SA 2024 | 用于增强模式发现的蛋白质-蛋白质相互作用网络深度表示学习
今天介绍的是发表于Science Advances的研究,聚焦于蛋白质-蛋白质相互作用网络中模式发现这一长期存在的核心问题。PPI网络结构复杂、非线性强,传统网络嵌入方法往往只能刻画局部邻域关系,难以同时反映节点在整体网络中的功能角色。该工作提出了一种自监督的深度表示学习方法DNE,通过对比节点邻居与远距离节点的表示,在同一框架下融合局部连接信息与全局网络结构,从而获得更具判别力的节点嵌入。系统评估结果表明,DNE在多个物种的PPI数据集中,在相互作用预测和功能模块识别任务上均显著优于现有方法。同时,DNE具备良好的鲁棒性,并可灵活整合来自蛋白质语言模型的序列特征,进一步提升预测性能。该研究不仅为高质量PPI网络分析提供了新思路,也为疾病基因预测和蛋白质功能注释等应用奠定了方法基础。

获取详情及资源:
0 摘要
蛋白质-蛋白质相互作用网络(PPI网络)以蛋白质作为节点,以不同形式的相互作用作为边,是理解生物系统内部动态机制的基础框架。尽管PPI网络在现代生物学研究中具有核心地位,但由于网络结构高度复杂且关系交织,从中可靠地识别潜在模式仍然是一项重要挑战。问题的关键在于,如何在整体层面刻画单个节点与网络中其他节点之间的关系,并将这些信息有效转化为可用于模式发现的表示。
该研究提出了一种自监督的网络嵌入框架,称为判别式网络嵌入(discriminative network embedding,DNE)。不同于传统方法主要依赖节点之间的直接邻接关系或有限阶的局部邻域信息,DNE通过对比相邻节点与远距离节点的表示,同时在局部和全局尺度上刻画节点特征,从而获得更具判别力的表示。
实验结果表明,DNE在多项关键网络分析任务中均优于现有方法,包括PPI关系推断以及蛋白质功能模块的识别。这些结果说明,DNE是一种稳健且有效的PPI网络节点表示策略,为多种生物医学应用提供了新的可能性。
1 引言
生物网络通过系统化刻画基因、蛋白质及其他细胞组分之间纷繁复杂的相互作用,为建模复杂生物系统提供了关键框架。这类网络通常将生物实体表示为节点,将从物理相互作用到功能关联等多种关系表示为边,为解析生物系统结构与过程的复杂性奠定了基础。以蛋白质-蛋白质相互作用网络(PPI网络)为例,其中高度交织的连接关系蕴含着理解细胞过程与疾病机制的重要信息。然而,如何从这些结构复杂的网络中提取有意义的生物学规律,始终是一项具有挑战性的任务。
网络嵌入是一种常用的分析手段,其目标是在保留网络结构特性和节点关系的前提下,将网络中的节点映射为低维向量表示,从而实现模式识别和下游分析。网络嵌入的准确性在很大程度上决定了后续分析和应用的效果。生物网络的底层结构普遍具有高度非线性,这是由复杂且非加性的相互作用所导致的,同时还同时包含局部结构,即节点的直接连接关系,以及高阶结构,如网络中的聚类特性。尽管已有大量研究致力于开发能够应对这种复杂性的网络嵌入方法,但在实际应用中仍难以获得理想的解决方案。
传统的网络嵌入方法多通过矩阵分解或浅层模型来刻画节点之间的邻近关系,但这类方法往往依赖低秩近似或对网络结构的简化假设,难以充分捕获高度非线性的网络模式,从而限制了嵌入表示的质量。近年来,基于深度学习的方法开始被引入网络表示学习领域,通过多层非线性变换来刻画复杂的网络结构。例如,变分图自编码器通过图神经网络聚合节点邻域信息,在一定程度上增强了节点表示对非线性结构的表达能力。然而,该类方法主要关注节点的局部邻域模式,在理解跨越整个网络的节点关系方面仍然存在局限。
为弥补这一不足,已有方法尝试引入全局结构信息。例如,Deep Graph Infomax通过将节点表示与全局图摘要对齐来保留整体结构特征,但其侧重全局信息,可能忽略局部层面的精细结构。Deep Graph Contrastive Representation Learning则通过对图结构进行数据增强,学习对扰动不变的节点表示,从而间接捕获全局信息,但其效果在一定程度上依赖于数据增强策略的质量。
在此背景下,该研究提出了一种通用的图表示学习框架,利用深度学习在低维空间中同时保留网络的非线性与多层次结构,以支持高性能的生物网络分析。该方法被称为判别式网络嵌入(discriminative network embedding,DNE),其核心思想是通过对比节点直接邻居与网络中远距离节点的表示,以非线性的方式刻画每个节点的特征。该框架能够从整体角度理解节点在网络中的角色,既突出节点的直接相互作用,如PPI网络中的蛋白质相互作用,也刻画其在更大尺度上的社区归属,例如蛋白质功能模块。
实验结果表明,DNE在多种网络和多项下游任务中均显著优于现有网络嵌入方法,包括链接预测,即PPI关系预测,以及节点聚类,即功能模块识别。此外,DNE具有良好的灵活性,能够将节点自身特征与网络结构信息相结合以进一步提升性能。通过引入基于蛋白质语言模型的蛋白质序列特征,DNE在下游任务中的表现相比传统方法获得了显著提升。研究还表明,DNE不仅适用于PPI网络,也可推广至其他类型的网络。总体而言,该方法为网络分析提供了一种新的思路,有望在生物医学数据科学领域产生广泛影响。
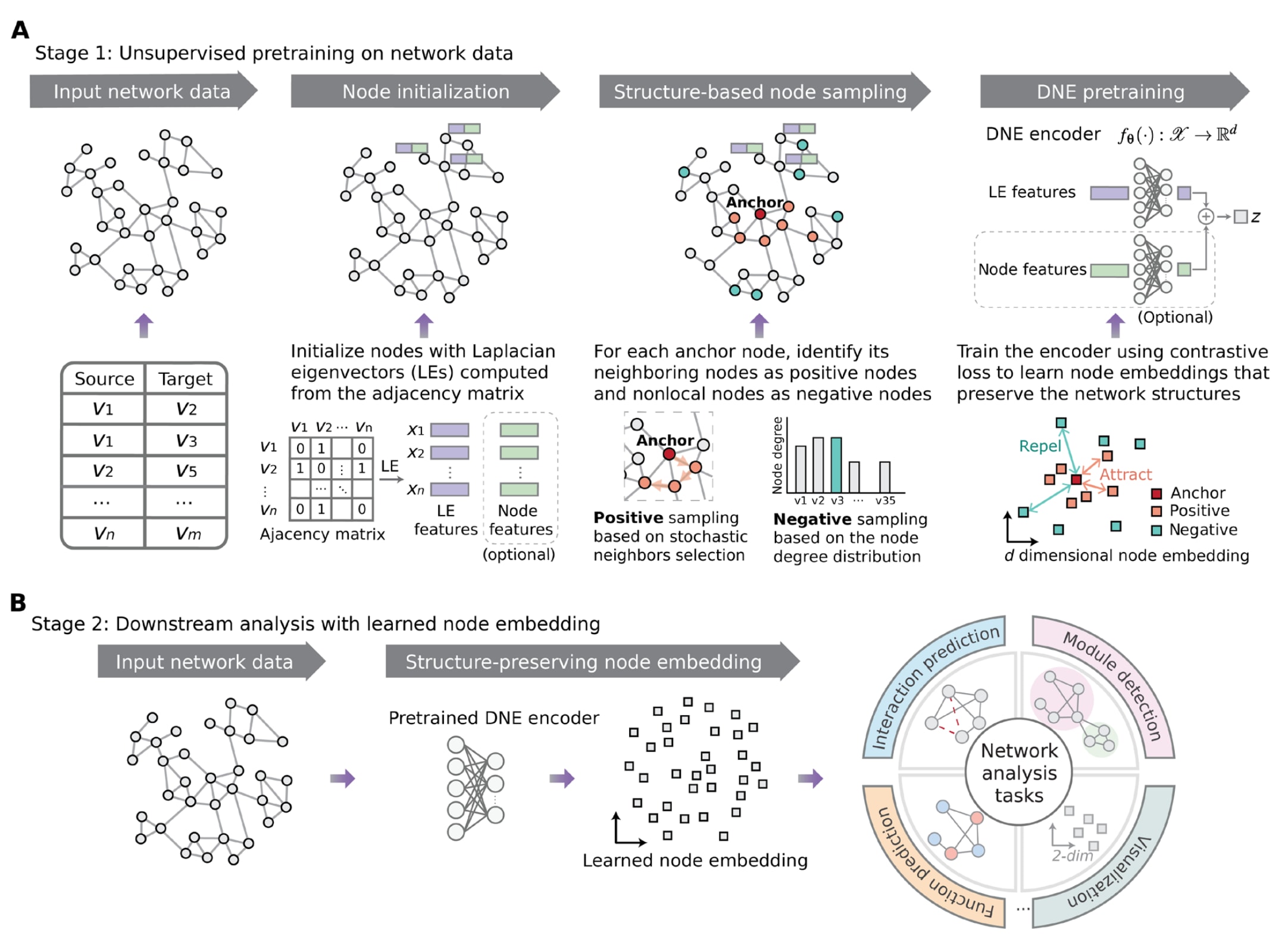
图1|DNE总体框架示意图。 (A)DNE主要包括三个步骤:(i)使用网络邻接矩阵的拉普拉斯特征向量对节点进行初始化,在节点特征可用的情况下,可将其与初始化表示进行拼接;(ii)通过随机化的邻居选择策略将节点的邻居定义为正样本节点,并根据节点度分布从网络其他区域选取负样本节点;(iii)利用深度学习编码器对每个节点进行嵌入表示学习,通过优化编码器参数,使得到的节点表示能够有效区分相邻节点与非局部节点。(B)将训练完成的编码器用于生成节点表示,以支持多种下游分析任务。
2 结果
2.1 DNE在PPI链接预测任务中持续优于现有网络嵌入方法
首先,研究通过链接预测任务验证了DNE的有效性。链接预测旨在仅基于已知的网络结构信息,评估网络中潜在边存在的可能性。为此,研究将DNE与多种主流算法在蛋白质-蛋白质相互作用网络中的预测性能进行了系统比较,所使用的PPI数据集包括四个典型互作组:(i)来源于拟南芥的植物互作组,包含2774个蛋白质和6205条PPI;(ii)基于秀丽隐杆线虫的线虫互作组,包含2528个蛋白质和3864条PPI;(iii)来源于酿酒酵母的酵母互作组,包含2674个蛋白质和7075条PPI;(iv)来源于HuRI的人类互作组,包含8272个蛋白质和52548条PPI。对于每一个互作组,随机抽取20%的边作为测试集并从网络中移除,其余数据用于训练,随后在训练数据上进行五折交叉验证以获得最优性能,整个过程重复10次独立实验。
研究将DNE与包括DGI、GRACE、VGNAE、VGAE、Node2Vec、GraRep、HOPE、LINE、NetMF、LLE和SVD在内的11种网络嵌入方法进行了对比,性能结果如图2所示。在拟南芥数据集的链接预测任务中,DNE在精确率-召回率曲线下面积(PR-AUC)和受试者工作特征曲线下面积(ROC-AUC)两项指标上均取得最高得分。具体而言,DNE在10次重复实验中的平均ROC-AUC达到88.05%,相比次优方法提升约4%。此外,DNE在所有测试的PPI网络中均表现出稳定优势,无论是在网络规模、节点度分布还是网络密度存在差异的情况下,均能保持较高的预测性能。相比之下,其他方法在不同数据集上的性能排名波动较大。DNE在多次实验中的一致性和整体性能提升,表明其能够有效捕获PPI网络的结构信息。
进一步地,研究还将DNE与五种基于启发式规则的链接预测方法进行了比较,包括公共邻居、Jaccard指数、Katz指数、优先连接和资源分配指数。这些方法主要依赖节点相似度进行预测,其整体表现未能达到理想水平。相比之下,DNE在拟南芥和秀丽隐杆线虫网络中的ROC-AUC指标均比这些启发式方法高出8%以上。这一显著差距说明,仅依赖预定义的相似性度量在预测新的蛋白质相互作用时存在明显局限性。例如,在复杂的生物网络中,节点拥有公共邻居并不必然意味着两者之间存在直接相互作用。
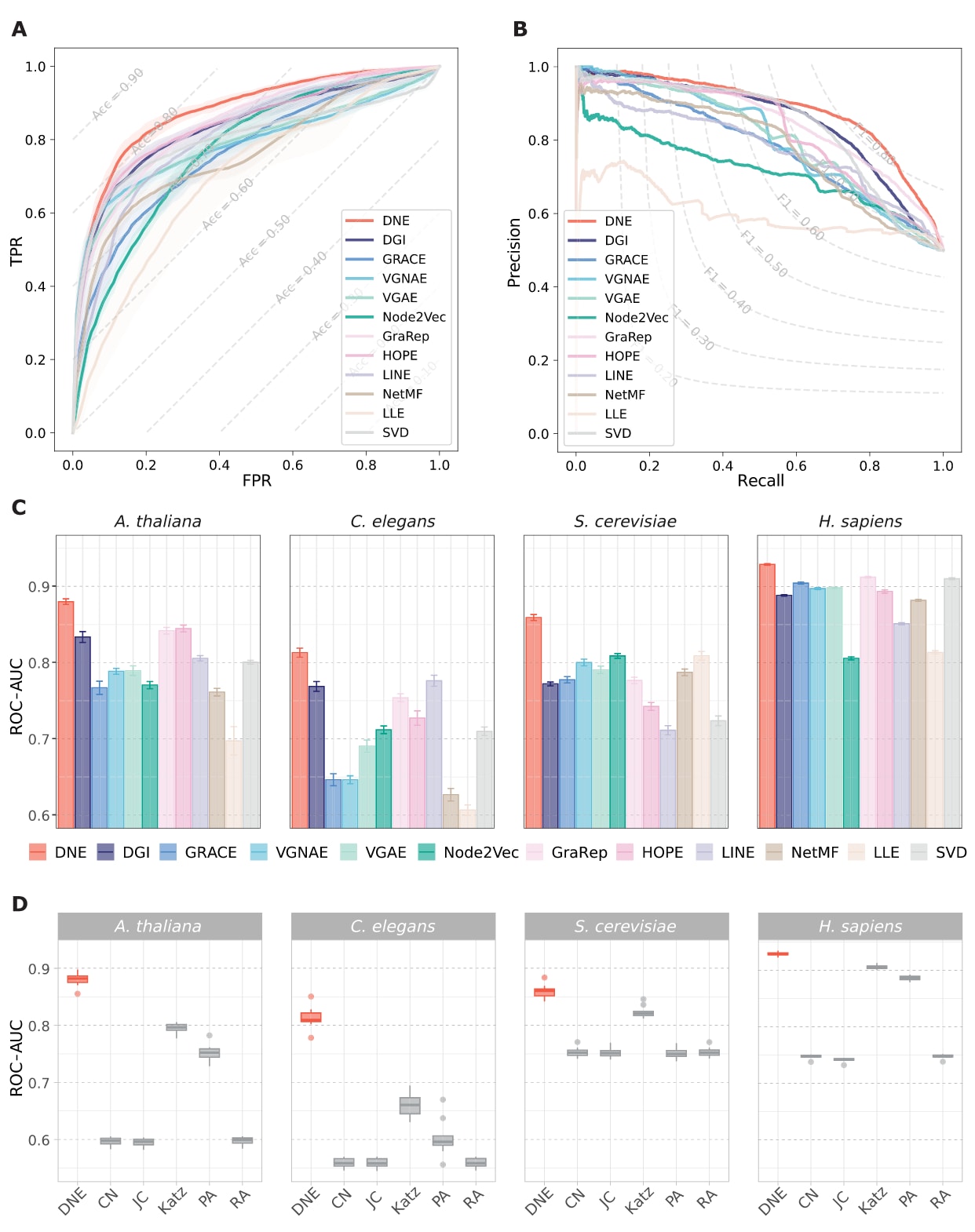
图2|不同方法在四个PPI基准数据集上的链接预测性能。 (A)在拟南芥数据集上,DNE与其他11种网络嵌入方法在PPI预测任务中的ROC曲线,(B)对应的PR曲线。(A)和(B)中的虚线分别表示准确率和F1分数的等值线。(C)DNE与多种网络嵌入方法在四个PPI基准数据集上的性能比较,结果展示了10次独立实验中ROC-AUC的均值及标准差。(D)DNE与基于相似性的链接预测方法在四个PPI基准数据集上的性能比较,展示了10次实验得到的ROC-AUC结果,箱线图中间的横线表示均值,箱体边缘分别表示第一和第三四分位数,须线延伸至四分位距的±1.5倍范围内。
2.2 DNE能够有效识别PPI中的功能模块
在PPI及其他生物网络中,模块识别是一项关键任务,其目标是发现由紧密相互连接节点构成的簇,其中每一个簇通常对应一组具有相似功能的蛋白质。研究以酿酒酵母的PPI数据为例,评估了DNE在功能模块识别中的表现。由于酿酒酵母的蛋白质复合物具有较为完善的生物学注释,该物种为网络聚类方法的评测提供了理想测试平台。研究采用IntAct蛋白质复合物、KEGG通路以及GO生物过程作为参考标准。图3对比了多种网络嵌入方法在上述三个模块检测基准上的表现。
在实验中,不同的网络嵌入方法首先将蛋白质表示为连续向量,随后基于这些表示采用层次凝聚聚类方法识别PPI中的功能模块。评估指标采用调整互信息(AMI),用于衡量预测得到的模块与酿酒酵母中已注释复合物之间的一致性。结果显示,DNE在蛋白质复合物预测任务中表现突出,其平均AMI得分显著高于其他方法。具体而言,DNE相比Node2Vec和NetMF在AMI上提升约2%,相较于其余基线方法的提升幅度更为显著,达到10%至50%的区间。此外,研究还利用Jaccard指数评估已知蛋白质复合物与预测模块之间的重叠程度,并比较了DNE与六种代表性基线方法在各个复合物上的差异。结果表明,DNE不仅能够识别更多的功能模块,而且在模块重叠度方面也具有更高的得分。
为进一步解析DNE在模块识别中的优势,研究对Retromer复合物进行了案例分析。该复合物由PEP8、VPS35、VPS29、VPS17和VPS5等基因组成,在液泡蛋白分选过程中发挥关键作用。DNE通过学习得到的嵌入表示成功捕获了该复合物的全部成员,而其他方法往往只能识别其中一部分成员,或引入无关蛋白。该结果表明,DNE能够生成具有生物学意义且较为准确的节点表示,有助于蛋白质功能的推断。
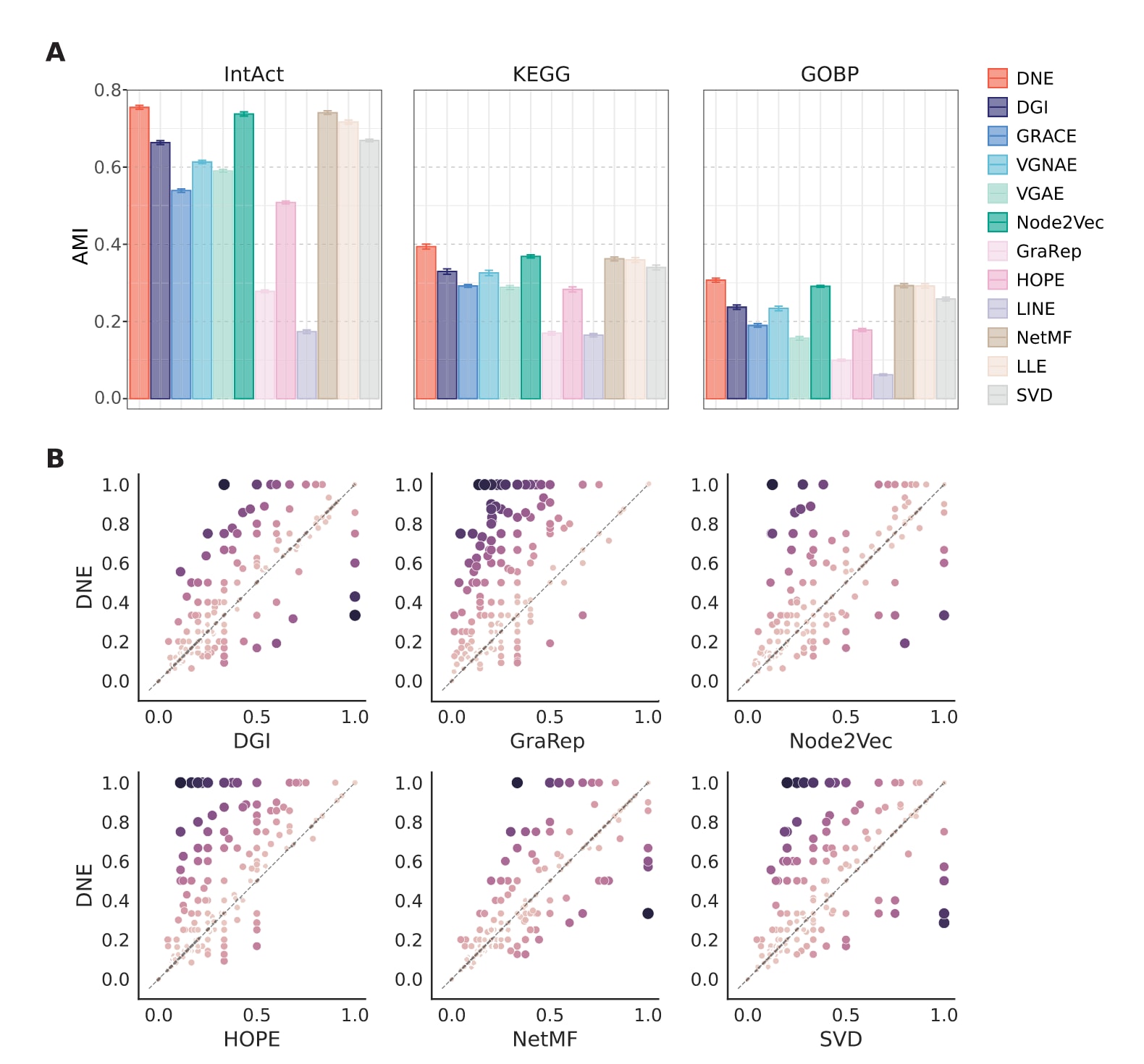
图3|不同网络嵌入方法在功能模块识别任务中的性能比较。 (A)以IntAct、KEGG和GOBP中注释的蛋白质复合物作为参考标准,基于10次独立实验计算得到的AMI得分,图中给出了平均值,误差线表示标准差。(B)DNE与六种代表性基线方法在单个模块层面的Jaccard得分比较,每一个点对应一个蛋白质复合物。横轴和纵轴分别表示指定基线方法和DNE得到的模块重叠度(Jaccard)得分,得分为0表示该复合物的成员未被识别,得分为1表示该复合物的所有成员均被完整捕获。点的颜色和大小表示DNE与其他基线方法在对应复合物上的Jaccard得分差异。
2.3 DNE支持整合来自蛋白质语言模型的蛋白特征
与许多仅关注网络结构信息的网络嵌入方法不同,DNE在设计上具备将节点特征融入嵌入过程的灵活性。在该研究中,作者从酿酒酵母基因组数据库获取对应的蛋白质序列,并利用预训练的蛋白质语言模型ESM-2将其转换为蛋白特征。这些特征包含丰富的蛋白质语义信息,随后作为节点特征引入PPI网络中。之所以选择该数据集进行评估,是因为其为网络中的每一个蛋白质提供了完整的序列信息,而其他基准数据集往往不具备这一条件。
为评估DNE整合节点特征的能力,研究设置了三种不同情形进行比较,分别仅使用ESM-2提取的蛋白特征、仅使用网络结构信息,以及同时结合网络结构与蛋白特征。结果显示,在酿酒酵母数据集的PPI预测任务中,相较于仅使用蛋白特征的情况,DNE在ROC-AUC指标上提升超过20%。同时,DNE在有无节点特征的两种设置下均稳定优于DGI、GRACE和VGNAE等基线方法。进一步比较表明,在引入蛋白特征后,DNE的ROC-AUC相较于不使用特征的情形仍提升约1.3%。这些结果说明,DNE能够有效融合蛋白质序列特征与网络结构信息,从而显著提升蛋白质表示的质量。
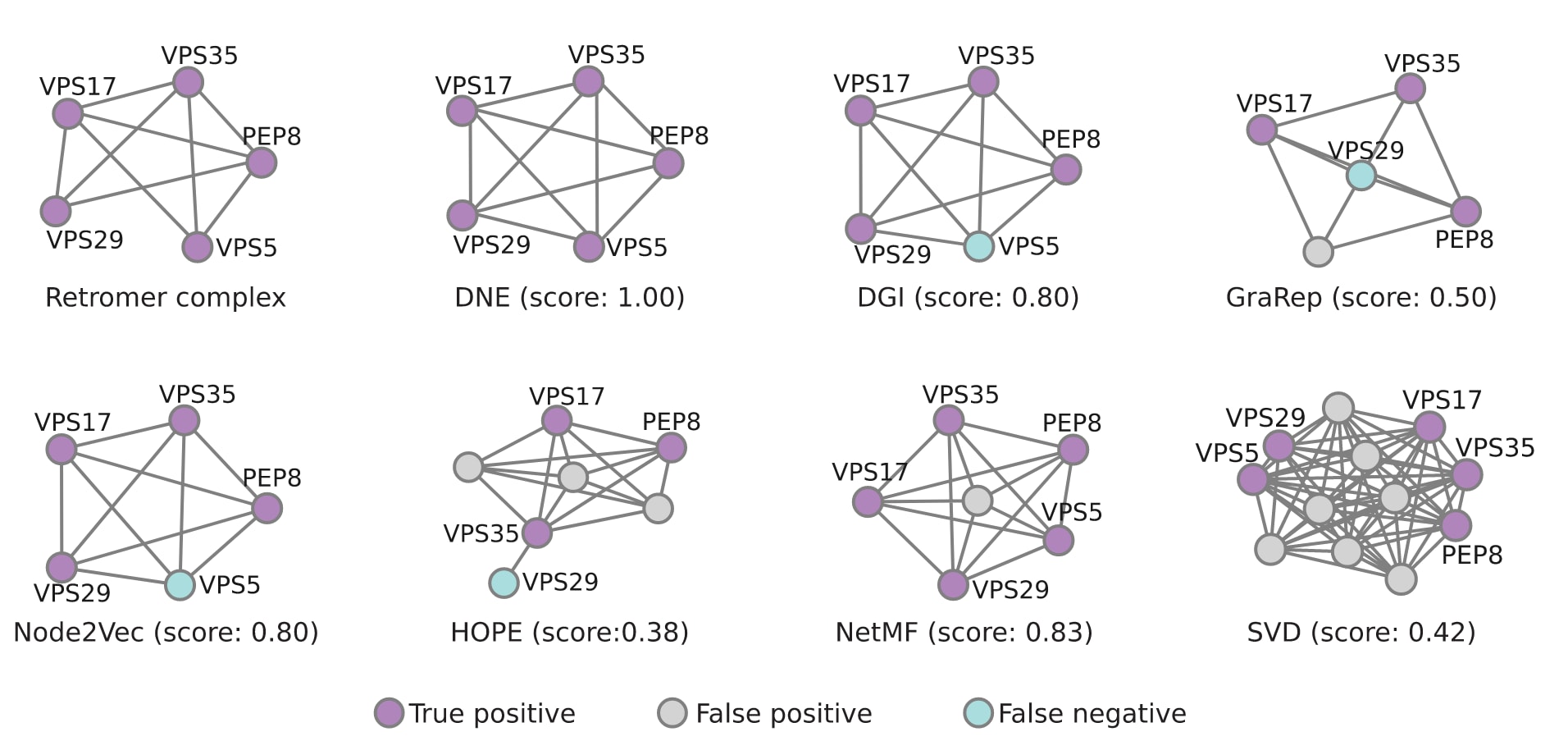
图4|预测复合物与标准Retromer复合物之间重叠程度的评估。 以IntAct注释的Retromer复合物作为基准,用于评估不同方法在功能模块识别任务中的表现。该标准复合物包含五个成员:PEP8、VPS35、VPS29、VPS17和VPS5。预测得到的复合物与标准复合物之间的重叠程度通过Jaccard指数进行衡量。图中紫色表示预测成员属于标准复合物,灰色表示预测成员不属于标准复合物,绿色表示标准复合物中的成员未被预测方法捕获。
3 讨论
研究提出了一种名为DNE的网络嵌入方法,用于从给定网络中学习具有判别力且富有生物学意义的节点表示。与传统主要关注有限阶节点邻近关系的方法不同,DNE通过对比节点直接邻居与网络中远距离节点的表示来刻画节点特征,从而同时考虑局部连接模式与节点在整体网络中的相对位置。这种设计使得节点关系能够在更全面的网络背景下被理解。
在多个PPI数据集上的系统评估表明,DNE在蛋白质相互作用预测和功能模块识别等任务中均显著优于现有方法。同时,DNE在面对网络扰动时表现出较强的鲁棒性,在不同扰动比例下均能保持稳定优势。此外,通过嵌入表示,DNE能够有效反映蛋白质在PPI网络中的n跳距离关系以及其在Gene Ontology功能相似性上的接近程度,说明该方法能够捕获具有生物学意义的信号。
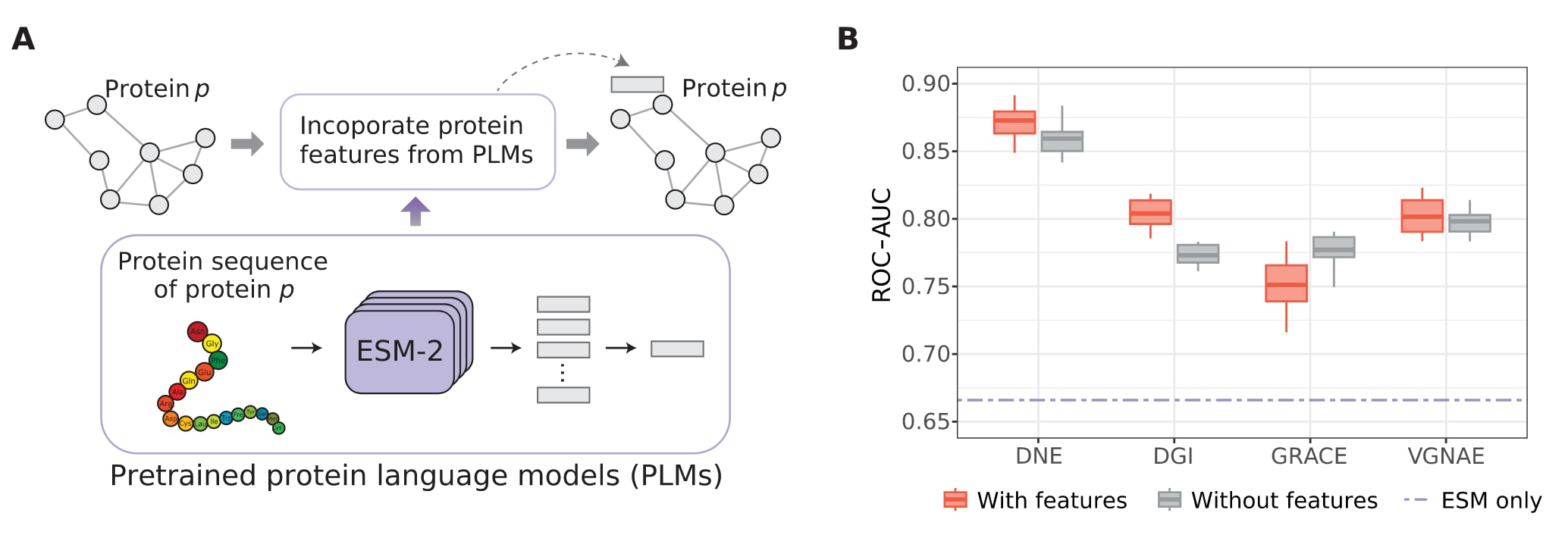
图5|融合蛋白质特征的链接预测性能比较。 (A)将来自蛋白质语言模型的蛋白质特征作为节点特征引入PPI网络,用于网络嵌入学习的示意。(B)在酿酒酵母数据集上,DNE及其他基线方法在10次独立实验中得到的ROC-AUC得分比较。紫色虚线表示仅使用ESM-2提取的蛋白质特征的情形(仅ESM)。灰色箱线表示仅考虑网络结构信息的情况,红色箱线表示同时结合网络结构与节点特征的情况。
尽管DNE可以仅依赖网络结构信息生成节点嵌入,但在节点特征可获得的情况下,该方法同样支持将其纳入嵌入过程。在PPI等生物网络中,节点通常对应蛋白质,其特征可来源于多种信息,包括氨基酸序列、蛋白质三维结构以及亚细胞定位等,这些信息能够补充网络拓扑之外的蛋白质属性。通过整合来自预训练蛋白质语言模型的序列特征,DNE在PPI预测中的性能显著优于仅依赖序列信息的方法,显示出该策略在提升预测精度方面的优势。
总体而言,DNE在网络分析中具备多方面优势。首先,该方法生成的嵌入具有更强的判别性,不仅刻画节点的局部连接特征,还能够将其与网络其他区域加以区分,从而更准确地反映节点的结构角色和社区归属,并降低对局部噪声的过拟合风险。其次,通过同时整合邻域信息与更广泛的网络上下文,DNE为网络提供了更加整体性的表征。再次,DNE能够同时利用网络结构和节点特征,生成信息更加丰富的嵌入表示。在该研究中,这些表示被用于推断蛋白质相互作用和识别功能模块,未来还可拓展至疾病相关基因预测以及蛋白质功能预测等应用场景。
值得注意的是,DNE的适用范围并不限于PPI网络。在引文网络、电力网络以及互联网服务提供商网络等多种类型的网络中,初步实验结果同样显示出其良好的通用性。因此,DNE在网络嵌入方法上代表了一项重要进展,为高性能网络分析提供了一种迫切需要的解决方案。
同时,该方法仍存在进一步改进的空间。首先,当前模型在设计上仍以网络结构信息为主,节点特征主要用于初始化嵌入表示。未来可以在上下文节点采样过程中,同时考虑节点特征相似性与网络连接关系,以更充分地融合多源信息。其次,现有方法采用多层感知机作为编码器,后续研究可探索引入图神经网络等结构,以进一步提升表示能力。
生物网络,尤其是PPI网络,是理解复杂生物系统的重要基础,但其内在复杂性也为分析和下游应用带来了挑战。该研究提出了一种自监督的网络嵌入方法,旨在为高维网络数据提供更加判别性的低维表示。该方法通过同时利用节点的局部环境和全局网络背景,有效捕获节点的内在特征。大量实验结果表明,这种双重视角能够生成全面且稳健的网络表示,从而支持可靠的模式发现和准确的下游分析。因此,DNE有望成为生物信息学和系统生物学研究中的一项重要工具。