性。该研究考察了GBP及其多组分晶体在100至400 MPa压缩压力范围内的压片性能(图S22)。为消除颗粒尺寸对压片性能的影响,实验前对粉末进行了粉碎处理,以确保颗粒尺寸分布一致(图S23)。GBP-4HA粉末在最高100 MPa的压缩压力下仍无法形成完整片剂,并出现分层和顶裂现象(图S24)。其余样品在整个压缩压力范围内均无需添加任何辅料即可制备成完整片剂,且在24 h松弛后仍保持完整,未观察到分层或顶裂现象。各粉末样品的可压片性能曲线如图9b所示。在相同压缩压力下,具有塑性特征的GBP-RS-MA、GBP-S-MA和GBP-R-MA的片剂拉伸强度显著高于GBP。其中,基于CrystalGAT筛选得到的两种塑性共晶GBP-S-MA和GBP-R-MA的片剂拉伸强度分别较GBP提高了852%和572%。此外,GBP-3HA、GBP-SA和GBP-1H2NA尽管为脆性晶体,其片剂性能同样优于GBP,表现出一定的改善效果,而GBP-CA和GBP-4HA的片剂拉伸强度则与GBP相近。
塑性晶体优异的压片性能进一步通过Heckel曲线得以验证。如图9c所示,具有塑性的三种共晶具有更大的斜率,均高于GBP(k=0.00489)(图S25),这表明其平均屈服压力更低、塑性变形能力更强,意味着在外加压力下片剂内部颗粒能够更加紧密结合,从而显著提升片剂的拉伸强度。
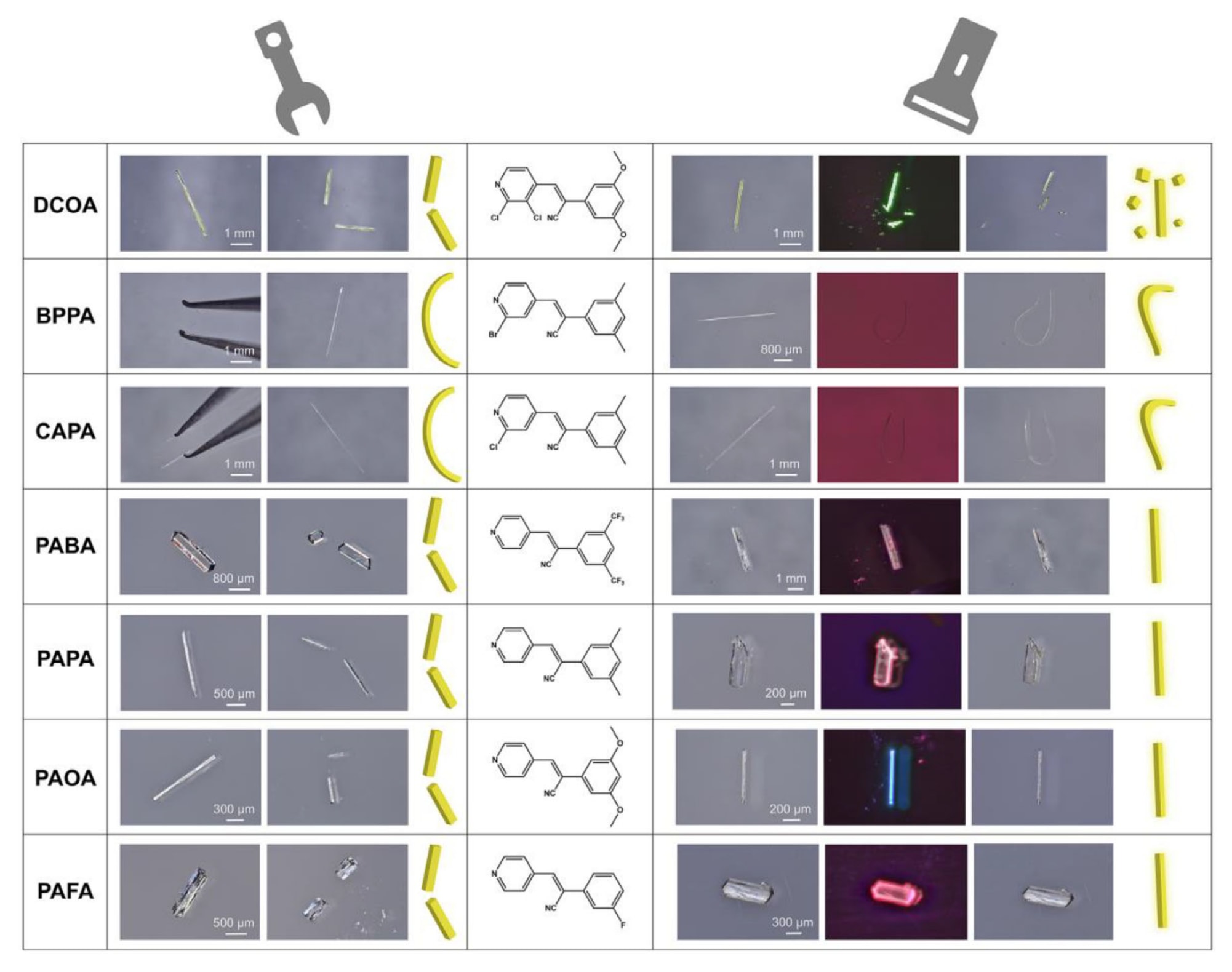
图6|PAPA分子及其修饰分子的结构示意图,左列对应受力诱导的力学行为,右列对应晶体的光响应行为。
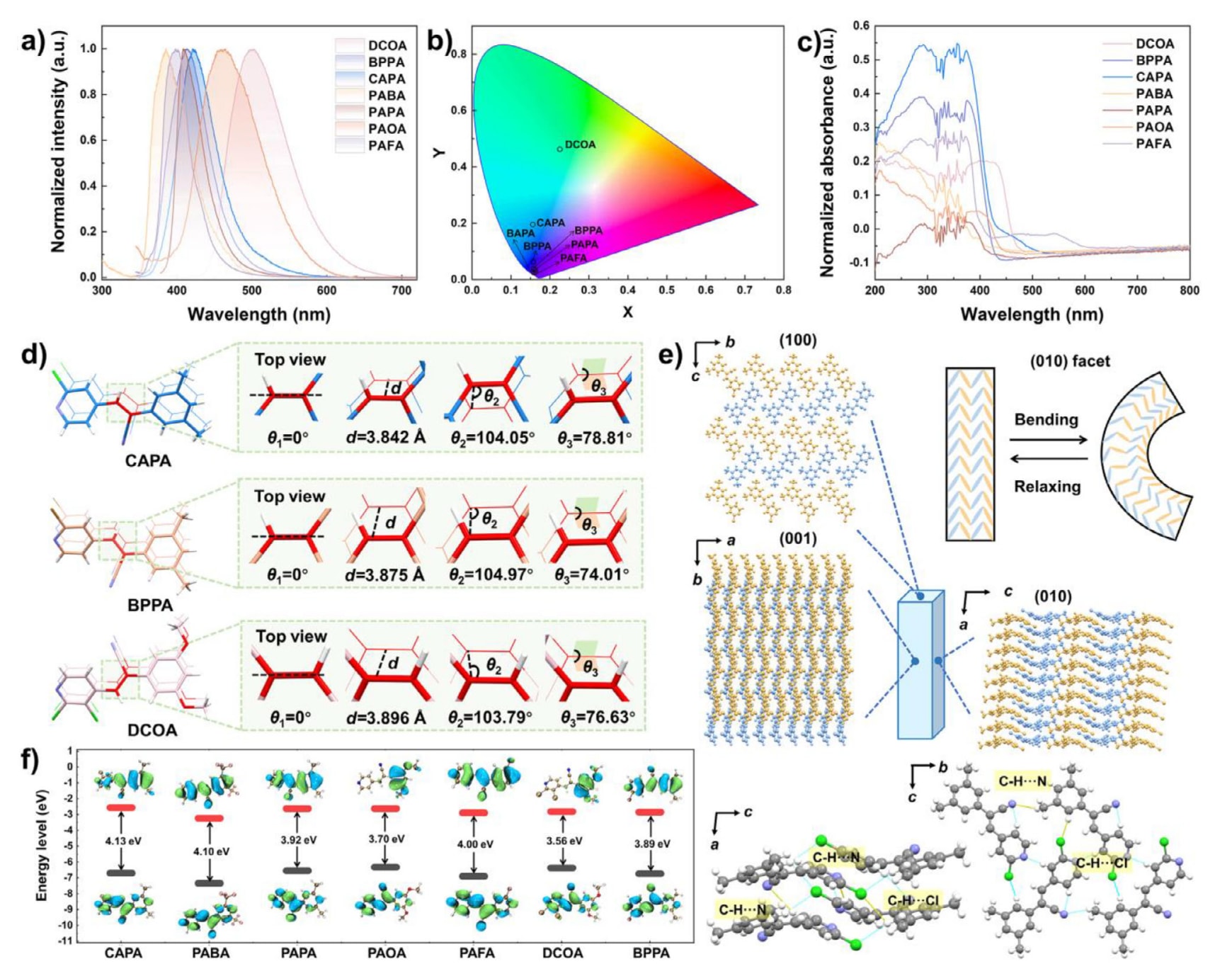
图7|a) 所有晶体在λex=365 nm条件下的荧光发射光谱。 b) CIE色坐标图。 c) 所有晶体的UV-vis吸收光谱。 d) CAPA、BPPA和DCOA晶体中的分子内π-二聚体结构。 e) 沿不同晶体取向观察的CAPA晶体分子间相互作用模式。 f) 所有晶体的HOMO和LUMO分子轨道示意图及其等密度表面分布。
2.5 Web服务器的用户界面
网站设计的主要目标在于实现分子SMILES字符串及其结构信息的快速查询、转换、性能预测与可视化。因此,整体框架在设计时兼顾了高并发请求下的稳定负载能力,并注重界面的友好性,以便不具备编程背景的研究人员也能够便捷使用。基于上述考虑,在完成模型及相关数据的部署后,设计了CrystalGAT的用户界面。该界面主要由“SMILES Input”和“Manual Molecules Construction”两个核心模块组成,图10通过界面截图展示了整体布局及部分功能细节。
其中,“SMILES Search”模块作为分子力学性能预测的主要入口。在计算页面中,用户可通过编辑器输入分子的SMILES字符串,随后点击“Predict”按钮提交任务。任务完成后,结果页面将显示分子在弹性、塑性和脆性方面的预测结果,以1或0的形式给出,同时在分子结构中以蓝色高亮显示各原子的相对重要性,直观反映其对预测结果的贡献。
对于尚未明确SMILES表示的分子,“Manual Molecules Construction”模块提供了一种直观的分子结构编辑工具。用户可在“Atom Type”下拉菜单中选择所需原子类型,点击“Add Atom”后,所选原子将按编号顺序列于下方表格中。通过“Add Bond”按钮,可在选定原子之间添加单键、双键或三键连接。在完成所有原子与化学键的构建后,点击“Generate SMILES”即可在画布中显示对应的分子结构,并自动生成相应的SMILES字符串。基于手动构建的分子,同样可以进一步进行力学性能预测以及原子重要性可视化分析。
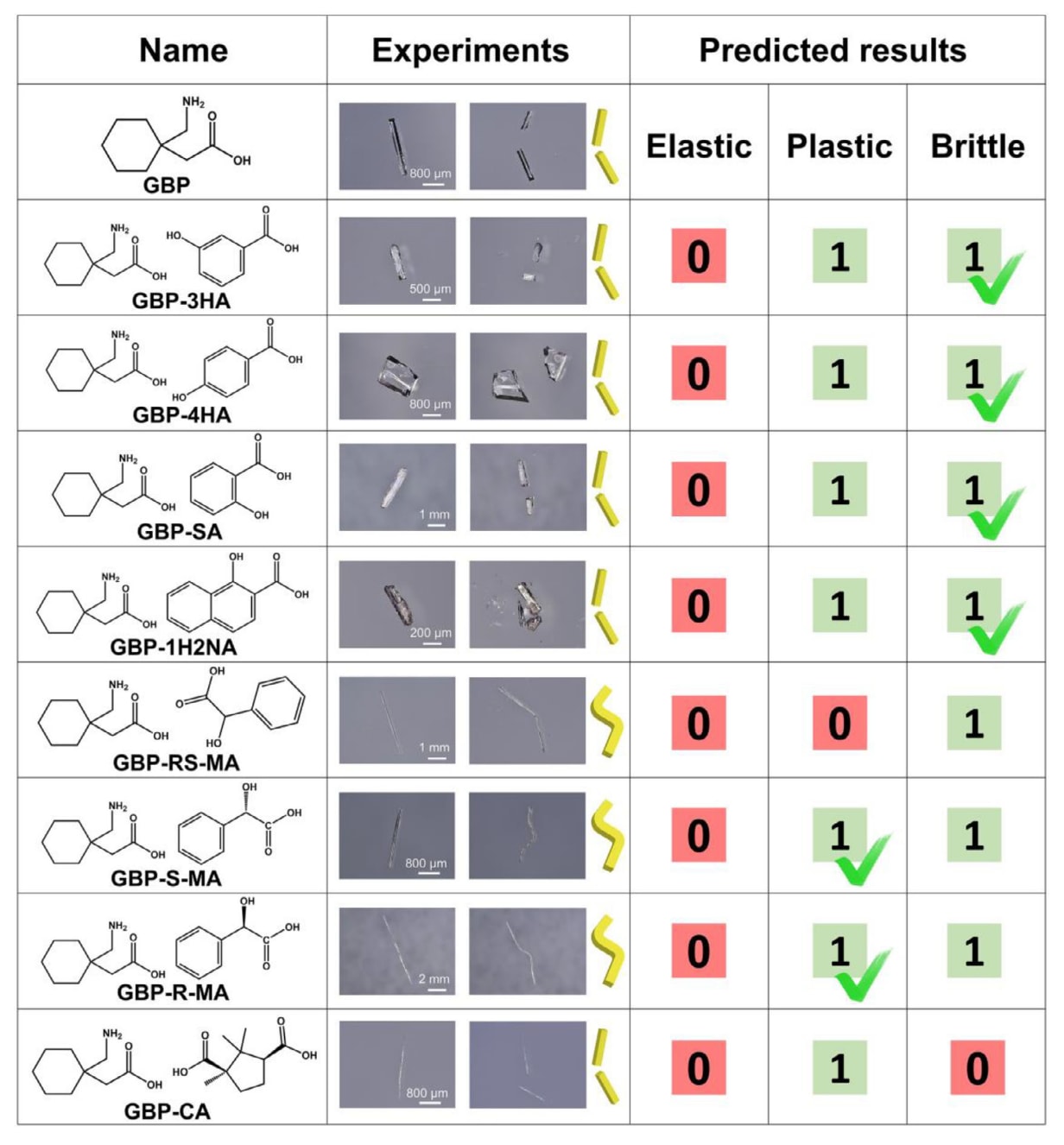
图8|GBP多组分晶体的实验结果与模型预测结果对比。
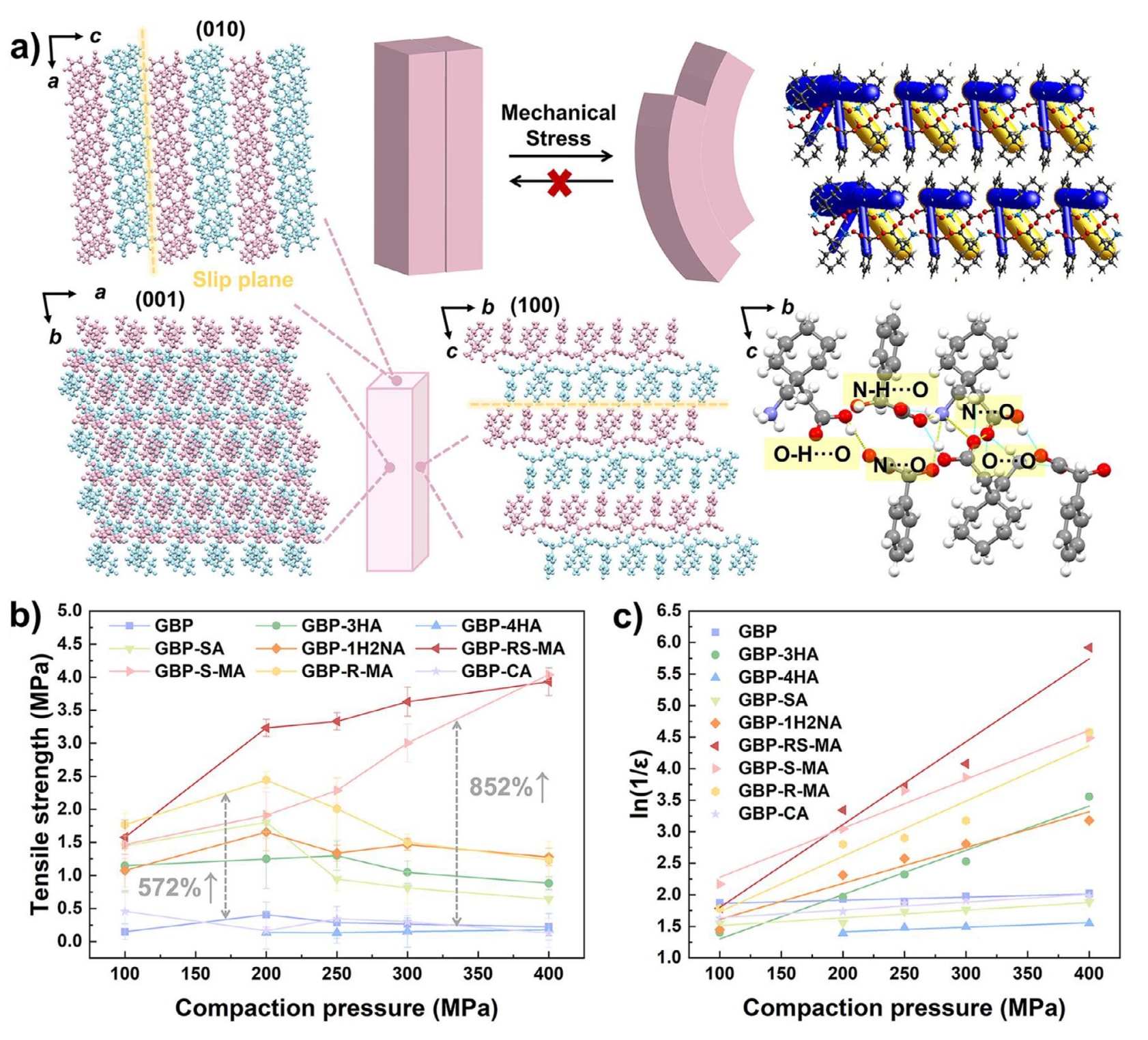
图9|a) 沿不同晶体取向观察的GBP-RS-MA晶体分子间相互作用模式。 b) GBP及其多组分晶体的可压片性能曲线。 c) GBP及其多组分晶体的Heckel分析结果。
3 结论
综上,从已发表文献中系统整理并构建了一个涵盖225种单组分晶体和143种多组分晶体力学性能的综合数据集。首先,基于SMILES字符串的序列特性引入数据增强策略,以降低模型过拟合风险。随后,训练并评估了多种模型在晶体力学性能预测任务中的表现,结果表明CrystalGAT在该任务中展现出稳健而优越的性能。具体而言,CrystalGAT在弹性模型和塑性模型上的验证集准确率均可达到90%,塑性模型的AUC最高可达0.98,而基线模型的准确率约为70%。这些结果表明,通过模型学习直接捕捉原子类型与化学键信息,能够有效表征结构-性能关系并实现高精度的力学性能预测,凸显了基于注意力机制的方法在计算化学研究中的潜力。
在此基础上,对不同力学性能相关的关键结构片段进行了可视化分析,并成功将其应用于实例研究,实现了将具有脆性晶体性质的分子修饰为兼具弹性和光响应行为的分子。此外,CrystalGAT还成功筛选出三种能够显著改善加巴喷丁(GBP)压片性能的塑性共晶,其中两种共晶GBP-S-MA和GBP-R-MA为该研究首次获得的新结构,模型预测结果与实验结果一致,其片剂拉伸强度分别较GBP提高了852%和572%。在多组分晶体样本仅占原始数据约三分之一的情况下,CrystalGAT仍准确预测了8个体系中的6个,充分展示了该模型在分类任务中的良好泛化能力。
进一步地,还为CrystalGAT设计并实现了一个用户友好的平台界面,使用户能够通过可视化报告快速、便捷地完成性能预测并获取分子中关键原子的贡献信息。该研究通过引入机器学习方法有效解决了晶体材料领域中的性能预测难题,为材料设计与发现提供了有价值的指导。机器学习与材料科学的深度融合有望在未来进一步推动材料科学的发展。
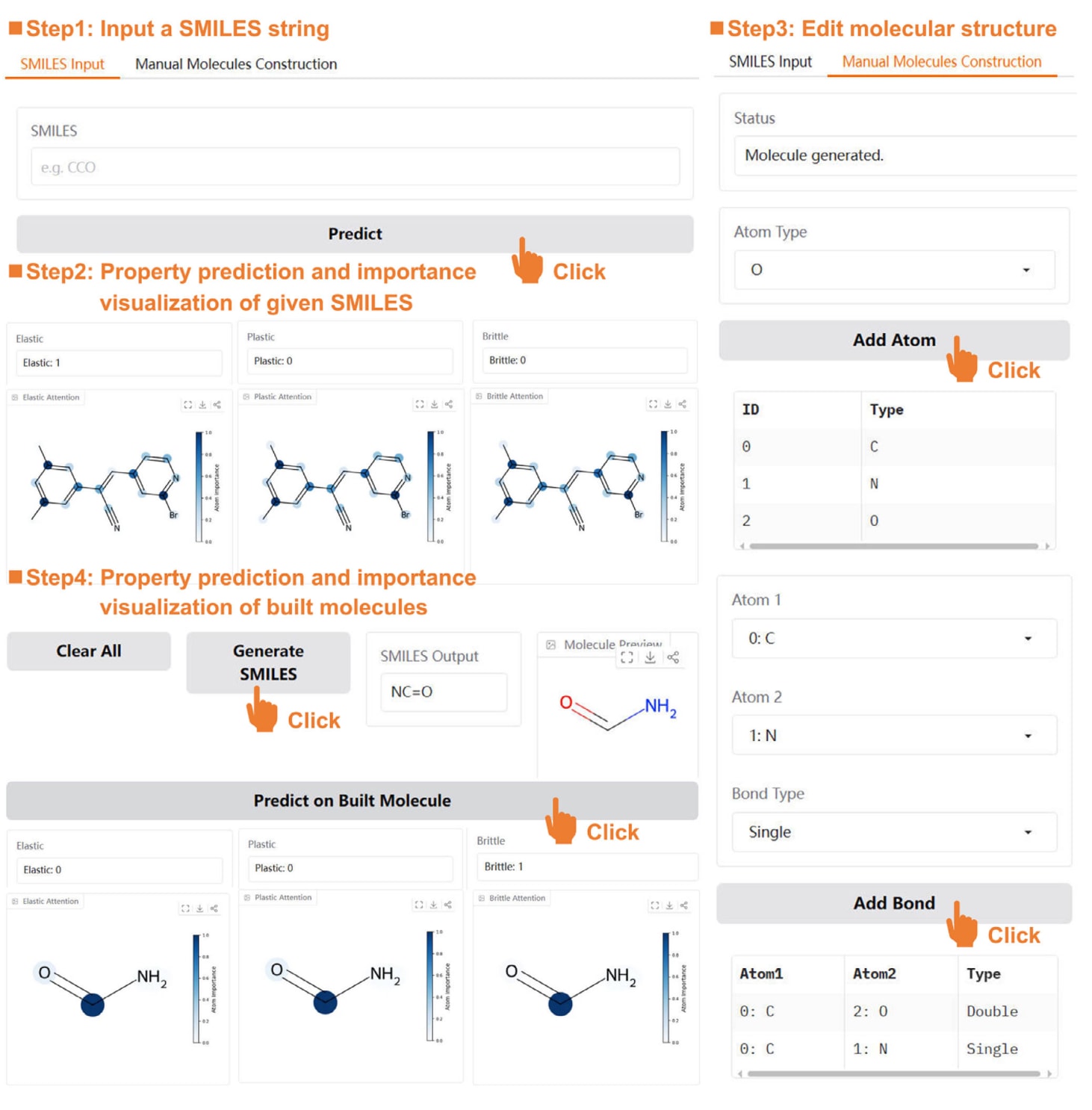
图10|CrystalGAT整体界面及部分具体功能的界面截图。