NCS 2026 | MAPLE: 一种基于成对学习的稳健计算框架,用于甲基化年龄与疾病风险预测
今天介绍的是发表在 Nature Computational Science(NCS) 上的一项重要工作——MAPLE:一种基于成对学习的稳健计算框架,用于甲基化年龄与疾病风险预测。该研究针对传统表观遗传时钟在跨平台、跨组织和跨研究应用中泛化能力不足的核心瓶颈,提出了以“相对关系”而非“绝对预测”为建模目标的全新思路。作者通过成对学习与孪生网络结构,有效削弱了 DNA 甲基化数据中普遍存在的批次效应,在大规模、多来源数据上实现了对表观遗传年龄的高精度预测,并进一步拓展至心血管疾病和 2 型糖尿病等衰老相关疾病风险评估。MAPLE 不仅在方法论上为表观遗传时钟提供了新的范式,也为甲基化数据在真实临床场景中的应用迈出了关键一步。

获取详情及资源:
0 摘要
传统表观遗传时钟在泛化能力方面面临诸多挑战,尤其是在训练数据集与测试数据集之间存在显著批次效应时,其在衰老评估中的临床应用受到明显限制。该研究提出了一种名为MAPLE的稳健计算框架,通过成对学习实现甲基化年龄与疾病风险预测。MAPLE利用成对学习策略,刻画两份DNA甲基化谱在年龄或疾病风险层面的相对关系,从而有效提取与衰老或疾病相关的生物学信号,并同时减弱数据中的技术偏差影响。在来自不同研究、测序平台、数据预处理方法及组织类型的31项基准测试中,MAPLE在整体性能上优于五种对比方法,其中位绝对误差仅为1.6年。此外,在衰老相关疾病风险评估中,MAPLE同样表现出色,其疾病识别的平均曲线下面积达到0.97,对疾病前状态的检测平均曲线下面积为0.85。总体而言,该研究表明MAPLE在临床层面评估表观遗传年龄及衰老相关疾病风险方面具有广阔的应用潜力。
1 引言
衰老以发病率不断升高及生活质量同步下降为主要特征,二者共同带来了沉重的社会与经济负担。近几十年的突破性研究表明,通过热量限制和部分表观遗传重编程等干预手段,在一定程度上延长寿命和健康寿命是可行的。然而,衰老干预在临床中的应用前提是对生物学年龄及衰老速率进行精确测量。DNA甲基化是指甲基共价修饰发生在胞嘧啶第五位碳原子上的过程,在哺乳动物中通常出现在胞嘧啶-鸟嘌呤二核苷酸位点。DNA甲基化改变被认为是衰老的重要标志之一。例如,在某些CpG岛中,尤其是在Polycomb靶基因以及肿瘤抑制基因启动子区域,DNA甲基化水平往往随年龄增长而升高。这些累积性的DNA甲基化变化可能逐步建立起与年龄相关的转录调控程序。因此,全基因组DNA甲基化特征可作为生物学年龄的估计指标。
然而,现有表观遗传时钟的性能容易受到多种因素影响,包括测序平台、数据预处理方法、组织类型以及人群差异,这在很大程度上限制了其临床应用。以Horvath时钟和HannumAge为代表的模型主要依赖LASSO等线性方法,在刻画复杂的表观遗传衰老模式方面存在一定局限。另一类研究如AltumAge虽然引入了深度学习模型,但在建模过程中并未充分处理不同数据集之间的批次效应问题。因此,这些模型可能在无意中学习到了与衰老无关的技术性偏差,从而削弱了其在复杂临床场景中对未知数据的泛化能力。此外,用于训练DNA甲基化模型的数据往往具有特征数量远大于样本数量的特点,显著增加了过拟合风险并损害模型的泛化性能,这在机器学习中被称为高维小样本问题。由此可见,亟需一种具有良好泛化能力的DNA甲基化时钟,能够在保留与衰老相关生物学信号的同时有效消除技术偏差,从而实现对人类衰老过程的精准测量。
尽管表观遗传时钟为评估表观遗传年龄提供了重要工具,但仅依赖表观遗传年龄本身仍不足以指导针对性的抗衰老干预。2型糖尿病和心血管疾病在全球范围内高度流行,而衰老被认为是这两类疾病最主要的风险因素。对这些疾病进行早期诊断和干预,有助于延缓其发生,提升高风险人群的生活质量,并可能降低相关死亡率。因此,利用DNA甲基化特征对衰老相关疾病如2型糖尿病和心血管疾病进行风险评估,对于实现个体化的衰老干预与健康管理具有重要意义。与表观遗传时钟类似,现有针对2型糖尿病和心血管疾病的风险评估模型多基于Cox比例风险模型等线性方法,在跨研究和跨测序平台应用时同样面临显著的泛化能力挑战。
深度成对学习,通常被称为孪生网络,是一类由两个共享相同参数和权重的子网络构成的神经网络架构,已在面部识别、语音识别和视觉跟踪等多个领域得到广泛应用。成对学习在应对高维小样本问题以及缓解模型对训练数据过拟合方面展现出良好潜力。该研究提出了一种名为MAPLE的稳健计算框架,基于成对学习实现甲基化年龄与疾病风险预测。MAPLE通过比较任意两个样本的DNA甲基化谱,预测它们在表观遗传年龄或疾病风险上的差异,从而将来源多样的DNA甲基化数据映射到统一的潜在空间中,实现对不同衰老状态或疾病状态样本的有效区分。基于这一策略,MAPLE在不同研究、测序平台、数据预处理方法及组织类型中均能稳定优于传统表观遗传时钟。同时,MAPLE还能够对衰老相关疾病风险进行精准评估,并准确识别疾病前状态。其在表观遗传年龄和疾病风险评估方面表现出的稳健性与准确性,为DNA甲基化测序在衰老评估与干预中的临床应用奠定了坚实基础。
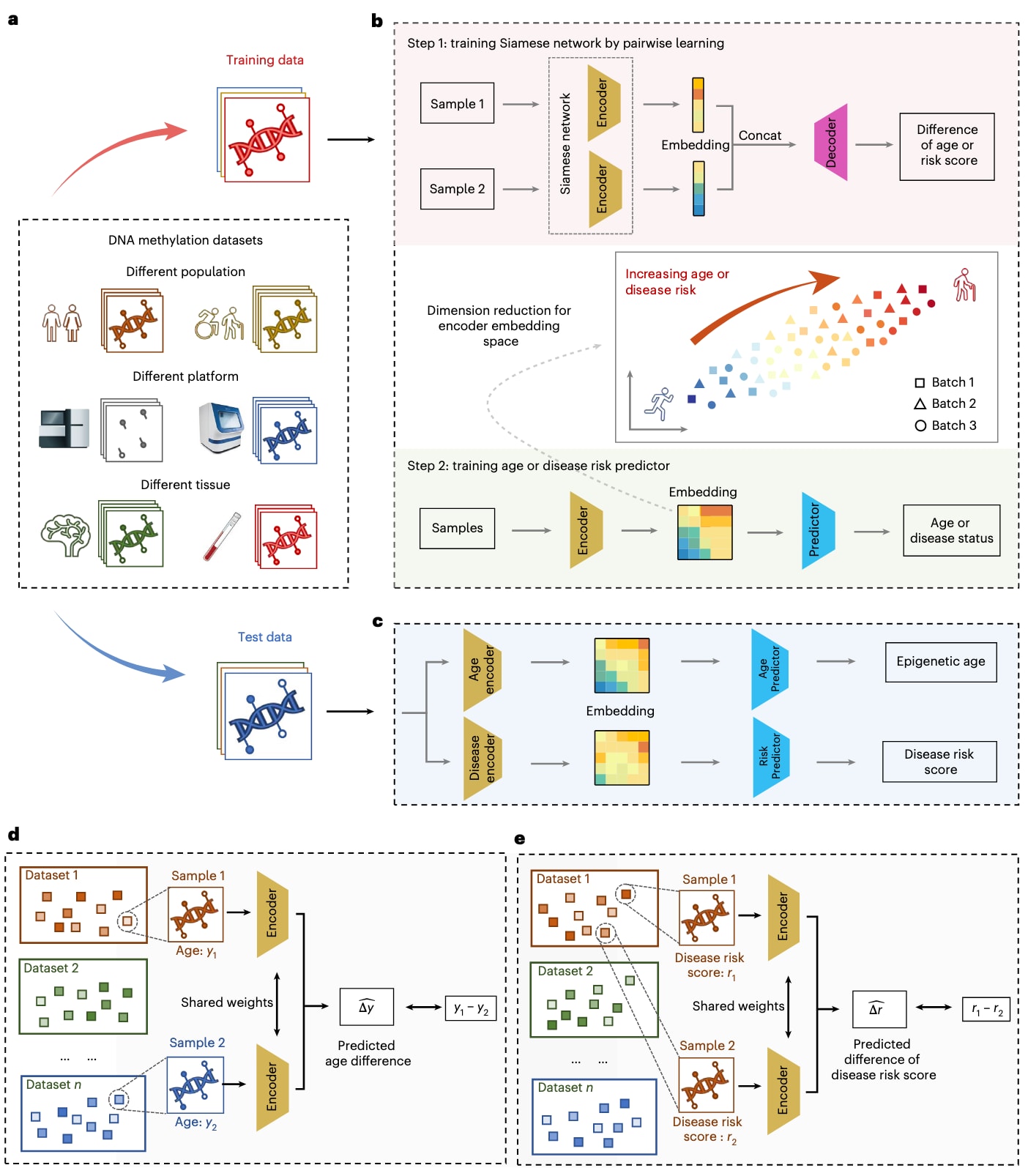
图1|MAPLE总体概览。 a,DNA甲基化数据的异质性。在临床应用场景中,甲基化谱来源于不同人群、测序平台和组织类型。b,MAPLE的训练流程。首先通过成对学习训练编码器,将来自不同来源的训练甲基化谱映射到统一的嵌入空间中。随后在该嵌入表示基础上训练预测器,用于估计表观遗传年龄或疾病状态。该嵌入空间在尽量降低混杂因素影响的同时,保留了与年龄或疾病相关的表观遗传信号。c,MAPLE的推理过程。训练完成的模型以测试集中的甲基化数据作为输入,输出对应的表观遗传年龄预测值和疾病风险评分。d,e,用于训练的成对学习策略:d,在表观遗传年龄预测中,通过成对学习预测来自不同数据集的两个样本之间的实际年龄差异;e,在疾病风险预测中,通过成对学习预测同一数据集中两个样本之间的疾病风险差异。
2 结果
2.1 MAPLE总体概述
表观遗传时钟在临床应用中的一个关键挑战在于,如何在不同测序平台、数据预处理方法、人群背景和组织类型存在差异的情况下,仍能对未见过的DNA甲基化数据进行稳健且准确的表观遗传年龄预测(图1a)。MAPLE通过成对学习策略有效缓解了不同数据集之间固有的批次效应,从而应对这一挑战。尽管由于数据来源或预处理流程不同,各数据集中的DNA甲基化谱分布可能存在显著差异,但来自不同数据集样本的实际年龄本身仍具有可比性。MAPLE通过预测任意两个DNA甲基化谱之间的年龄差异,将DNA甲基化数据编码到一个统一的潜在空间中,在消除数据异质性带来的批次效应的同时,保留与衰老相关的关键信息。
此外,成对样本构建方式使训练样本数量呈二次增长,从而显著提升了模型的训练充分性并降低了过拟合风险。MAPLE采用完全基于训练数据的两阶段训练流程实现表观遗传年龄预测。首先,利用成对学习训练一个孪生网络作为编码器,将来自不同来源的DNA甲基化谱整合至统一的潜在空间中。具体而言,从训练数据中随机选取两个样本,将其DNA甲基化谱同时输入同一编码器,生成对应的潜在空间嵌入(图1b)。随后,基于这些嵌入预测两个样本之间的实际年龄差(图1d)。当编码器训练完成后,进一步构建年龄预测器,根据单个样本的嵌入向量估计其年龄(图1b)。在推理阶段,未参与训练的测试样本DNA甲基化谱被输入已训练好的编码器和年龄预测器,从而获得其预测的表观遗传年龄(图1c)。
在疾病风险预测模型的训练中,首先采用传统线性模型生成疾病风险评分,并将其作为成对学习的优化目标。由于这些风险评分仅在同一数据集内部具有可比性(补充图1),因此MAPLE通过成对学习的方式,在同一数据集中学习两个样本之间的风险评分差异(图1e)。类似于表观遗传年龄预测流程,模型首先在训练数据上学习一个编码器以提取与疾病相关的表观遗传信号。随后构建疾病预测器,基于生成的嵌入向量对疾病类型进行分类,从而将监督式疾病标签信息整合进模型中(图1b)。在推理阶段,未见过的测试样本经编码器生成嵌入表示,并基于嵌入空间中的距离计算其疾病风险评分(图1c;具体方法见方法部分)。
2.2 MAPLE在表观遗传年龄预测中的系统性基准评估
具有临床应用价值的表观遗传时钟,需要在不同测序平台、数据预处理流程和组织类型的未见DNA甲基化数据上,实现稳定且准确的年龄预测。为系统评估MAPLE的泛化能力,研究设计了涵盖31项基准测试的两类对比场景,覆盖多种生物学和技术条件。在第一类对比场景中,评估对象为包含显著技术偏差的血液DNA甲基化数据集。来自Illumina Infinium Methylation450和MethylationEPIC芯片的6个独立测试数据集,分别采用SWAN、BMIQ和GMQN三种常用预处理方法进行处理,共形成24项基准测试。在第二类对比场景中,重点考察非血液样本中的表观遗传年龄估计能力,该能力对于不同组织衰老生物学研究尤为重要。模型性能通过预测年龄与实际年龄之间的中位绝对误差和皮尔逊相关系数进行评估。
为分析成对学习的贡献,研究将MAPLE与一个多层感知机基线模型进行比较,该基线模型与MAPLE具有相同的网络结构,但直接预测绝对年龄而非相对年龄。在第一类场景中,MAPLE的平均中位绝对误差为1.45年,相关系数为0.97,而多层感知机基线模型的平均中位绝对误差为4.14年,相关系数为0.88(图2a及补充图2a)。在非血液数据集中,MAPLE同样显著优于该基线模型(图2b及补充图2b),充分体现了成对学习在提升表观遗传年龄预测性能中的关键作用。
进一步地,研究将MAPLE与五种当前主流的表观遗传时钟进行比较,包括HorvathAge、HannumAge、PhenoAge、AltumAge和cAge。在第一类对比场景中,针对6个原始数据集,MAPLE取得了最佳表现,平均中位绝对误差为1.40年,相关系数为0.97;相比之下,HorvathAge和PhenoAge由于无法适配EPIC芯片数据,表现最差(图2a及补充图2a)。MAPLE在不同预处理流程下依然保持高度稳定,在SWAN、BMIQ和GMQN处理数据上的中位绝对误差分别为1.61、1.47和1.29年(图2a)。相反,HannumAge、AltumAge和HorvathAge在预处理方法变化时性能大幅下降,例如在BMIQ处理数据中,其误差分别上升至6.12、7.32和8.36年。
在第二类对比场景中,MAPLE在非血液数据集上同样表现最优,平均中位绝对误差为2.30年,平均相关系数为0.95(图2b及补充图2b)。相比之下,AltumAge和HorvathAge的平均误差分别为5.22年和5.75年,而cAge、HannumAge和PhenoAge的平均误差均超过10年。在全部31项基准测试中,MAPLE取得了最低的整体平均中位绝对误差1.61年,显著优于排名第二的cAge和排名第三的AltumAge,充分证明了其在不同数据集、预处理方法和组织类型下的高精度与高稳健性。
为进一步评估批次效应的消除效果,研究采用主成分分析和主方差成分分析。对MAPLE生成的嵌入进行主成分分析后,可观察到清晰的年龄相关梯度分布,不同组织和研究来源的样本在潜在空间中良好混合(补充图3a–d)。相比之下,原始DNA甲基化数据的主成分分析结果主要由批次效应主导,呈现明显分组(补充图3e,f)。主方差成分分析结果进一步表明,经MAPLE整合后,由批次效应解释的方差比例从0.88显著下降至0.53,而由年龄解释的方差比例则从不足0.01显著提升至0.42(补充图3)。综上所述,这些结果表明MAPLE能够有效削弱批次效应并强化与年龄相关的生物学信号,从而在高度异质的数据条件下实现稳健可靠的表观遗传年龄预测。
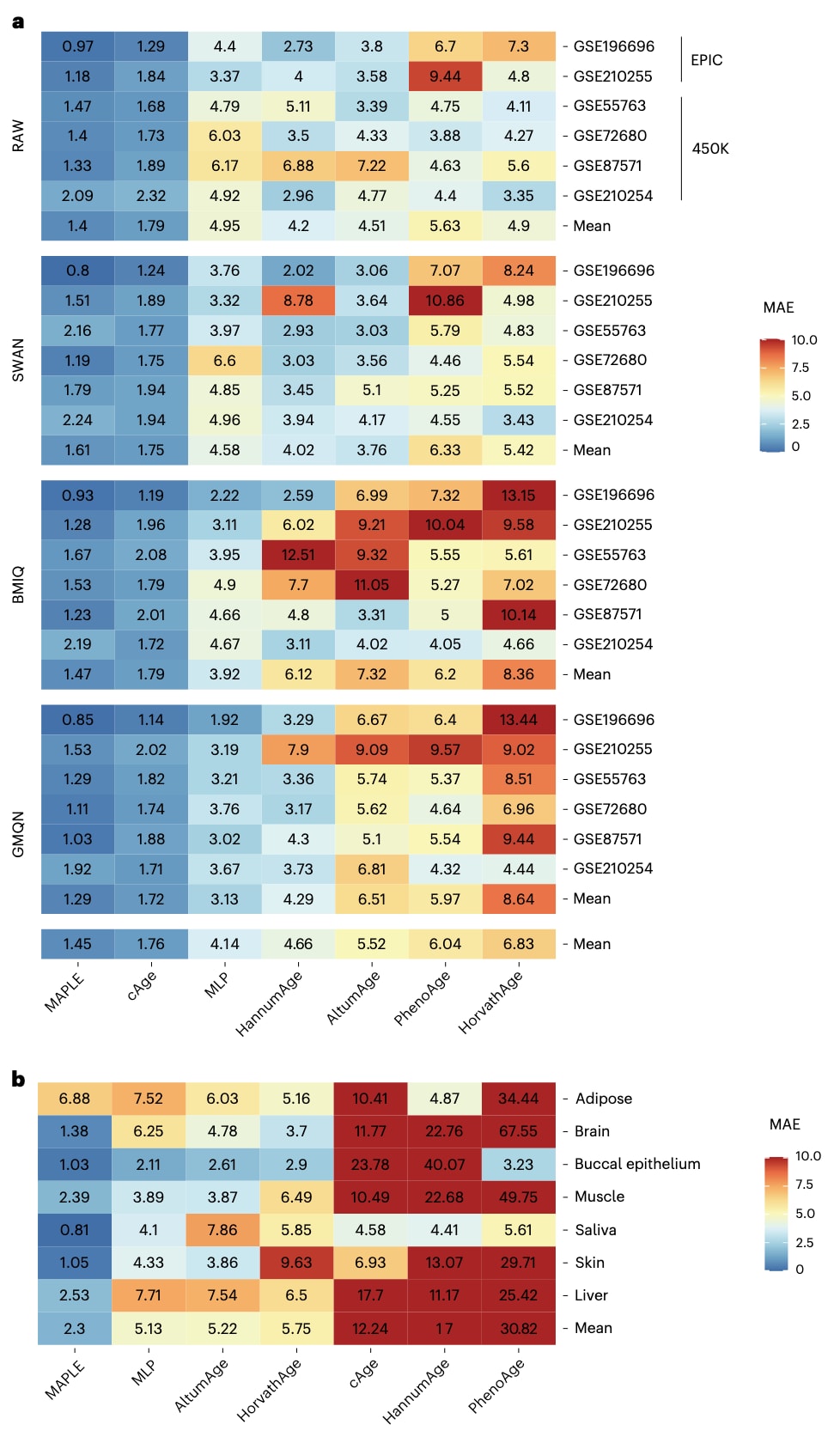
图2|MAPLE与六种对比方法在31项基准测试中的性能比较。 a,热图展示了在血液来源测试数据集上的表观遗传年龄预测中位绝对误差,数据集以其GEO登录号标识。其中,GSE196696和GSE210255基于EPIC芯片生成,其余数据集均采用450K芯片。甲基化数据的预处理方法标注于左侧。七种方法按平均中位绝对误差从小到大由左至右排列。b,热图展示了在七种非血液组织类型中的年龄预测中位绝对误差。测试数据集以组织类型表示,各方法同样按平均中位绝对误差从小到大由左至右排列。
2.3 MAPLE识别的衰老相关CpG位点
表观基因组全关联研究(epigenome-wide association study,EWAS)的目标是系统考察全基因组范围内的表观遗传变异(主要为CpG位点的DNA甲基化水平),以检测与目标表型在统计学上相关的差异。EWAS有助于解析疾病发生机制并鉴定疾病特异性生物标志物。传统上,年龄相关的CpG位点通常通过EWAS加以识别。该研究选取了两项相互独立的衰老相关研究^24,25,并通过EWAS Atlas数据库^26获取其对应的年龄相关CpG集合。尽管这两组CpG位点之间存在显著重叠(优势比OR=58.2,P<10^−16),但这些重叠CpG共定位的基因仅在与细胞黏附相关的通路中显著富集(补充图4a,b)。相比之下,将MAPLE与集成梯度(integrated gradients,IG)方法应用于同样的两套数据后,来自两个数据集的前2000个信息量最大的CpG位点之间呈现出显著更高的重叠程度(OR=4823.1,P<10^−16)(补充图4a;CpG位点与归因方法的选择策略见方法学部分)。这些重叠CpG共定位的基因在多种Gene Ontology(GO)条目中显著富集,包括细胞黏附、器官发育与形态发生、认知功能、细胞因子产生等(图3a)。上述通路富集结果与衰老生物学领域的既有研究高度一致^27–31。综上所述,这些结果表明,MAPLE通过优先关注在机制层面参与衰老过程的、具有生物学意义的CpG位点,实现了对表观遗传年龄的精准预测。
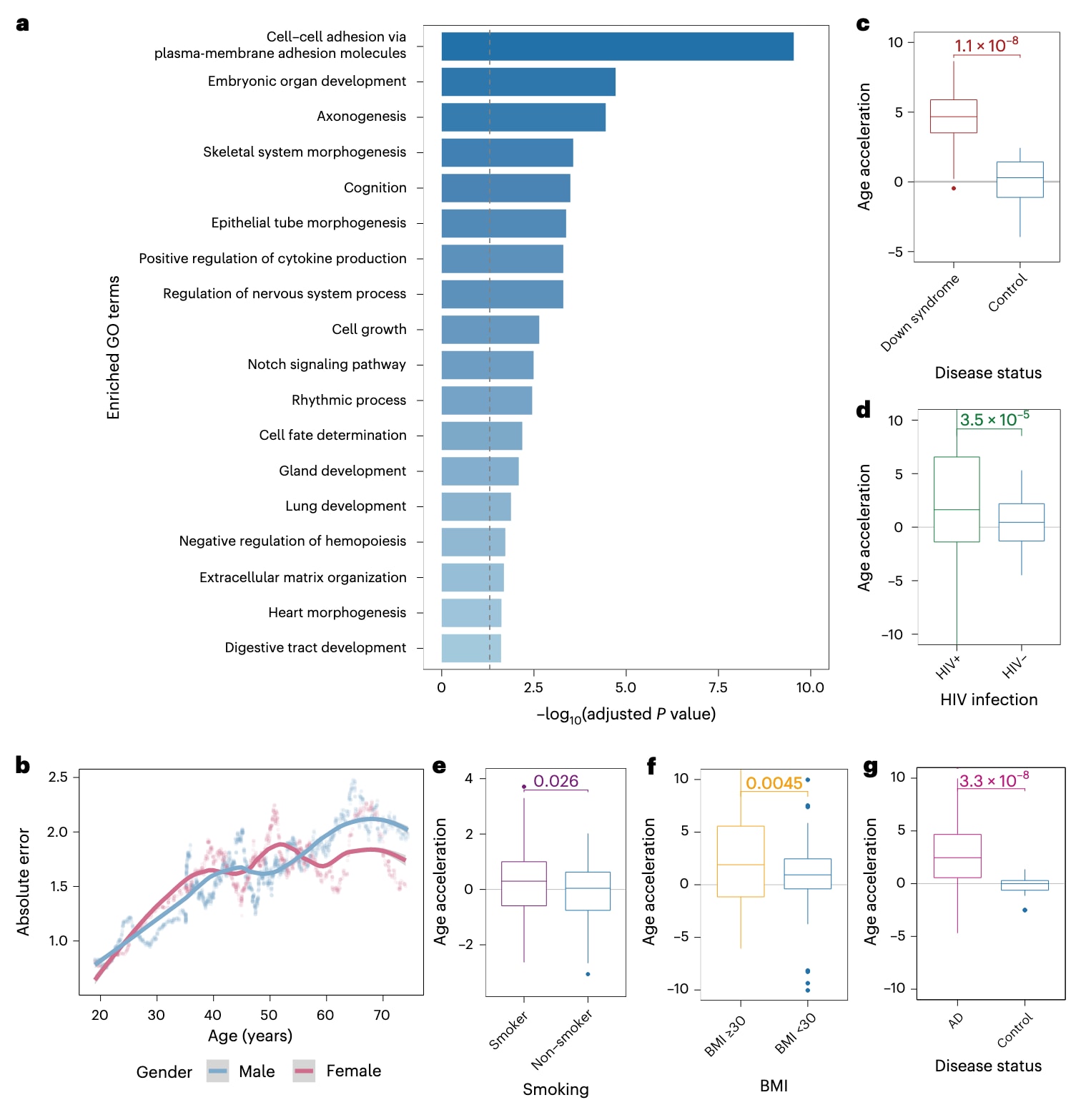
图 3|MAPLE 捕获衰老相关生物学过程并识别加速衰老。 a,柱状图显示由 MAPLE 识别的年龄相关 CpG 位点所共定位基因的通路富集结果。通路富集采用单侧超几何检验进行评估,P 值使用 Benjamini–Hochberg 方法进行多重检验校正。灰色虚线表示校正后的显著性阈值 0.05。b,散点图展示了在整个生命周期范围内,表观遗传年龄与时间年龄之间绝对误差的 100 个样本滚动平均轨迹。每个点表示一个包含 100 个样本的滑动窗口内的平均绝对误差,颜色表示性别。实线为基于局部加权散点平滑(LOESS)的拟合结果,灰色阴影区域表示 95% 置信区间。c–g,箱线图展示了唐氏综合征(c)、HIV 感染(d)、吸烟(e)、肥胖(f)以及阿尔茨海默病(AD)(g)人群相对于各自对照人群的年龄加速水平。每个点代表一个独立生物学样本(即单个个体的 DNAm 图谱)所估计的年龄加速值。c–d 中样本量如下:唐氏综合征,病例 n = 29、对照 n = 19(血液);HIV,病例 n = 229、对照 n = 45(血液);吸烟,吸烟者 n = 72、非吸烟者 n = 80(血液);肥胖,病例 n = 84、对照 n = 82(肌肉);AD,病例 n = 64、对照 n = 63(脑)。箱线图中,中线表示中位数,箱体上下边界分别表示第 25 和第 75 百分位数,须线延伸至 1.5 倍四分位距(IQR)。统计显著性采用单侧 t 检验评估。
2.4 MAPLE识别的性别特异性衰老轨迹与疾病状态相关的加速衰老
既往研究报道了衰老轨迹中的性别差异,尤其是绝经与血液组织中表观遗传衰老加速密切相关,且约50岁左右的女性会经历与衰老相关的显著生理变化^32,33。这一现象可能反映了绝经期表观遗传衰老的急剧转变,而时间年龄的变化相对平稳。因此,处于绝经期年龄段的女性预计会表现出表观遗传年龄与时间年龄之间更大的不一致性。为验证该假设,该研究使用MAPLE对留出的血液组织数据集进行表观遗传年龄预测,并计算预测表观遗传年龄与时间年龄之间绝对误差的滚动平均值,以量化这种不一致程度。如图3b所示,约52岁的女性个体出现了明显的绝对误差升高,而男性亚组中未观察到类似模式。值得注意的是,大多数女性在45–55岁之间经历绝经,平均年龄约为52岁。这种性别特异性趋势表明,MAPLE能够有效捕捉与绝经相关的关键衰老生物学过程。
此外,已有广泛共识认为,表观遗传时钟估计的年龄可用于计算“年龄加速”,即估计年龄与实际时间年龄之差,用以反映个体既往的衰老速率^6,9,10,22。较高的年龄加速值意味着更快的衰老速率以及更高的死亡和年龄相关疾病风险。为评估MAPLE预测的表观遗传年龄是否能够真实反映衰老速率,该研究选取了若干在生物学上公认的加速衰老人群作为例证,包括唐氏综合征、人类免疫缺陷病毒(HIV)感染者、吸烟人群、肥胖人群以及阿尔茨海默病(AD)患者^30,34–37。与衰老生物学的既有发现一致,MAPLE预测上述人群相较对照组具有显著更高的年龄加速水平(图3c–g及补充图5a–c)。值得注意的是,MAPLE在AD患者的脑组织中检测到显著的年龄加速,而在其血液样本中未观察到这一现象(图3g及补充图5d)。这一结果表明,非血液组织能够揭示独特且具有生物学意义的衰老信号,而这些信号可能仅依赖血液分析时被忽略,进一步凸显了跨组织表观遗传年龄预测在衰老生物学研究中的重要性。
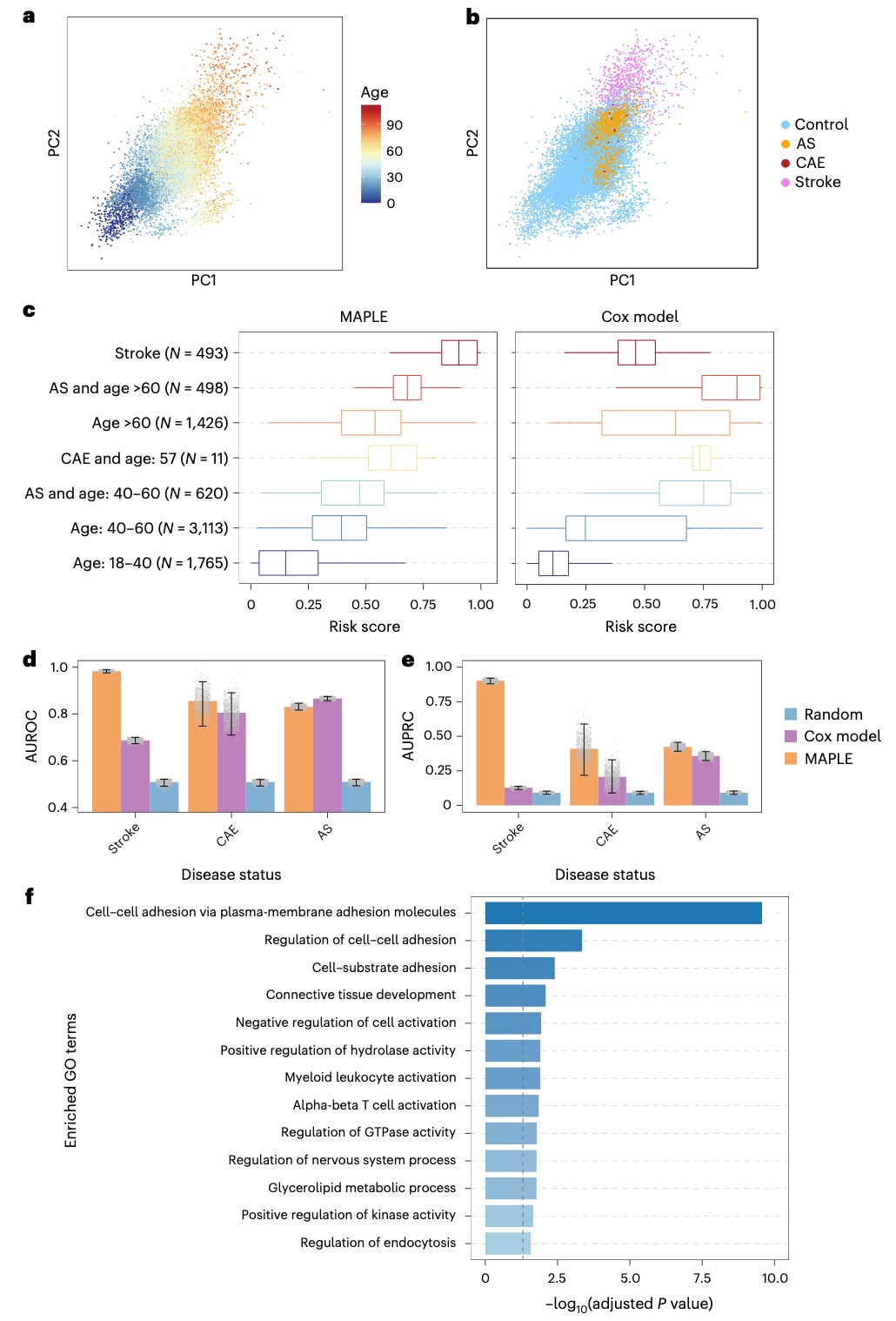
图 4|基于 DNAm 数据的 MAPLE 心血管疾病(CVD)风险评估。 a,b,训练集与测试样本甲基化嵌入的 PCA 可视化。在 a 中,点的颜色表示样本年龄;在 b 中,点的颜色表示与 CVD 相关的疾病状态(N_control = 13,321,N_AS = 1,118,N_CAE = 11,N_stroke = 710)。c,箱线图比较了 MAPLE(左)与 Cox 模型(右)对不同测试样本群体预测的 CVD 风险评分。每个点表示一个独立生物学样本(单个个体 DNAm 图谱)的预测风险评分。各组样本量标注于图中。箱线图显示中位数(中心线)、第 25 和第 75 百分位数(箱体边界),须线延伸至 1.5 倍 IQR。d,e,柱状图分别显示区分卒中、CAE 和 AS 样本与对照样本的 AUROC(d)和 AUPRC(e)值。数据以均值形式呈现,误差线表示通过 1,000 次自助法(bootstrap)重采样估计的 95% 置信区间(2.5–97.5 百分位)。灰色散点表示每一次 bootstrap 迭代中获得的单个 AUROC 或 AUPRC 值,用以展示估计分布的完整情况。f,柱状图显示由 MAPLE 识别的 CVD 相关 CpG 位点所共定位基因的通路富集结果。通路富集采用单侧超几何检验评估,P 值使用 Benjamini–Hochberg 方法校正,灰色虚线表示校正后的显著性阈值 0.05。
2.5 MAPLE在心血管疾病风险评估中的应用
心血管疾病(CVD)和2型糖尿病(T2D)等复杂慢性疾病通常受遗传因素与环境因素的共同影响。DNA甲基化作为一种重要的表观遗传修饰,是遗传变异和环境暴露影响疾病风险的关键途径,能够为个体疾病风险提供动态预测信息。在CVD的发生发展过程中,年龄是一个强有力且不可改变的风险因素^38。动脉粥样硬化(atherosclerosis,AS)是一种以动脉壁内斑块沉积为特征的病理状态,可导致血流减少并显著增加心肌梗死和卒中的风险^39。冠状动脉扩张(coronary artery ectasia,CAE)表现为冠状动脉异常扩张,多由AS引起,且通常预后较差^40。卒中是一种严重疾病,定义为由血管原因导致的中枢神经系统急性局灶性损伤所引发的神经功能缺损,多数源于动脉粥样硬化斑块的栓塞^41。
MAPLE能够有效整合来自多项研究的DNAm数据并预测CVD风险。在其嵌入空间的主成分分析(PCA)图中,年轻的健康对照样本聚集于左下方,而年龄较大或患有卒中、CAE或动脉粥样硬化的不健康样本则分布于右上方(图4a,b)。来自不同研究的数据样本充分混合(批次效应的PVCA由0.78降至0.53;补充图6),表明MAPLE能够将异质批次映射到一个统一的嵌入空间,从而捕捉与CVD相关的表观遗传信号。基于这些嵌入,MAPLE为每个样本计算CVD风险评分。在七个测试人群中,卒中样本的风险评分最高,而18–40岁人群最低(图4c)。相比之下,Cox模型在不同数据集间产生不一致的风险评分,难以泛化至独立队列(图4c)。
进一步比较两种模型在区分疾病及疾病前状态与健康对照方面的能力时,MAPLE在卒中识别中取得了0.98的ROC曲线下面积(AUROC)和0.90的精确率–召回率曲线下面积(AUPRC),显著优于Cox模型(AUROC 0.69,AUPRC 0.13;图4d,e)。在疾病前状态(包括CAE和AS)的识别中,MAPLE同样获得更高的AUPRC(CAE为0.41,AS为0.42),而Cox模型分别为0.20和0.35(图4d,e)。值得注意的是,即便在阳性与阴性样本的年龄分布被平衡的情况下,MAPLE仍保持优越性能(补充图7a,b)。此外,在区分三种非健康状态与健康对照的分析中,MAPLE在卒中分类中的AUPRC超过0.99,而Cox模型仅为0.80(补充图7c,d),表明MAPLE在降低假阳性方面更为有效。总体而言,MAPLE在检测与CVD相关的疾病状态及疾病前状态方面始终优于Cox模型。
对MAPLE识别的CVD相关CpG位点所共定位基因进行富集分析显示,这些基因显著富集于细胞黏附相关条目、“水解酶活性正向调控”“髓系白细胞活化”“αβ T细胞活化”等通路(图4f),与既往关于CVD发病机制的研究结果高度一致。
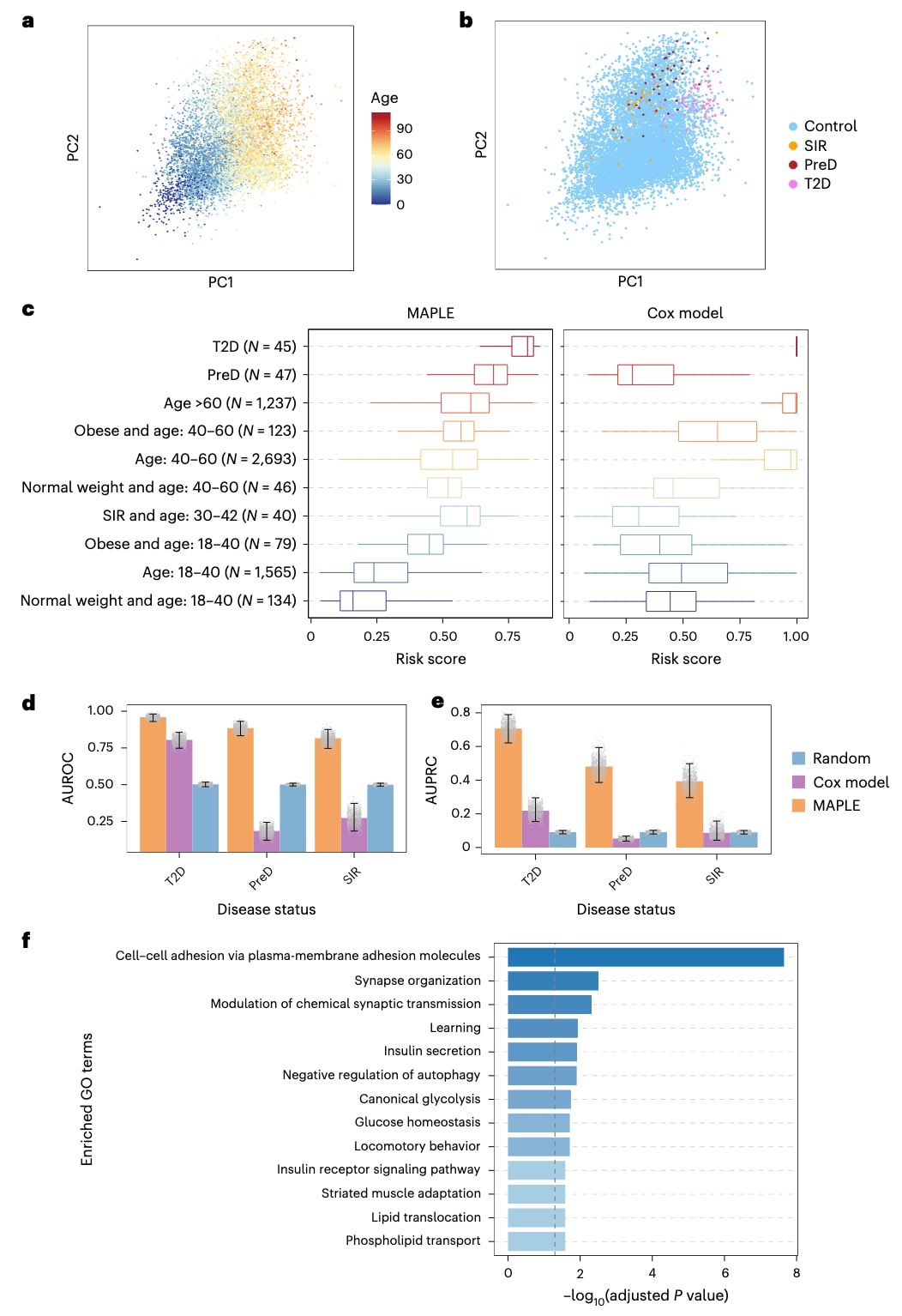
图 5|基于 DNAm 数据的 MAPLE 2 型糖尿病(T2D)风险评估。 a,b,训练集与测试样本甲基化嵌入的 PCA 可视化。在 a 中,点的颜色表示样本年龄;在 b 中,点的颜色表示与 T2D 相关的疾病状态(N_control = 12,576,N_SIR = 40,N_PreD = 47,N_T2D = 74)。c,箱线图比较了 MAPLE(左)与 Cox 模型(右)对不同测试样本群体预测的 T2D 风险评分。每个点代表一个独立生物学样本(单个个体 DNAm 图谱)的预测风险评分,各组样本量标注于图中。箱线图显示中位数、第 25 和第 75 百分位数,须线延伸至 1.5 倍 IQR。d,e,柱状图分别显示区分 T2D、PreD 和 SIR 样本与对照样本的 AUROC(d)和 AUPRC(e)值。数据以均值呈现,误差线表示通过 1,000 次 bootstrap 重采样估计的 95% 置信区间(2.5–97.5 百分位)。灰色散点表示每次 bootstrap 迭代得到的单个 AUROC 或 AUPRC 值,展示其分布情况。f,柱状图显示由 MAPLE 识别的 T2D 相关 CpG 位点所共定位基因的通路富集结果。通路富集采用单侧超几何检验评估,P 值经 Benjamini–Hochberg 方法校正,灰色虚线表示校正后的显著性阈值 0.05。
2.6 MAPLE 在 2 型糖尿病(T2D)风险评估中的应用
T2D 的风险评估同样可以采用与 CVD 模型一致的建模思路。年龄和肥胖是 T2D 发生发展的重要风险因素。系统性胰岛素抵抗(systemic insulin resistance,SIR)是一种机体对胰岛素反应减弱、无法有效将葡萄糖转化为能量的病理状态;糖尿病前期(prediabetes,PreD)则表现为血糖水平高于正常但尚未达到 T2D 诊断标准。SIR 和 PreD 个体更容易进展为 T2D。T2D 本质上是一种以碳水化合物、脂质和蛋白质代谢失调为特征的疾病。
在 T2D 风险预测中,MAPLE 将 DNAm 图谱编码到一个统一的嵌入空间中,该空间能够捕获与 T2D 相关的变异,同时最大限度减少技术偏倚。在 PCA 可视化中,年轻的健康对照样本主要聚集在左下角,而年龄较大或不健康的样本(包括 SIR、PreD 和 T2D)则分布在右上角(图 5a,b)。来自不同研究的数据在嵌入空间中良好混合(批次效应的 PVCA 从 0.84 降低至 0.60;补充图 8),表明 MAPLE 能够稳健地整合跨数据集信息。
MAPLE 为每个样本计算 T2D 风险评分,其中 T2D 个体的评分最高,其次为 PreD。在按年龄分层的对照人群中,风险评分随年龄增加而升高,且肥胖个体的评分高于体重正常者;SIR 样本相对于年龄匹配的对照也表现出更高的风险评分(图 5c)。相比之下,Cox 模型在不同数据集之间产生的风险评分不一致且不可比。
在分类性能方面,MAPLE 在 T2D 识别中取得了 AUROC 0.96 和 AUPRC 0.71,显著优于 Cox 模型(AUROC 0.80,AUPRC 0.22)(图 5d,e)。对于 PreD 和 SIR,MAPLE 分别获得了 0.89 和 0.82 的 AUROC,以及 0.48 和 0.39 的 AUPRC,而 Cox 模型的表现甚至低于随机水平(图 5d,e)。即使在平衡正负样本年龄分布后,MAPLE 仍保持明显优势(补充图 9a,b)。虽然 MAPLE 和 Cox 模型在区分 T2D 与健康样本时均表现良好,但在 PreD 和 SIR 的识别中,MAPLE 明显优于 Cox 模型(补充图 9c,d)。这些结果表明,MAPLE 在跨数据集泛化能力和减少健康人群误判方面具有显著优势。
进一步的通路富集分析显示,MAPLE 能够有效识别与 T2D 相关的 CpG 位点。这些 CpG 位点共定位的基因显著富集于细胞黏附、“学习”、胰岛素分泌、经典糖酵解、葡萄糖稳态、运动行为以及胰岛素受体信号通路等功能通路(图 5f),与既往关于 T2D 发病机制的研究结果高度一致。
3 讨论
由于 DNAm 数据的高维特性(CpG 位点数量远大于样本数量),以及测序平台、预处理流程和组织来源差异所引入的异质性,开发具有临床应用价值的表观遗传时钟仍面临重大挑战。MAPLE 通过成对学习策略同时应对了这两大难题:成对样本输入在训练过程中显著扩展了有效样本数量,从而降低了过拟合风险;同时,MAPLE 不直接预测绝对年龄,而是预测样本之间的相对年龄差异,使模型能够聚焦于真实的衰老相关信号,并忽略组织类型或平台等混杂因素。这一设计也使 MAPLE 在疾病风险预测中显著优于传统的 Cox 模型。
大规模队列研究通常积累了丰富的 DNAm 数据和纵向临床信息,为慢性疾病风险预测提供了重要机会。然而,出于隐私保护考虑,这类研究往往仅公开风险评估模型,而不发布原始 DNAm 和临床数据,从而限制了更先进预测框架的开发。该研究中,MAPLE 通过整合公开 DNAm 数据与已发布的风险模型,实现了对 CVD 和 T2D 两种慢性疾病风险的预测,展示了在保护隐私前提下开展基于甲基化的风险评估的可行性。此外,包括神经系统疾病和自身免疫性疾病在内的多种疾病进展均与 DNAm 改变密切相关。作为一种通用的计算框架,MAPLE 有望拓展至更广泛的健康结局预测,从而提升基于甲基化评估的临床实用价值。
近年来,计算表观基因组学领域涌现出多种有价值的方法,如基于序列的 DNAm 预测算法 iDNA-ABF 和 Methyl-GP,以及基础模型 MethylGPT。这些方法为 MAPLE 提供了互补的发展方向。基于序列的方法直接从 DNA 序列推断甲基化模式,侧重于甲基化的内在基因组决定因素;而 MAPLE 以 DNAm 图谱为输入,综合遗传和环境因素来评估衰老和疾病风险。未来,可在 MAPLE 框架中联合使用 DNAm 与序列数据,例如先利用序列模型生成预测的 DNAm 图谱,再与实测 DNAm 数据整合,以提升下游预测性能,从而区分遗传决定与环境诱导的甲基化变化,更深入地解析个体健康轨迹背后的机制。
此外,像 MethylGPT 这样的基础模型能够从大规模数据中学习具有生物学语义的 DNAm 表征,若与 MAPLE 结合,可形成互补优势:MethylGPT 提供单样本层面的生物学表征,而 MAPLE 则刻画样本间差异,实现更精准的表观遗传年龄和疾病风险预测。这种协同策略有望显著提升基于甲基化预测模型的准确性和鲁棒性。