AS 2026 | Ouroboros:融合分子构象空间与药效团相似性的基础模型
今天介绍的这项工作来自 Advanced Science。Ouroboros是一种采用正交架构的分子基础模型,将分子表征、结构重构与性质建模解耦:表征模块以分子图为输入,通过GNN相似性学习并引入构象空间-药效团相似性CSPS等约束,在较小规模预训练下仍获得可泛化分子表示,在DUD-E与LIT-PCBA等大规模基准上实现强虚拟筛选表现,并在多靶点药物发现中结合相似性筛选与对接从超大化合物库中筛得多靶点命中分子,其中酶活性实验展现出通过构象变化适配不同靶点残基的先导潜力。重构模块用自回归Transformer将1D分子表示还原为SMILES,支持随机传播与以性质损失引导的定向化学演化,用于溶解度、膜通透性等性质优化,以及骨架跃迁与化学融合式双靶点设计,在对接、

获取详情及资源:
0 摘要
大规模化学预训练模型的出现,显著提升了对分子结构与功能之间复杂关系的刻画能力。尽管分子基础模型因其通用表征与对下游分子优化任务的支持而受到关注,但在预训练阶段显式融入化学知识(例如构象信息与药效团信息)的工作仍然相对有限。考虑到小分子在溶液中的高度动态特性、与靶标结合时可能发生的构象变化,以及药效团互补性在分子识别中的关键作用,将这些因素纳入分子基础建模具有重要意义。该研究提出一种分子基础模型,在预训练过程中引入构象空间与药效团相似性的投影约束,以此对表示空间进行正则化,从而让模型在学习通用分子表征时更贴近真实化学规律。整体框架采用类似衔尾蛇(Ouroboros-like)的架构:首先利用图神经网络将分子图编码为1D表示向量,随后通过自回归Transformer模块将该潜在表示重建为SMILES序列。这样的双模块设计在同一潜在空间中同时支持表征学习与分子生成,并具备较强的灵活性与可扩展性。大量实验结果显示,该模型能够有效应对多类实际化学问题,包括基于相似性的虚拟筛选、面向特定多靶点作用的分子设计、化学性质预测,以及有方向的分子优化等任务,体现出将构象与药效团信息融入预训练所带来的综合收益。
1 引言
人工智能已被广泛应用于化学研究的多个方向,包括材料设计、药物发现与计算生物学等。以数据驱动为核心的方法正在重塑分子性质、相互作用、折叠与组装的预测范式。相关研究一方面大量借鉴自然语言处理与计算机视觉领域的发展经验,另一方面也推动了更契合分子结构特征的图神经网络体系,从而更有效地刻画化学结构信息。
近年来,小分子通用基础模型逐渐成为热点,其目标通常是通过预训练获得更强的通用表征,以提升下游性质预测等任务的表现。分子基础模型的核心原则在于学习化学结构的有意义表示空间,类似深度神经网络对数据内在结构的抽象建模。已有研究表明,借助深度神经网络可以学习到更具可解释性的分子表征,这些表征对分子性质预测以及基于相似性的虚拟筛选尤为关键。围绕化学预训练模型,不同预训练策略也在持续引入多种化学知识,以增强其在多类下游任务中的适用性与迁移能力:自监督预训练通常面向不同化学表示格式的统一理解,例如SMILES、分子图、InChI与分子指纹等;监督式预训练则更强调将化学结构与多模态信息、功能或性质之间的对应关系显式整合进模型能力之中。
生成式预训练的兴起进一步赋予分子基础模型以生成能力,使其不仅能够生成分子结构,也能生成或刻画相关化学性质。这类方法往往利用带标注的数据集训练统一的表征与生成模型,但高质量、带实验标注的数据极为稀缺,实践中常需借助化学信息学工具生成大量相对简单的标签,从而限制模型在预训练阶段获得更复杂化学知识的能力。与此同时,生成式预训练通常依赖可被分词的“化学语言”,这在一定程度上阻碍了模型直接通过分子图学习表示;而一些研究已经指出,分子图在表示学习方面具备更强的优势。
针对上述矛盾,该工作提出在单一模型中设置两个相互正交的模块:一个模块负责将分子结构(分子图)映射为1D表示向量,另一个模块负责将表示向量转换为分子SMILES(化学语言)。这种设计使得表示学习与分子生成能够相对独立地训练,从而可针对不同模块分别选择更合适的神经网络结构、数据集与训练策略。与部分分子生成框架不同,其重构模型不需要额外的prompt或噪声输入,而是以表示空间为依据对化学结构进行忠实重建。基于这一正交框架,模型(命名为Ouroboros)可作为药物小分子预测器的基础模块,使得新模型具备对小分子结构进行优化乃至从头分子设计的潜力。实验结果表明,Ouroboros不仅在相似性虚拟筛选与面向特定多靶点作用的药物设计中展现了表征能力,还能够覆盖多种药代与毒理性质的建模需求,并可进一步服务于分子结构优化任务。
2 结果
2.1 表征模块与重构模块的正交架构
2.1.1 通过表征学习获得构象空间与药效团知识
既有研究已经强调药效团信息对分子表征的重要性,但常见做法是将药效团作为输入特征喂给模型,而不是让模型在学习过程中掌握药效团的构建与比较机制。该工作将药效团相似性直接用于表征学习,以帮助模型在不同2D结构之间建立分子形状与性质的联系,从而感知不同化学骨架之间更内在的相关性。同时,工作中引入了系统性搜索得到的分子构象空间,并在构象层面对药效团相似性进行比较,使模型能够捕捉分子在溶液与结合过程中的动态行为,这一点也符合分子可能以不同构象结合不同靶标的化学常识。
在Ouroboros框架中,表征模块旨在构建一个结构化的表示空间来解释化学分子结构,其基本假设是化学结构相近或药理性质相近的分子在表示空间中应当彼此接近。此前相关结果已显示,引入构象空间相似性有助于获得面向配体发现任务的通用分子表征。为了进一步提升表征学习效果,该工作在方法上引入了两类分子间相似度信号:一类是通过更强空间搜索扩展得到的构象空间-药效团相似性CSPS,另一类是分子指纹相似性(具体定义与构建方式见Methods)。
该工作提出一种新的相似性学习策略,将分子表示同时投影到多种分子相似度上,以增强表征模块学习到通用且信息量更高的分子表示的能力。表征模块使用图神经网络将分子图转换为1D表示向量(图1a),并在由查询分子与参考分子两两相似度构成的相似度矩阵上进行预训练。通过CSPS学习(图1b),表征空间被正则化到更有意义的结构中。这里的CSPS矩阵规模很大,可视为由少量化学结构集合构建出的超大规模相似度监督信号,其维度为
在训练过程中,得到的1D表示会通过两个外部投影头分别映射到分子指纹相似性与CSPS,训练目标由均方误差损失
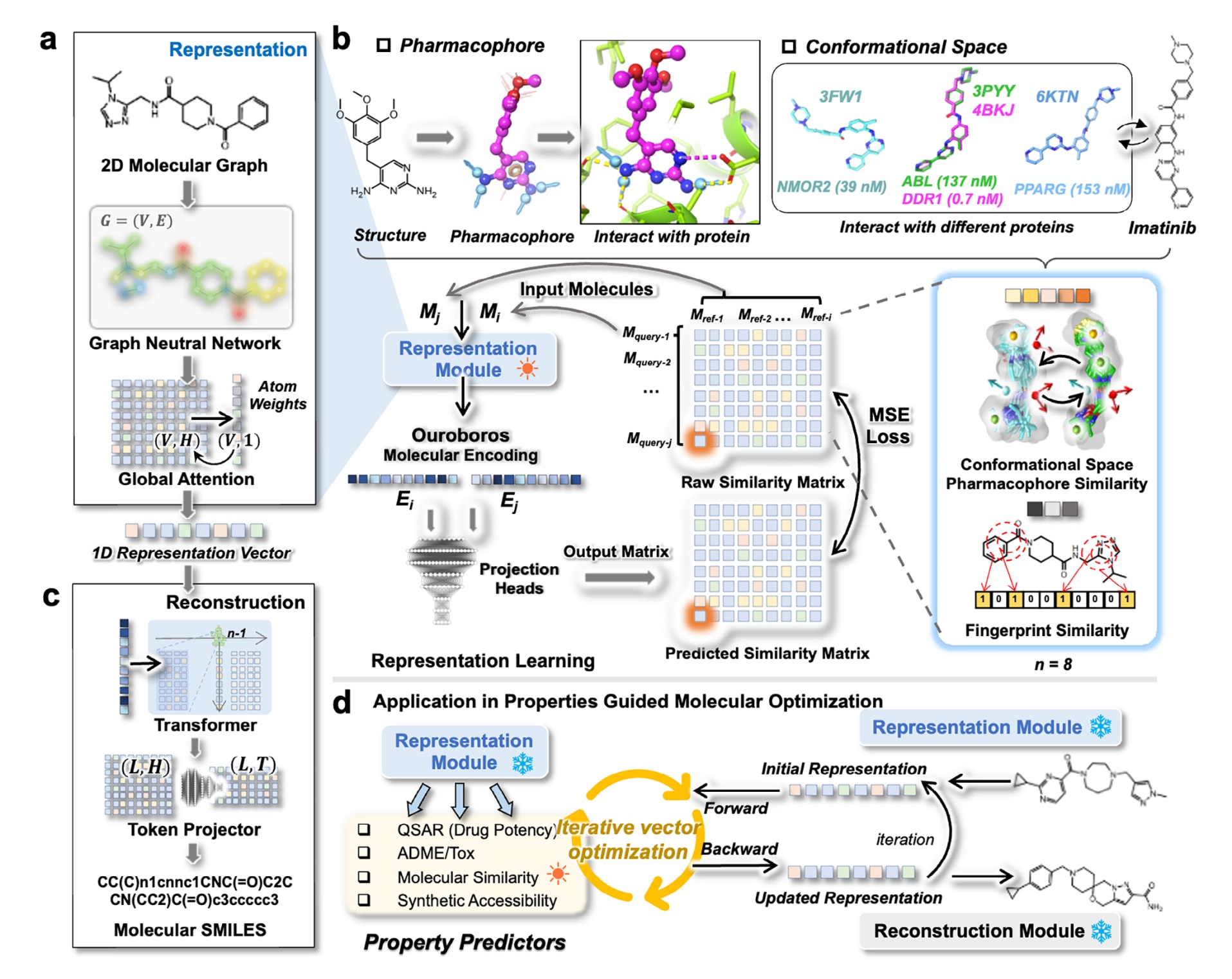
图1 | 展示了Ouroboros分子基础模型的动机与整体架构。 (a)表征模块通过图神经网络与原子级注意力机制将分子图转换为1D表示向量。(b)Ouroboros表征学习的动机在于,小分子的药效团与构象空间体现其执行药理功能的方式:在药效团视角中,氢键供体标记为天蓝色箭头,氢键受体标记为红色箭头,芳香环标记为橙色圆形或球体,疏水基团标记为绿色球体;在配体-口袋结合构象中,氢键以黄色虚线表示,盐桥以品红色虚线表示;伊马替尼的实验配体结构对应的PDB编号标注在分子构象旁;其中
2.1.2 从分子表示空间重构化学结构
以往研究中,化学语言的编码器与解码器通常采用联合训练,以保证能从隐藏层准确重构分子表示。但这种强耦合会给生成式AI中的分子表征学习带来困难,其中一个关键问题是:编码器-解码器模型中间隐藏层学到的信息,往往难以在多样的分子性质维度上保持良好的泛化能力。为了解决这一矛盾,该工作构建了一个独立的重构模块,使用基于Transformer的架构将编码得到的1D向量重构回分子的原始SMILES表示(图1)。具体而言,通过训练一个自回归Transformer解码模块,实现从1D表示向量到SMILES化学结构的转换(图S3)。该模块在大规模无标注化学结构数据上训练,数据量达到4820万,其目标是促使模型充分探索表示空间,从而提升分子生成能力。训练结果显示模型在单个epoch内即可快速收敛(图S4),这表明自回归Transformer解码器能够在token级别完成对SMILES的稳定重构。
2.1.3 验证集中超过80%的结构可被快速恢复
为了评估重构效果,图S5给出了验证集中真实分子结构与Ouroboros重构结构之间的AtomPairs指纹相似性分布。随着batch数量增加,平均相似度曲线很快稳定在80%的水平,说明该模型能够从1D表示向量中高效且稳健地恢复分子结构,并在结构重构任务上具有较强的可靠性。
2.2 Ouroboros的可解释性
2.2.1 原子注意力权重可视化
化合物中的官能团往往决定其关键性质,因此在表征模块中考察不同原子的注意力权重,有助于模型聚焦于重要官能团,并刻画不同原子对整体分子表示的差异性贡献。即使属于同一元素类型,只要药效团特征不同,图神经网络也会赋予不同的权重。例如,带正电的氮原子与处于酰胺基团中的氮原子会被区别对待(图S7a)。此外,当分子中出现体积较大的疏水基团时,模型会表现出更强的关注,从而强调这类基团在药效团表达与构象柔性中的重要作用。
2.2.2 表示向量的t-SNE分析
由于Ouroboros在表征学习阶段融合了构象空间与药效团信息,因此理论上能够区分具有不同构象动态或不同药效团特征的分子。基于这一动机,研究在不进行任何任务微调的前提下,对三个数据集上的Ouroboros表示向量进行降维分析。结果如图S7b所示,模型能够区分在血脑屏障渗透性、眼刺激性与腐蚀性方面潜力不同的分子。进一步来看,血脑屏障相关性质与分子柔性及形式电荷密切相关,而眼腐蚀与眼刺激则更多与酸碱性相关,这些关联提示了模型区分上述性质类别时可能依赖的内在化学机制。
2.2.3 扰动下相似度下降快于有效性
为了进一步评估重构模块在表示空间中探索潜在分子结构的能力,研究通过在解码过程中引入随机性,检验模型是否能够生成既有效又多样的分子结构。如图S6所示,随机解码采用了两类策略:其一是在分子表示向量上按照表示空间分布加入噪声进行扰动;其二是在token采样阶段引入随机性,使用Gumbel-max方法进行采样。随着温度与噪声水平的提高,原始分子与解码分子之间的相似度会逐步降低,这一趋势符合预期。更关键的是,相似度曲线的下降速度明显快于有效性曲线,其中有效性用于衡量解码分子中化学价键是否合理的比例。这表明模型能够在保持结构有效性的同时生成更新颖、更多样的分子结构,也再次体现出该重构模块能够从1D表示向量中准确重构分子结构,并具备良好的生成扩展能力。
2.3 将表征模块用于基于相似性的虚拟筛选
2.3.1 基准任务与数据集
该部分首先通过零样本相似度评估来检验表征模块的表示能力与泛化能力。零样本相似度评估的目标是识别结构相近且可能具有相似生物活性的分子。在小分子药物发现中,相似性评估可直接用于相似性虚拟筛选,通过检索与已知活性化合物相似的新分子来发现新的化学骨架。因此,分子表示向量的通用性与可迁移性,直接影响Ouroboros在药物发现与计算生物学任务中的应用能力。
在实验设定中,表征模块仅在相对有限的分子结构子集上训练,规模为126248个分子,但需要在更大规模的基准数据集上保持良好的泛化,包括DUD-E中的1224678个唯一分子与LIT-PCBA中的383893个唯一分子。评估时通过计算表示向量之间的余弦相似度,将Ouroboros与其他分子表征方法和分子基础模型、传统分子指纹,以及基于实验结构的药效团叠合基线方法进行对比,以检验其是否能够泛化到大规模基准集。
2.3.2 在117个单靶点上的相似性虚拟筛选
在两个虚拟筛选基准上,即DUD-E的102个靶点与LIT-PCBA的15个靶点,使用早期富集指标BEDROC对比多种方法后,Ouroboros相对6种基线方法展现出更强的表示能力(图2a)。结果显示,尽管训练所用结构数据集较小,该表征模块仍能在两个基准上取得较为均衡且较高的富集分数。除BEDROC外,在AUPRC、AUROC、富集因子EF与logAUC等指标上,Ouroboros在多数情况下同样优于或不弱于基线方法(表S2)。在分子基础模型基线中,MolFormer表现最佳,而ChemBERTa-2与MolT5的整体水平与分子指纹相近。由于MolFormer依托更大规模的预训练数据(如ZINC与PubChem)而具备竞争力,该工作进一步在DUD-E与LIT-PCBA上对Ouroboros与MolFormer进行逐靶点对比(图2b,c)。结果表明,Ouroboros在DUD-E上与MolFormer整体相当,且在不同靶点上呈现互补优势;在源自高通量实验筛选的LIT-PCBA上,Ouroboros在多数靶点上超过MolFormer。值得注意的是,Ouroboros仅使用约12万规模的分子数据进行预训练,这要求模型从相对少量结构中学习到可推广到广泛化学空间的原则,与依赖超大规模预训练数据的策略存在本质差异。
此外,由于引入了构象空间相似性,GeminiMol与Ouroboros在LIT-PCBA上能够超过分子指纹以及依赖构象的实验结构药效团方法PhaseShape。整体结果提示,Ouroboros的表示更擅长在表示空间中将具有相似药理特征的分子聚类到邻近区域,从而支持其作为独立工具用于小分子相似性筛选。
2.3.3 多靶点抗癌抑制剂的设计
基于表征模块中学习到的构象空间信息,以及其在DUD-E与LIT-PCBA上的表现,该工作将模型用于更具挑战性的多靶点药物发现情景,以评估其实用价值。考虑到晚期癌症常因异质性而难以治疗,该场景聚焦于多个癌症驱动基因相关靶点的多靶点抑制剂筛选与设计,例如KRAS、TP53、PI3Kα、SMAD4与ARID1A,目标包括合成致死或促进细胞增殖相关通路。其中特别强调PI3Kα与PI3Kγ作为原癌基因,并在KRAS下游增殖信号通路中发挥关键作用。AURKA与SMARCA4、ARID1A、RB1以及SMAD4等致癌基因存在合成致死关联,而这些基因突变在多种癌症中起驱动作用。
该工作期望Ouroboros能够从EnamineREAL多样性集合中识别结构新颖的候选抑制剂,并同时命中至少两个上述癌症驱动相关靶点。实验共设置10个驱动相关靶点(KRAS、PI3Kα与PI3Kγ、MEK1/2、PLK1、WEE1、CHK1/2、AURKA、PRMT5、PARP1/2)以及119个参考化合物。流程上先通过相似性筛选富集潜在活性分子,再结合分子对接挑选进入实验测试的候选物。结果显示,候选物不会与所有靶点的参考化合物同时相似,通常只对其中2到3个靶点表现出相似性;在这些靶点中,激酶类靶点更容易找到具有多靶点潜力的候选物。
2.3.4 实验鉴定潜在先导化合物
在酶活性测定中,共有18个分子成功合成并完成测试,其中7个化合物(38%)显示活性,3个化合物(16%)如预期表现出多靶点抑制行为(表S3)。这一命中率高于虚拟筛选命中率中位数13.3%。图S8汇总了3个命中分子的结果,这些分子与关注靶点(包括PI3Kα、PI3Kγ与AURKA)的活性参考化合物之间的最大相似度均超过0.5,并在酶学实验中体现出可观的抑制活性。
其中,化合物#13在三个靶点上均表现出更强活性,对AURKA的
为了进一步验证#13的活性与安全性,研究考察了其在乳腺癌细胞系MDA-MB-468、乳腺上皮细胞系MCF10A以及胚胎肾细胞系HEK293中的生长抑制作用。图2e显示,#13对癌细胞的
2.3.5 回顾性分析
为理解Ouroboros发现化合物的原因,以及不同靶点是否会在其构象空间中选择不同构象,该工作对#13在多个靶点中的结合模式进行了回顾性分析,以凸显引入构象空间信息的意义。图2f展示了#13在三个靶点口袋中的结合构象:1,3,8-三氮萘结构在所有靶点中均与铰链区形成氢键;而2-噁唑烷酮基团会发生翻转,并在不同靶点中与不同残基形成氢键,分别为AURKA中的T217、PI3Kα中的R770以及PI3Kγ中的K883。进一步对配体构象叠合(图2g)可见,2-噁唑烷酮在三种构象中均发生显著翻转,以支持其与不同残基侧链形成氢键。该现象体现了学习到构象空间信息的构象无关AI模型在多靶点药物发现中的独特优势,相较依赖单一构象的构象相关方法,更能解释并支持多靶点作用所需的构象适配。
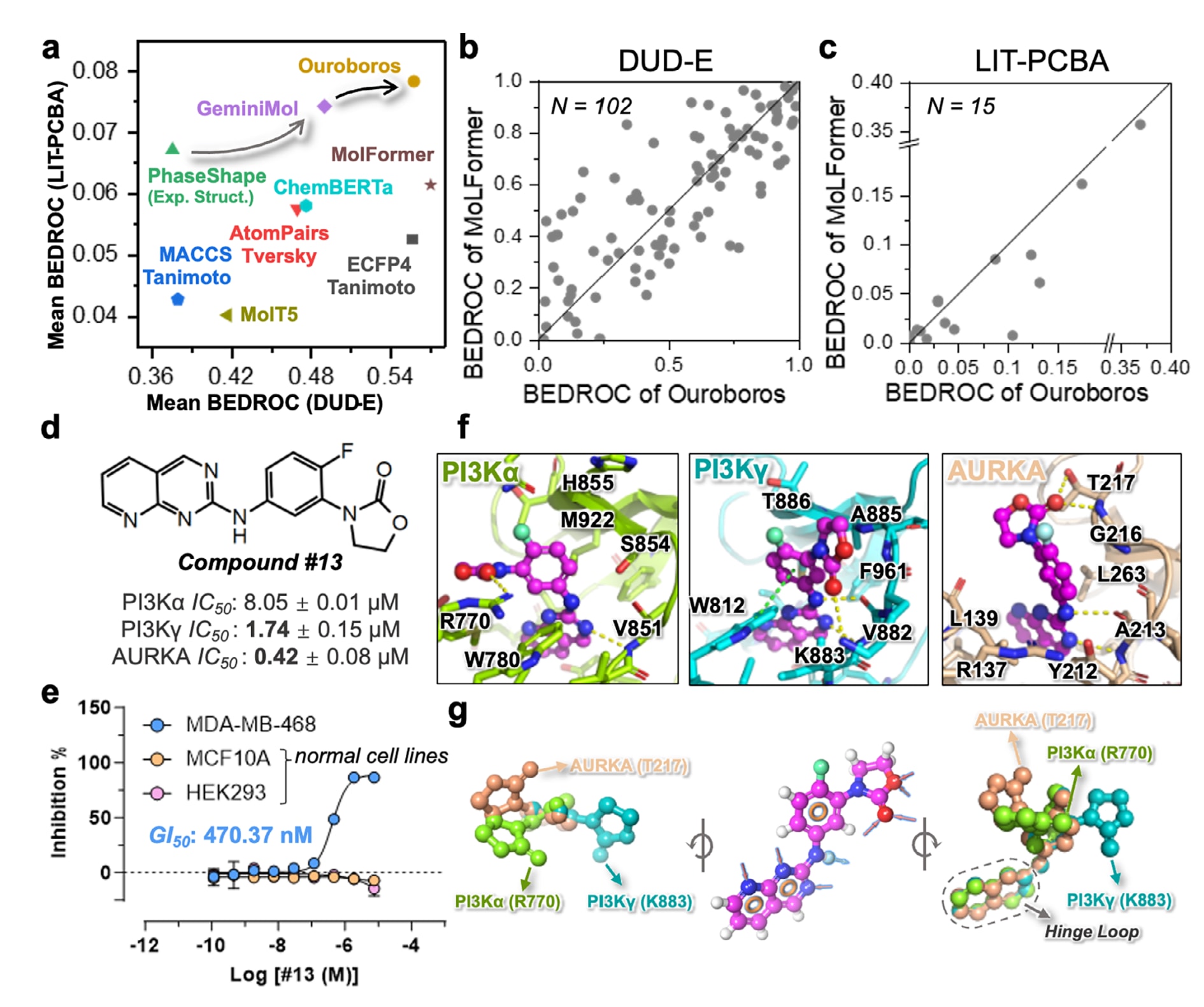
图2 | 展示了使用Ouroboros表示相似性进行虚拟筛选与定向多靶点设计的结果。 (a)在DUD-E的102个靶点与LIT-PCBA的15个靶点上,使用平均BEDROC比较多种方法的相似性虚拟筛选性能,包括Ouroboros、GeminiMol、PhaseShape(构象依赖)、ChemBERTa(ChemMLM表现最佳版本)、AtomPairs、ECFP4与MACCS。(b)在DUD-E的102个靶点上,Ouroboros与MolFormer逐靶点对比。(c)在LIT-PCBA的15个靶点上,Ouroboros与MolFormer逐靶点对比。(d)命中化合物#13对PI3Kα、PI3Kγ与AURKA的
2.4 基于表征模块的化学性质建模
2.4.1 通过投影进行分子性质建模
在Ouroboros中,预测得到的化学性质可作为优化过程中的损失函数,用于引导分子在表示空间中的定向迁移。基于回归的性质预测器能够提供连续的性质强度梯度,从而在优化时给出更直接、更可用的方向信号。对应实现方式是将分子表示向量输入深度神经网络,并映射到具体性质(图3a)。在评估上,该工作在10个与药代动力学与毒理学相关的分子性质数据集上,以相同数据划分对比Ouroboros与多种基线方法的测试集预测能力。图3b给出了在10个性质任务上的平均Spearman相关系数
2.4.2 与CombineFP的逐项对比
组合分子指纹被认为是性质建模中较为强力的传统方案之一,许多近期研究也表明,分子指纹与有效的机器学习模型结合后,能够展现出较强的泛化与预测能力。作为对照,图3c给出了Ouroboros与CombineFP的逐项对比结果。可以看到,Ouroboros不仅在脂溶性与溶解度等相对简单任务上表现突出,在半衰期与清除率等更具挑战性的任务上也取得了明显提升。这些结果进一步强调了Ouroboros表示的泛化性,而这种泛化性对于在多样化下游性质任务上训练有效的性质预测器至关重要。
2.4.3 通过微调进行分子性质建模
在独立下游任务上对预训练模型进行微调是一种常见的基准评测策略,但这一策略会牺牲Ouroboros在分子生成方面的独特优势。原因在于微调过程中,每个性质预测器会独立地优化表征模块,使得预训练得到的重构模块不再可用(图3d)。为了与其他化学语言类基线方法对齐比较,该工作也在微调模式下评估了Ouroboros的表现,结果见图3e与表S5。尽管模型本身并非为微调而设计,Ouroboros仍取得了所有方法中最高的平均表现0.687,其后为Uni-Mol的0.676。与此同时,Ouroboros也超过了专为分子性质建模设计的FP-GNN,后者通过将多种指纹与GNN融合来提升预测性能。进一步的逐项对比显示,Ouroboros与Uni-Mol在不同任务上各有优势,其中Ouroboros在清除率与血浆蛋白结合率方面表现更优(图S3f)。
2.4.4 与GeminiMol在细胞响应建模上的逐项对比
鉴于GeminiMol在药物诱导细胞响应建模中的优势,该工作进一步在药物反应预测任务上对比Ouroboros与GeminiMol,具体任务为NCI/DTP的60种细胞系上的细胞生长抑制
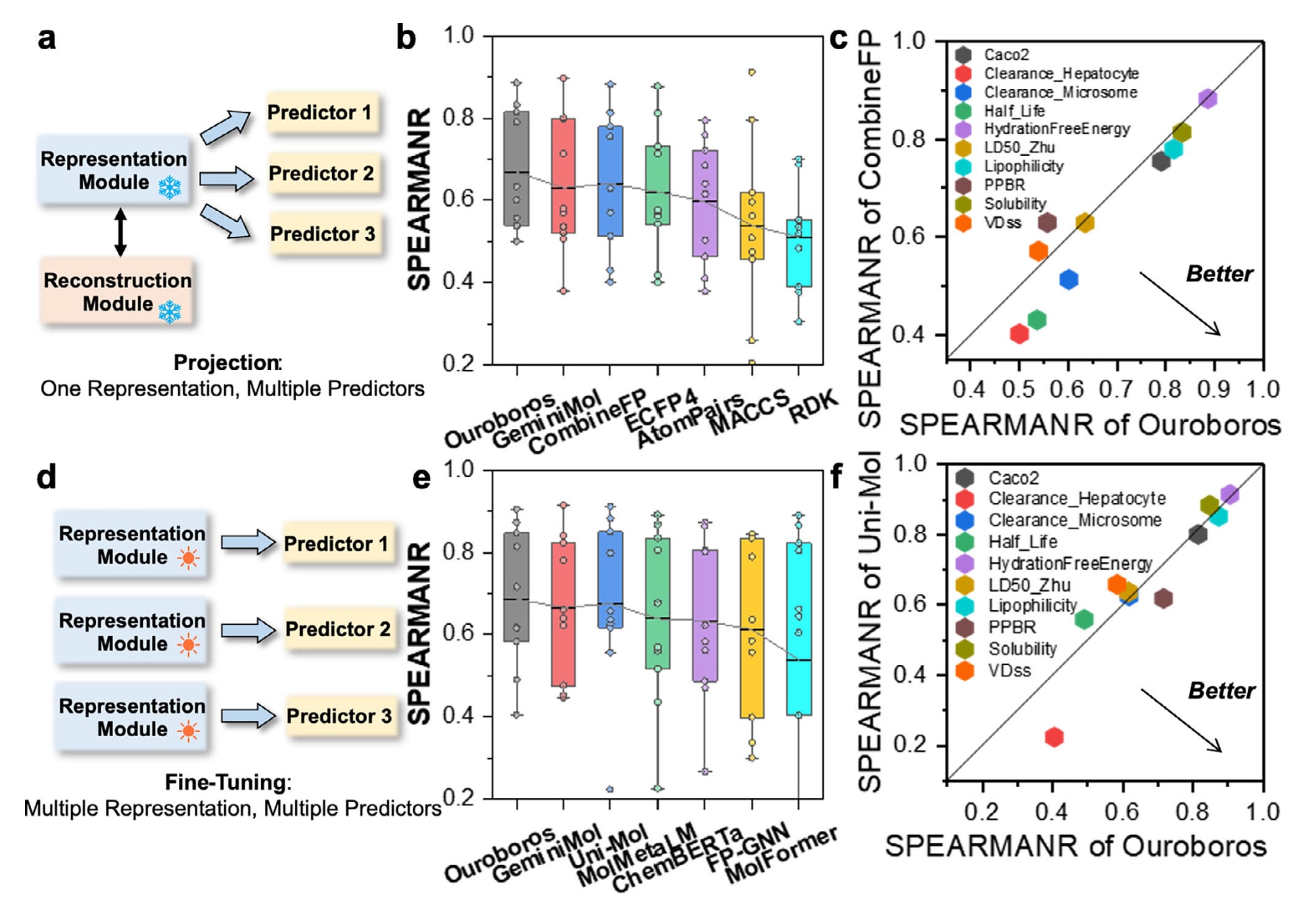
图3 | 展示了基于Ouroboros表征模块训练性质预测器的流程与结果。 (a)性质预测器的投影策略。(b)7种分子表示方法在10个回归性质任务上的Spearman相关系数对比,其中GeminiMol与Ouroboros的性质预测器均使用多层感知机MLP构建;CombineFP将AtomPairs、TopologicalTorsion、ECFP4与FCFP6四类指纹组合,并使用神经网络构建性质预测器。(c)Ouroboros在10个性质回归任务上的Spearman相关系数(以不同任务区分)与CombineFP的对比。(d)性质预测器的微调策略。(e)微调模式下,Ouroboros与GeminiMol、几何预训练基线Uni-Mol、三种化学语言基线MolMetaLM、ChemBERTa与MolFormer,以及QSAR基线FP-GNN的SpearmanR对比。(f)Ouroboros与Uni-Mol的逐项对比。
2.5 在表示空间中进行定向化学演化以实现分子生成
2.5.1 化学结构传播
为评估定向化学演化的有效性,该部分首先考察随机传播是否能够逐步改造起始分子的结构,并生成新的化学骨架(图S10a)。以阿司匹林为示例,图S10b显示,通过随机传播生成的分子结构会随着传播推进而逐渐偏离初始分子,但仍保持可观察到的相似性;与此同时,表示相似度会随传播过程持续下降。这说明在Ouroboros的表示空间中探索起始分子邻域,可以发现其结构类似物,从而支持将Ouroboros用于分子生成。
2.5.2 定向化学演化
与随机传播相对的是定向演化,其核心是通过损失函数引导表示向量的定向优化,进而实现化学结构的定向优化(图S10c)。作为示例,该部分展示了由两种特定性质引导的定向分子优化:水溶性与膜通透性。图S10d显示,从一个疏水分子出发,Ouroboros能够在保留整体结构相似性的同时生成溶解度更高的分子。类似地,从一个高度柔性且强亲水(因此通透性较差)的分子出发,Ouroboros生成了柔性更低、并带有正电荷的结构,其预测膜通透性得到提升。该结果表明,在演化过程中模型能够选择性地优化分子的特定性质。
2.5.3 定向迁移用于探索化学结构的转化路径
依托表征学习建立的表示空间,可以在两个化合物之间探索一条以药效团表征为线索的结构转化路径,用于药物骨架跃迁(scaffold hopping)。图4a给出了一个示例:Ouroboros在两个具有不同骨架的抑制剂之间构建表示向量迁移路径,两者分别为[1,3,5]-三嗪衍生物与5-杂环吡唑并吡啶,共同靶向
由于该框架强调重构模块对化学空间的忠实建模,因此在迁移路径上生成分子的
2.5.4 化学融合促进双靶点分子设计
在先前结果中,AURKA与PI3Kγ被证明属于更适合实现同时抑制的激酶靶点,这一发现促使研究进一步探索Ouroboros是否能够融合两组参考分子的药效团特征,从而生成新的双靶点抑制剂。具体做法是,一方面在EnamineREAL多样性集合上进行相似性筛选以寻找AURKA与PI3Kγ的潜在双靶点抑制剂,该集合同时也是重构模块的训练数据集;另一方面将相同的参考化合物输入Ouroboros进行化学融合,以便与相似性筛选结果进行直接对比。评估时保留化学融合过程中产生的部分融合分子,并要求其对两个靶点的最大相似度均超过0.65;在此基础上选取合成可及性排名前500的分子,与基于Ouroboros与ECFP4相似性筛选得到的前500个候选进行逐项对比。为进一步评估候选分子的开发潜力,研究进行了分子对接并计算
2.5.5 通过定向迁移进行先导化合物优化
得益于Ouroboros的正交架构,各类性质预测器都可以直接用于定向分子优化。该部分使用第三方评估工具QikProp对比随机传播与定向化学演化在性质优化上的表现。图4d,e展示了更大规模上的水溶性与膜通透性优化结果,选取了四个具有代表性的有机分子:两种膜通透性较低的分子(阿莫西林与地奥司明),以及两种疏水且水溶性较差的分子(起始结构见图中标注)。损失函数被设计为在保持与原始分子结构相似性的同时,共同提升溶解度、膜通透性与脂溶性(具体见Methods)。尽管对两类性质欠佳的分子使用相同损失函数,定向迁移仍能生成多种在膜通透性或溶解度上显著改善的新分子,并将表示相似度维持在0.6以上。这些结果体现了Ouroboros定向迁移在多目标分子性质优化中的通用性与有效性。
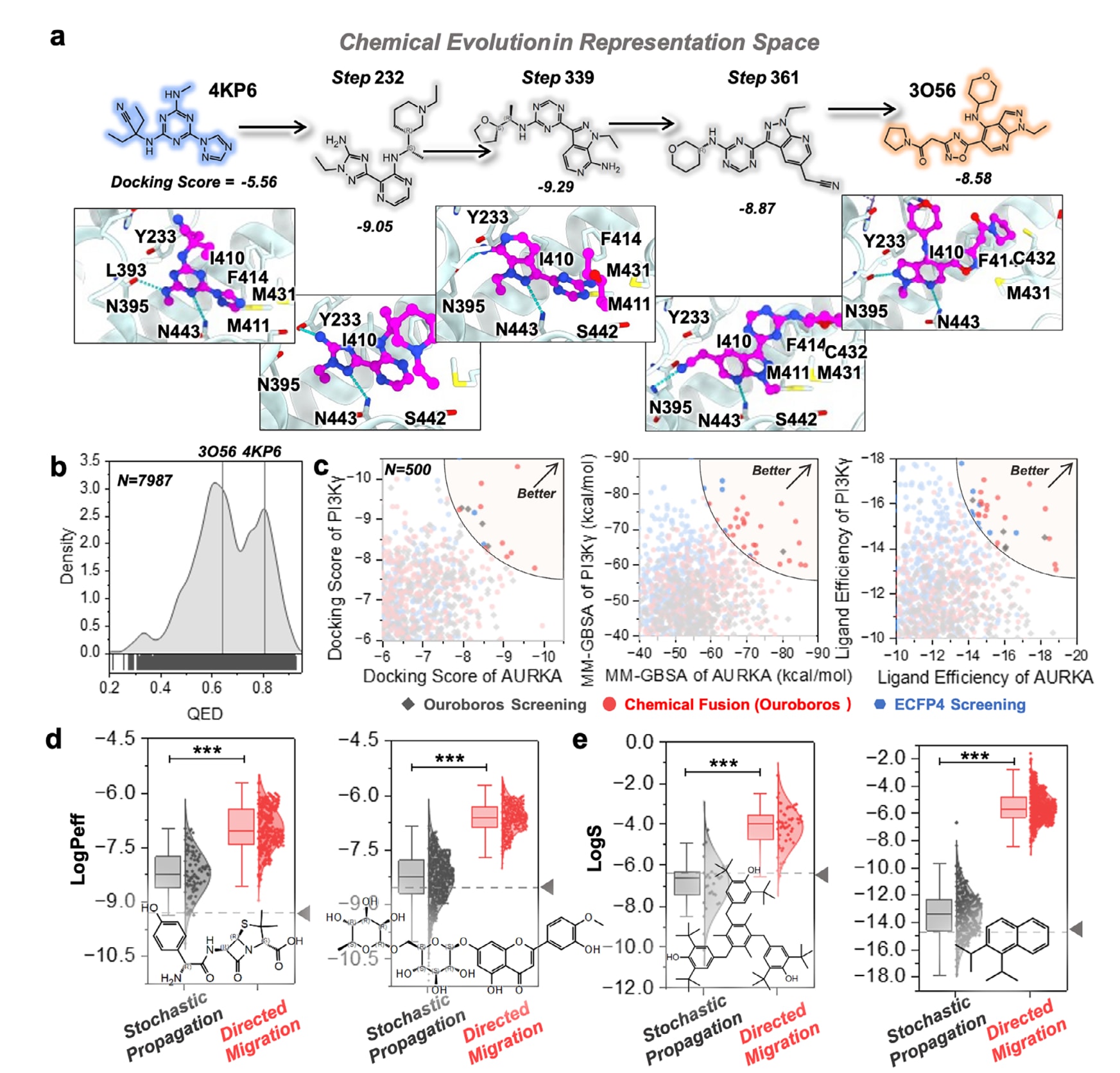
图4 | 展示了在Ouroboros中探索表示空间的实例。 (a)从一个PDE4B抑制剂向另一个抑制剂的化学迁移,对接分数使用GlideSP计算,配体的C原子以品红色显示。(b)定向迁移过程中生成分子的
3 总结
该工作提出分子基础模型Ouroboros,其核心特点是采用正交架构,将三个相对独立的模块拆分开来并分别选择更合适的训练策略与模型结构,包括化学分子表征、由表示向量重构分子结构,以及面向特定生物问题的化学性质关系建模。基于这种拆分,表征模块可以使用GNN与相似性学习,重构模块则采用基于自回归Transformer的化学语言模型。随后,模型能够围绕实际应用训练性质预测器,并将这些预测器用于构造损失函数,以梯度方式优化表示向量,最终再重构得到经过优化的分子结构。由于各模块彼此解耦,性质预测器也能针对不同任务灵活选择合适模型;虽然基准测试中统一使用深度神经网络,但后续仍可通过引入更多信息进一步扩展,例如加入靶标嵌入等以增强性质建模能力。
在表征层面,该工作引入的相似性学习策略显著提升了数据效率,使模型能够在不足150000个分子的紧凑数据集上完成有效预训练,同时在DUD-E与LIT-PCBA等超过百万分子规模的大型基准上取得突出的虚拟筛选表现。其现实可用性也通过多靶点药物发现得到进一步验证:在4820万规模的多样性化合物库中成功识别出3个新的多靶点抑制剂,显示出跨化学空间的强泛化能力。其中化合物#13被认为具有多靶点先导潜力,其分子量较低但能通过构象变化与不同靶点残基发生差异化相互作用,从而实现预期的多靶点效应。
在生成层面,重构模块使用自回归Transformer解码器从1D表示向量重建SMILES序列,并由此支持两种生成策略:随机传播与定向化学演化。基于分子性质优化与双靶点抑制剂设计的基准评估表明,该模块具备更优表现。在这一框架下,Ouroboros在分子表征学习与生成式AI之间建立了更直接的连接,并在表示空间中提出连续化学演化的新范式。除了文中展示的两类策略外,该框架还能够整合其他AI驱动模型,将其作为损失函数来进一步优化分子性质,从而扩展到更广泛的生成与优化任务。
尽管取得了显著进展,该框架仍存在若干挑战与发展方向。首先,Ouroboros并不直接预测分子的动态3D构象,而这被认为是一个值得进一步推进的方向。该设计选择与小分子构象本身的动态性与多样性有关,因为可旋转键数量与环境条件差异会带来丰富的构象集合,因此对分子动态构象空间的预测仍是重要且有潜力的后续工作。其次,虽然模型在多种分子性质预测上表现突出,当前基准仅覆盖10种性质,后续仍有较大拓展空间。与此同时,针对特定生物靶点生成具有强结合亲和力的分子也正受到越来越多关注,但当前Ouroboros并不直接预测药物-靶标结合亲和力,而是依赖分子对接在迁移路径上筛选对接得分与结合姿势更优的分子。这也进一步凸显出将蛋白质表征纳入训练、构建药物-靶标亲和力预测模型的需求,以持续增强Ouroboros框架的能力边界。