JACS 2026 | MetalloDock:通过物理感知深度学习解析金属蛋白-配体相互作用以推动金属蛋白靶向药物发现
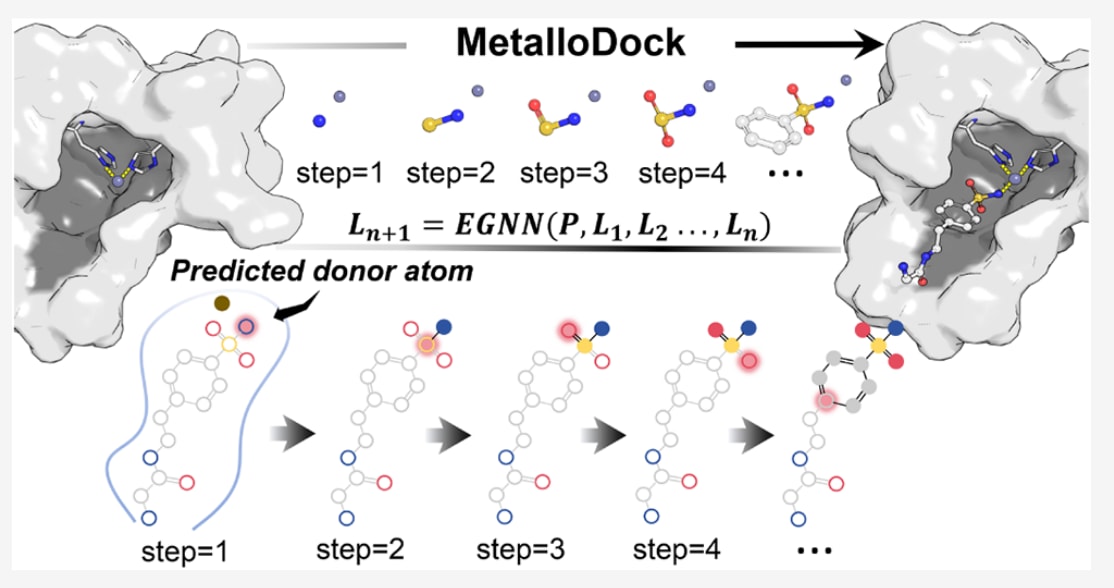
今天介绍的这项工作来自 JACS。该研究聚焦于金属蛋白靶向药物发现中一个长期存在却尚未被充分解决的核心难题,即如何在计算层面准确刻画金属离子参与下的蛋白-配体相互作用。金属配位作用具有明显区别于常规非金属相互作用的几何与电子特征,这使得传统分子对接方法以及通用深度学习模型在该类体系中普遍表现受限。针对这一问题,作者提出了MetalloDock这一面向金属蛋白的专用深度学习对接框架,将物理约束的几何建模思想与自回归构象生成策略有机结合,从方法层面系统性地解决金属配位几何难以精确重建的问题。该文章不仅在模型设计上引入了金属配位感知与多尺度结构表示,还通过严格构建的大规模高质量金属蛋白-配体数据集,对模型的对接精度、泛化能力以及虚拟筛选性能进行了全面评估。结果显示,该方法在多种金属离子环境、单金属与双金属配位体系中均展现出稳定且领先的性能优势。更为重要的是,研究将方法验证延伸至真实药物发现场景,通过前列腺特异性膜抗原虚拟筛选和流感病毒酸性聚合酶内切酶的理性设计实例,展示了该框架在实际应用中的可行性与实用价值。整体而言该文不仅提出了一种针对金属蛋白体系高度契合的新型对接方法,也构建了系统化的数据与评测标准,为后续金属蛋白靶向药物研发及相关算法研究提供了具有参考意义的技术基础与研究范式。

获取详情及资源:
- 📄 论文: https://pubs.acs.org/doi/abs/10.1021/jacs.5c15876
- 💻 代码: https://github.com/SII-ZhangHui/MetalloDock
0 摘要
金属蛋白-配体相互作用的准确预测对于金属蛋白靶向药物发现至关重要。然而,传统分子对接工具以及现有的深度学习模型难以可靠地刻画金属-配体相互作用,从而制约了高效金属蛋白抑制剂的发现。针对这一问题,该作则提出了MetalloDock,这是首个专门为金属蛋白靶点设计的深度学习分子对接框架。该方法通过创新性地融合自回归空间解码引擎与物理约束的几何生成范式,能够精确重建金属配位几何结构,并准确捕获金属-配体相互作用,从而同时提升金属蛋白-配体对接精度与结合亲和力预测的可靠性。在自建的基准数据集上的系统评估结果表明,MetalloDock在对接成功率和虚拟筛选性能方面均优于包括AlphaFold3在内的现有方法。在实际应用中,该方法在以前列腺特异性膜抗原为靶点的虚拟筛选中成功鉴定出多种新型命中化合物,并在酸性聚合酶内切酶靶点上实现了理性药物设计,发现了具有较强活性的抑制剂。这些结果表明,MetalloDock在加速金属蛋白靶向药物发现方面具有广泛的适用性,同时也为未来金属蛋白特异性对接算法的评估提供了标准化的研究框架。
1 引言
金属蛋白是指在结构或功能上与一个或多个金属离子发生配位的蛋白质,在核酸修饰、蛋白质降解以及酶催化等多种生物过程中发挥着不可或缺的作用。由于金属蛋白在调控关键生化通路中占据核心地位,其功能失调往往与多种疾病的发生和发展密切相关。例如,基质金属蛋白酶的过度表达会促进肿瘤转移,而超氧化物歧化酶的错误折叠则与神经退行性疾病的发生有关。因此,靶向金属蛋白逐渐成为一种极具前景的治疗策略,并为高选择性抑制剂的开发提供了重要方向。
靶向金属蛋白药物的独特作用机制来源于金属-配体相互作用,具体表现为金属结合药效团与靶蛋白中金属中心之间的配位作用。现有的金属蛋白相关数据库已经系统刻画了金属蛋白结合位点中金属中心与配体之间的复杂相互作用关系,凸显了金属配位在生物体系中的重要性。这些研究进展表明,金属-配体相互作用在药物发现中具有关键作用,并为理性药物设计提供了重要依据。一个具有代表性的实例是已获批准的帽依赖型内切酶抑制剂Baloxavir,该药物通过在催化位点与两个
尽管金属蛋白是重要的疾病治疗靶点,当前针对金属蛋白体系的计算方法在准确性方面仍然存在明显不足,持续制约着金属蛋白靶向药物的研发进程。目前仅有少数分子对接程序是专门为金属蛋白设计的,且大多集中于锌配位体系。传统分子对接方法在准确描述金属离子多样化的配位几何结构及其与配体之间复杂相互作用时面临显著挑战。金属离子的配位几何形式会随其氧化态和自旋态发生变化,通常表现出灵活的配位数和明显的方向性成键偏好。此外,金属离子与周围残基及配体之间的相互作用往往伴随着强烈的极化效应和电荷转移效应,而这些效应难以通过简单的成对加和势函数加以描述。这些复杂因素使得传统打分函数和固定电荷力场难以同时准确刻画金属-配体相互作用的能量学特征和几何特征,从而限制了传统对接方法在更广泛金属蛋白靶点中的适用性。相关研究也进一步证明了现有对接方法在金属蛋白-配体对接任务中的适用范围较为有限。
近年来,蛋白质结构数据库中高质量结构数据的快速积累为解析金属-配体相互作用的复杂本质提供了前所未有的机遇。与此同时,深度学习分子对接模型在蛋白-配体相互作用建模方面展现出卓越能力,推动了一系列先进方法的发展。然而,现有基于深度学习的对接方法并未针对金属蛋白-配体对接进行专门优化,往往忽略或过度简化了金属离子及其配位键的处理,从而削弱了对金属蛋白结合口袋复杂配位环境的刻画能力。此外,大规模高质量金属蛋白-配体复合物数据集的匮乏也成为制约深度学习对接性能的重要因素。当前多数模型依赖于包含有限金属蛋白样本的数据集进行训练,这在很大程度上限制了其在金属介导对接场景中的泛化能力。因此,开发专门面向金属蛋白-配体相互作用的深度学习方法,并构建更加全面且高质量的数据集,对于提升金属靶向药物发现的整体效率具有关键意义。
该研究提出了MetalloDock这一面向金属蛋白靶点的智能分子对接框架。该框架将自回归空间解码引擎与物理约束的几何生成范式相结合,实现了对金属蛋白-配体复合物中结合构象与结合亲和力的高精度预测。为高效刻画多尺度相互作用,构建了一种层次化的多尺度表示架构,在降低计算开销的同时,通过跨粒度特征融合,将蛋白的原子-残基混合表示与配体的原子-片段表示有机整合,从而有效捕获结合口袋中的多尺度相互作用特征。针对金属-配体配位作用的精细建模,引入了配位感知模块,利用蛋白与配体的编码特征对潜在供体原子进行概率预测,从化学机理层面增强了金属-配体相互作用结构描述的真实性。在此基础上,提出了一种基于自回归策略的金属引导拓扑生长算法。该算法以金属中心为起点,借助E(3)等变图神经网络模块优先预测供体原子的空间坐标,并显式引入金属配位几何的物理先验信息。随后,构象生成过程受配体共价拓扑结构引导,以逐步组装的方式完成,从而在整个生成过程中始终保持化学合理性。在精心构建的高质量金属蛋白-配体复合物数据集上,MetalloDock取得了当前领先的对接成功率,整体性能优于现有方法。该框架在包含多种金属离子的蛋白结合口袋中表现出良好的泛化能力,能够有效应对复杂配位几何以及多金属协同配位环境。在独立金属蛋白测试集上的虚拟筛选评估进一步验证了其在准确性和计算效率方面相对于主流分子对接工具的优势。作为概念验证,相关研究展示了该方法在前列腺特异性膜抗原靶向小分子发现中的实际应用价值,并基于金属结合药效团实现了针对酸性聚合酶内切酶的理性药物设计。这些结果表明,MetalloDock在金属蛋白靶向药物发现中具有变革性的潜力,为高通量筛选和理性药物设计提供了前所未有的技术能力。
2 结果与讨论
2.1 方法概述
MetalloDock采用模块化的图神经网络框架,整体由双编码器、亲和力预测模块以及构象生成模块构成。其中,双编码器包括图Transformer和几何向量感知器,用于特征提取与多尺度建模;混合密度网络用于结合亲和力预测;E(3)等变图神经网络模块用于对接构象生成(图1)。对接模块与打分模块共享同一套具备几何感知能力且融入化学先验的多尺度表示框架。在蛋白结合口袋表示方面,首先利用图Transformer在原子层级提取特征,随后通过散射机制将原子特征聚合至残基层级,并与几何向量感知器生成的残基级特征进行融合,从而实现精细结构信息由原子层级向残基层级的有效传递。在配体表示方面,配体首先依据单键被拆分为若干片段,再通过图Transformer对片段层级的化学语义特征进行编码,并借助索引映射将这些特征广播至对应的原子节点,实现功能语义信息向原子层级的下传。该多尺度表示框架中节点与边特征的具体定义以及维度设置在补充材料的表S1至表S3中给出。
在此多尺度表示的基础上,供体原子感知模块通过分析金属中心与潜在配位异原子(如N、O或S)之间的特征嵌入,以概率方式确定最可能参与金属配位的原子,并将其指定为自回归对接过程中首先生成的配体原子。分子对接模块采用金属引导的自回归几何生成框架,以金属离子作为初始锚点,逐步完成配体构象的组装。为保证化学合理性,引入了由共价图驱动的广度优先搜索算法,依据配体的配位拓扑结构确定原子生成顺序,从而严格遵循化学成键约束。
随后,模型通过金属引导拓扑生长算法对构象推断过程进行控制,在自回归几何生成框架下,沿着父子依赖链逐步构建配体原子的三维坐标。每个新生成的原子都会被视为其共价连接后续原子的父节点,而子节点的初始空间位置则通过在父节点局部坐标系中进行随机球面采样得到。为保证序列生成过程的有效性,训练过程中采用渐进式掩蔽策略,动态屏蔽尚未生成的下游原子,以避免信息泄漏。最终,MetalloDock利用混合密度网络对蛋白-配体复合物中氨基酸残基与配体原子之间距离的概率密度分布进行建模,并将其转化为统计势函数,用于量化蛋白-配体的结合强度。
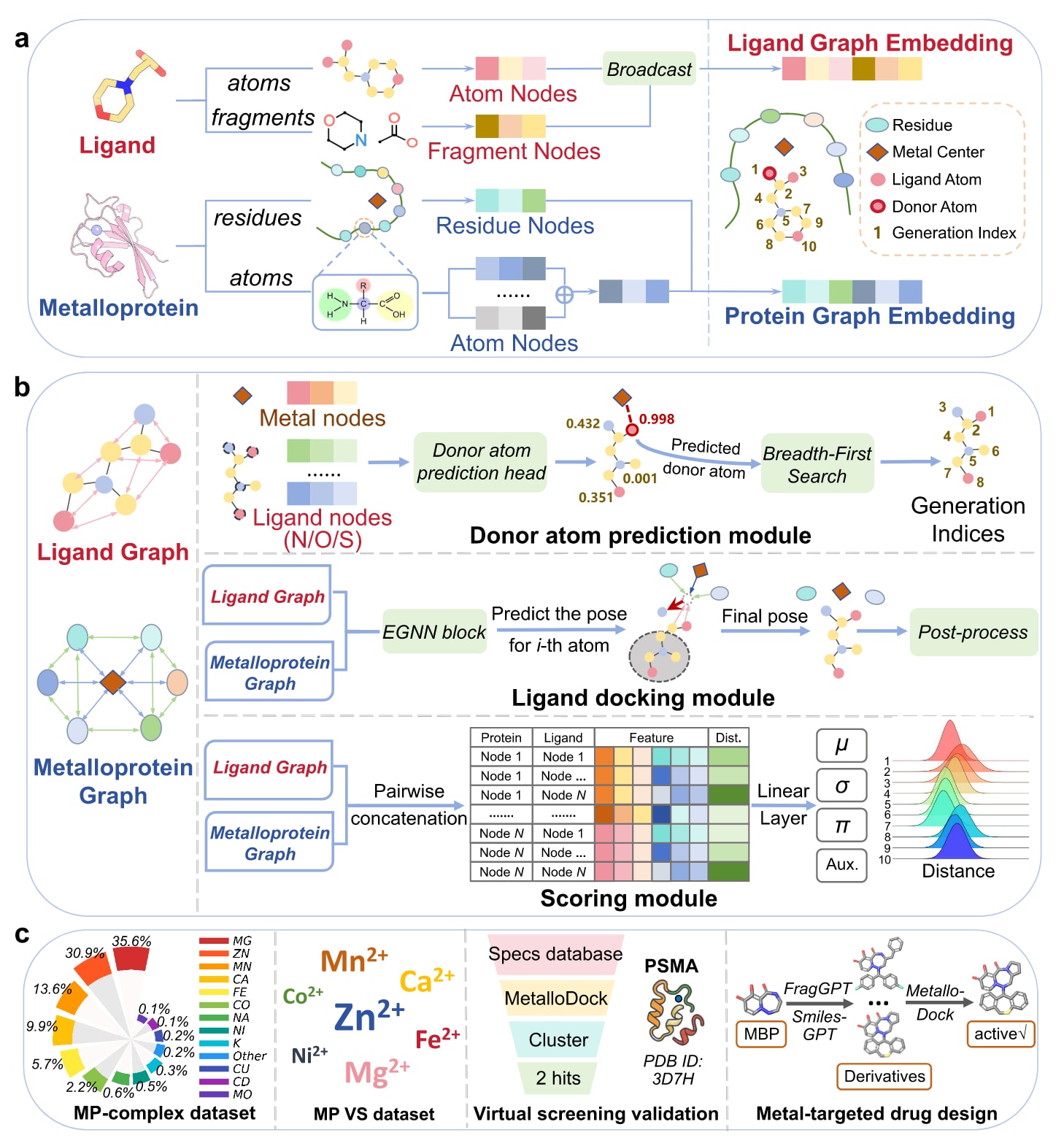
图1|MetalloDock体系结构概览。 (a) MetalloDock中层次化多尺度表示架构的示意图。(b) MetalloDock整体框架示意。模型以金属蛋白和配体作为输入,首先通过供体原子预测模块识别与金属中心发生配位的配体原子,并利用广度优先搜索算法确定配体原子的生成顺序。随后,对接模块基于父原子预测下一个原子的空间坐标,待预测原子在父原子周围随机初始化,其坐标通过E(3)等变图神经网络模块不断更新。生成的结合构象可进一步通过后处理方法进行优化。最后,结合亲和力由混合密度网络打分模块进行预测。(c) 该研究构建的金属蛋白-配体复合物数据集与金属蛋白靶向虚拟筛选数据集的示意图,以及针对前列腺特异性膜抗原的虚拟筛选流程和针对酸性聚合酶内切酶的理性药物设计流程。
2.2 基准数据集的构建
鉴于目前缺乏大规模且高质量的金属蛋白-配体复合物数据集,该研究基于蛋白质结构数据库构建了迄今为止规模最大、筛选最为严格的金属蛋白-配体复合物数据集。数据集的构建遵循了一系列严格的筛选标准,包括:仅纳入截至2024年6月30日前提交至数据库的晶体结构;结构分辨率不高于2.5Å;结合口袋中仅包含一个配体且配体至少含有8个重原子;结合口袋内至少存在一个同时与蛋白残基和配体发生配位作用的金属离子;对于同一结构条目,仅保留配体两两分子相似性不超过0.8的结合口袋。对于满足条件的每一个复合物,以任一配体原子为中心10Å范围内的蛋白残基及金属离子共同定义为蛋白结合口袋。最终构建的数据集共包含8,836个高质量金属蛋白-配体复合物。数据集采用基于时间分层的划分策略,以2023年1月1日为时间节点,划分为训练验证集与测试集。其中,2023年之前提交的8,296个复合物构成训练验证集合,再通过比例分层抽样划分为7,453个样本用于模型训练,842个样本用于验证。2023年1月1日之后提交的541个复合物被单独保留,作为独立测试集用于最终性能评估。数据集中金属离子类型及配体理化性质的分布情况在图S1中给出,显示了该数据集在化学组成和结构层面上的良好多样性与代表性。
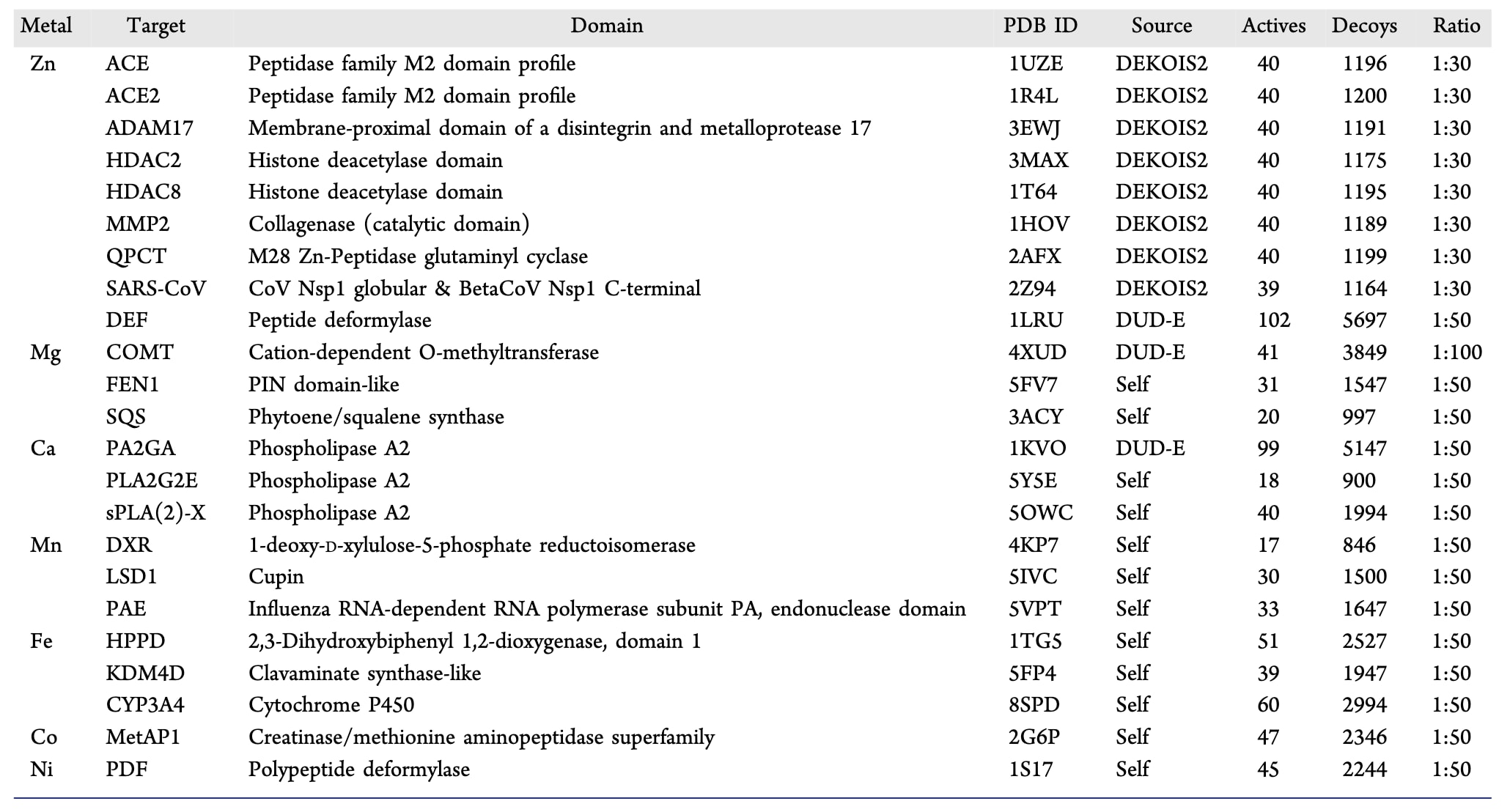
尽管现有基准数据集为分子对接虚拟筛选性能评估提供了基础框架,其金属蛋白靶点的覆盖明显偏向锌配位体系,这一偏倚限制了模型在不同金属辅因子环境下性能评估的泛化性。为弥补这一不足并系统评估MetalloDock的虚拟筛选能力,该研究对传统虚拟筛选数据集中金属蛋白相关子集进行了系统扩展 (表1)。
具体而言,对现有虚拟筛选数据集进行了全面分析,筛选出11个满足条件的金属蛋白靶点。对于其余靶点,活性化合物来源于公开生物活性数据库,而诱饵分子则按照既有规则人工生成,为每个活性分子匹配50个理化性质相似的诱饵。最终构建的虚拟筛选数据集共包含23个高分辨率金属蛋白靶点,其中包括CYP3A4等具有挑战性的体系,涵盖了Zn2+、Mg2+、Ca2+、Mn2+、Fe2+、Ni2+和Co2+七种在生物体系中具有重要功能的金属离子。该综合性的金属蛋白虚拟筛选数据集为评估分子对接模型在不同金属蛋白靶点中的适用性和筛选性能提供了多维度的验证平台。
表1|金属蛋白虚拟筛选数据集概览
2.3 MetalloDock在金属蛋白-配体对接中的性能表现
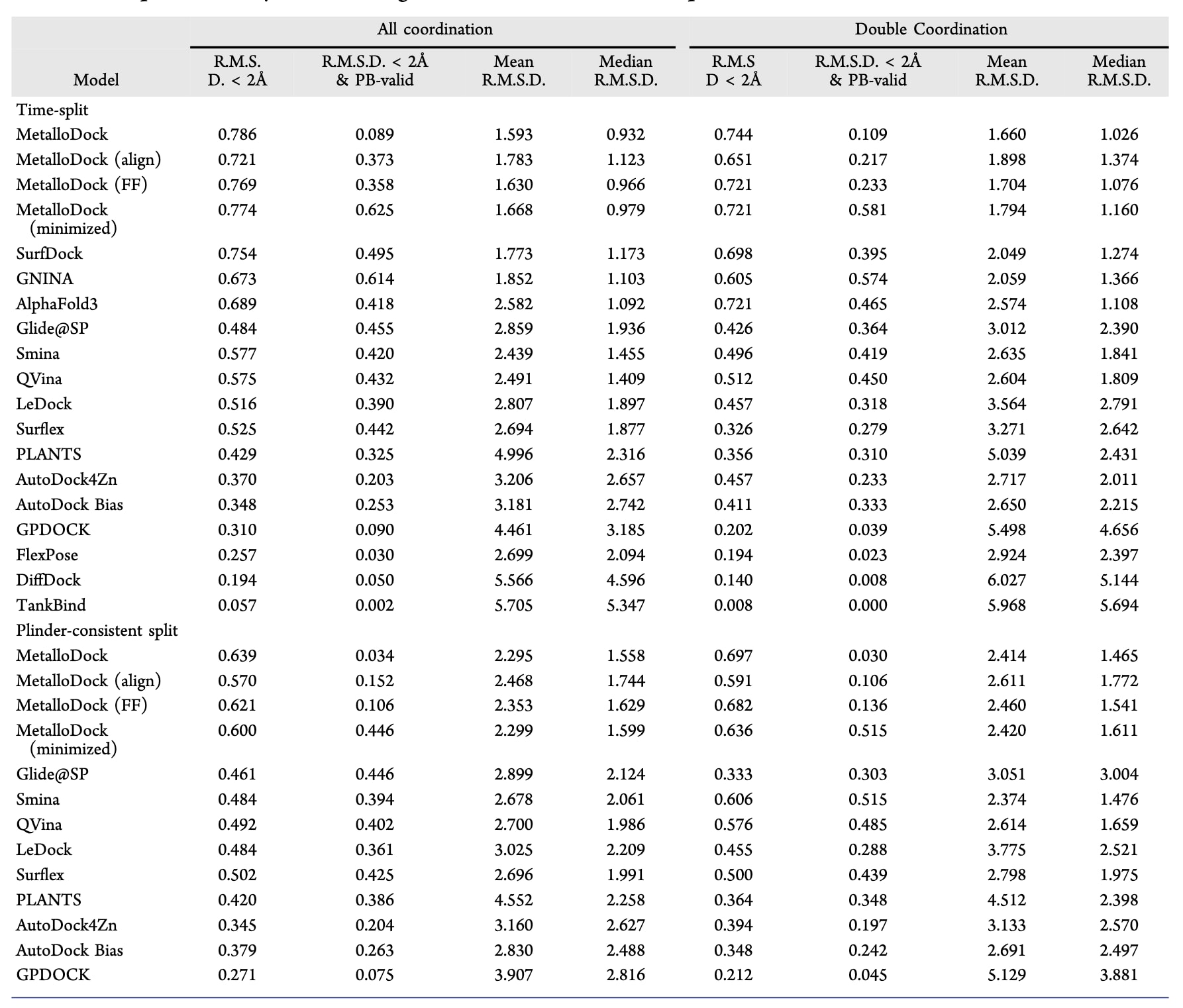
为严格评估MetalloDock在未来未知数据上的泛化能力,相关研究在基于时间分割的金属蛋白-配体复合物测试集上对其性能进行了系统评测。对比方法涵盖九种常用的传统分子对接工具,以及多种先进的深度学习对接模型。其中,部分方法针对金属蛋白体系或金属结合位点进行了专门设计,其余方法则代表了当前主流的通用对接策略。为保证比较的公平性,对于原本采用盲对接策略的方法,在评测中统一提供了预定义的结合口袋。对接成功率被定义为预测构象与对应晶体结构之间的均方根偏差小于2Å的比例。评测结果显示 (表2), MetalloDock在该基准测试中取得了78.6%的对接成功率,显著优于所有对比方法,表明其能够稳定生成接近天然构象的结合模式。在更具挑战性的双金属配位体系中,由于两个金属中心与配体之间存在复杂的空间和电子协同作用,对接难度显著增加,但MetalloDock仍保持了74.4%的成功率,明显高于其他方法。这一结果表明,该框架能够有效刻画双金属协同配位所固有的复杂空间相互作用,从而实现高精度建模。此外,在进一步严格筛选、剔除了与训练集高度相似样本的时间分割测试集中,MetalloDock依然保持了最优性能,充分体现了其在超越相近训练样本条件下的稳健性与泛化能力。
表2|金属蛋白数据集上不同分子对接方法性能的对比分析
需要指出的是,对接成功率主要反映预测构象在几何层面上与晶体结构的接近程度,并不能完全保证其理化合理性。为此,研究进一步利用构象合理性评估工具对预测结果进行了结构有效性分析。结果表明,与其他深度学习对接方法类似,MetalloDock直接生成的部分构象仍存在不合理的结构特征,说明在对接后引入结构优化流程具有必要性。针对这一问题,研究探索了多种后处理策略,包括基于力场和基于RDKit的键长与键角校正,以及在蛋白结合口袋环境中进行的力场驱动能量最小化 (图2a)。评测结果显示,所有后处理策略均在不同程度上提升了构象的物理合理性,其中基于能量最小化的策略改进最为显著,其结构质量已接近传统基于物理模型的对接方法。这些结果表明,合理的对接后优化流程能够有效缓解自回归对接模型在物理合理性方面的局限。
为更加全面地评估MetalloDock的泛化能力,研究还在近期发布的Plinder数据集上对其性能进行了测试。该基准数据集通过最大程度降低蛋白结合口袋之间的结构相似性,为模型在显著偏离训练数据分布条件下的表现提供了严格检验。基于该数据集的设计原则,评测仅将MetalloDock与传统对接工具进行比较,以避免深度学习模型之间潜在的数据泄漏风险。尽管在这一高难度测试集上的整体性能低于时间分割基准 (图2b),MetalloDock仍持续优于物理模型驱动的传统对接方法,表明其能够在分布外场景中有效学习并迁移蛋白-配体相互作用模式。与此同时,自回归生成架构在构象构建过程中可能引入误差累积,使模型性能对训练数据的质量和多样性较为敏感,这一现象提示后续研究需进一步扩展训练数据在结构和化学空间上的覆盖范围,以增强模型在复杂对接任务中的稳健性和泛化能力。
此外,研究系统评估了MetalloDock在不同金属离子体系中的泛化表现,涵盖Zn2+、Mg2+、Ca2+、Mn2+、Fe2+和Co2+等在蛋白催化与结构稳定中具有重要作用的金属类型。结果显示,在不同金属配位环境下,MetalloDock整体保持了稳定的对接性能 (图2c, d),仅在含Mn2+体系中观察到轻微性能下降。尽管这些金属离子在配位数目和偏好配位几何方面存在显著差异,模型仍能维持较高预测精度,说明其不仅学习到了通用的蛋白-配体相互作用模式,还有效捕获了金属特异性的几何约束与结合偏好。这一特性对于金属靶向药物发现与筛选具有重要价值。综合比较结果表明 (图2e),在所有测试金属类型上,MetalloDock的表现均与当前先进对接方法具有竞争力,凸显了其作为通用金属蛋白对接框架的实际应用潜力。
作为首个基于自回归生成框架的分子对接模型,MetalloDock在多种金属蛋白靶点上展现了当前领先的性能。这些结果充分证明了该方法在复杂且异质金属配位环境下的广泛适用性,并突出了其在跨金属体系中的优异泛化能力。模型在准确刻画金属离子介导的配体结合几何方面所体现出的稳定性与精确性,为其在金属酶抑制剂筛选以及金属蛋白靶向先导化合物优化等关键药物发现任务中的实际应用奠定了坚实基础。
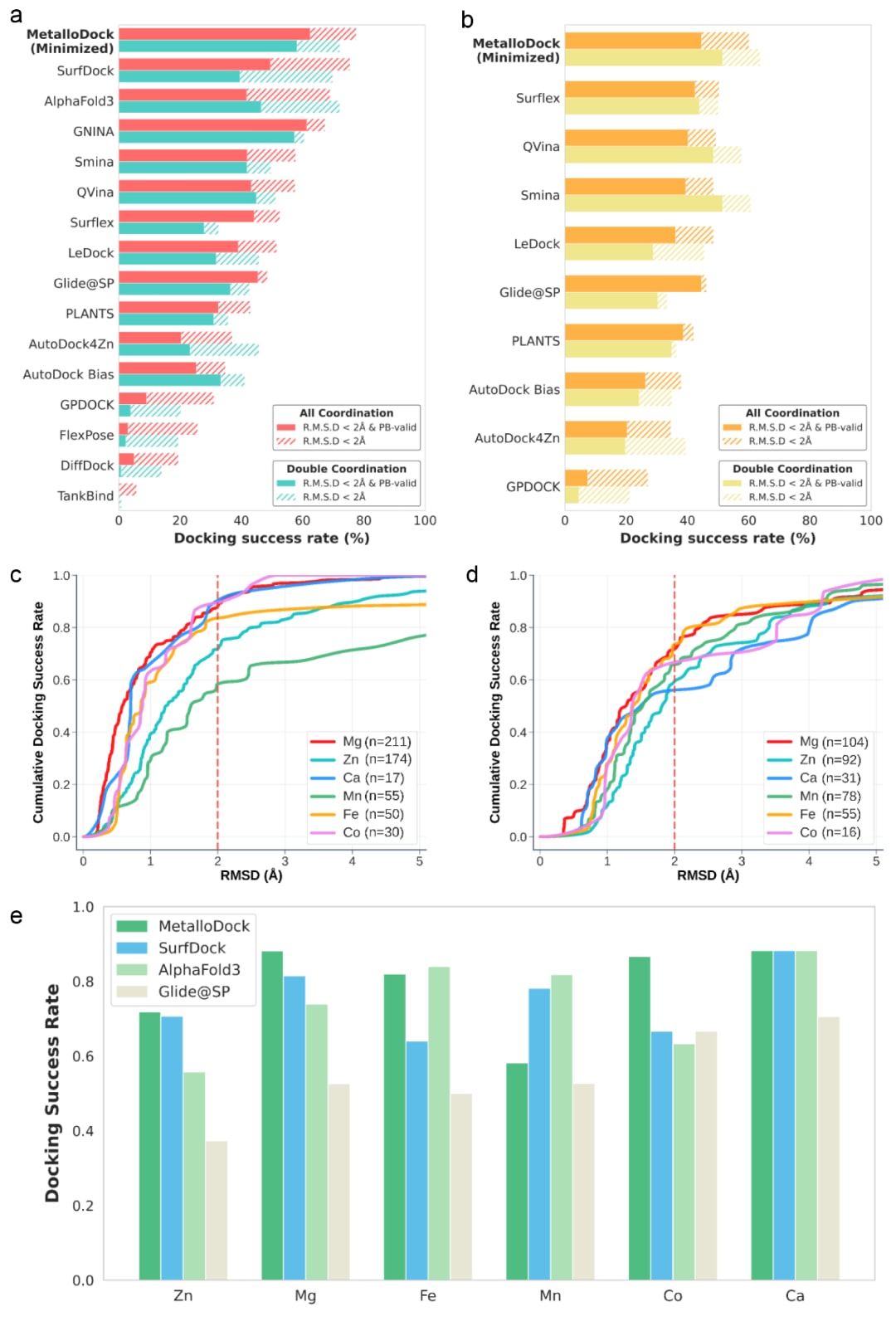
图2|不同分子对接方法的性能比较。 (a) MetalloDock与代表性方法在基于时间分割的金属蛋白-配体复合物子集上的性能表现。(b) MetalloDock与代表性方法在Plinder一致性分割的金属蛋白-配体复合物子集上的性能表现。(c) 在时间分割测试集中,MetalloDock在不同金属类型体系上的累积对接成功率。(d) 在Plinder一致性分割测试集中,MetalloDock在不同金属类型体系上的累积对接成功率。(e) MetalloDock、Glide@SP、AlphaFold3和SurfDock在不同金属类型体系上的对接成功率对比。
2.4 准确识别供体原子是可靠构象生成的关键
为进一步评估MetalloDock对关键金属-配体相互作用的刻画能力,该研究系统分析了其在配体中识别供体原子的准确性。由于MetalloDock采用自回归构象生成框架,对接整体性能在很大程度上依赖于对初始配位原子的正确判定,该原子作为后续构象生成的起始种子,对最终预测结果具有决定性影响。
在基于时间分割的测试集中分析结果表明,MetalloDock在供体原子预测任务中达到了88.9%的准确率。为深入探究供体原子预测置信度、预测结果、潜在配位原子数量以及对接性能之间的内在关系,相关数据通过三维散点图进行了可视化分析(图3a)。结果显示,预测置信度较高的供体原子通常对应较低的RMSD值,而初始供体原子识别错误往往会导致预测构象与晶体结构之间出现显著偏差。值得注意的是,配体中潜在配位原子的数量并未对预测准确性产生显著影响,这表明即使在存在多个候选原子的情况下,MetalloDock仍能够优先识别最具化学特征、最可能参与金属配位的官能团。这一现象反映了模型对金属结合化学基本规律的内化能力,也为其在结构多样的配位环境中保持稳健性能提供了重要基础。
为进一步考察MetalloDock对金属配位环境中供体原子分布特征的感知能力,研究引入基于在线平台预测的供体原子标注,用于评估不同对接模型在再现金属中心配位原子数量及类型偏好方面的表现。结果表明,MetalloDock在与晶体结构的一致性方面表现最佳,在配位数目和供体原子类型识别上均具有更高的准确性(图3b)。对不同供体类型预测偏差的定量分析也显示,MetalloDock整体性能优于对比方法。这种对供体原子分布特征的准确感知在一定程度上为模型生成高质量结合构象提供了有效引导。
在实际应用中,研究人员往往需要根据已有化学或结构知识明确指定与金属中心发生配位的供体原子,并基于这些预设配位模式对对接结果进行评估。为满足这一实际需求,MetalloDock支持在对接过程中引入用户指定的供体原子作为锚点。当启用该功能时,模型在不同体系中表现出不同程度的性能提升(图3c)。这一结果表明,在对金属-配体相互作用精确建模至关重要的应用场景中,将先验的金属配位知识融入对接流程,有助于进一步提升分子识别和结合亲和力预测的可靠性。
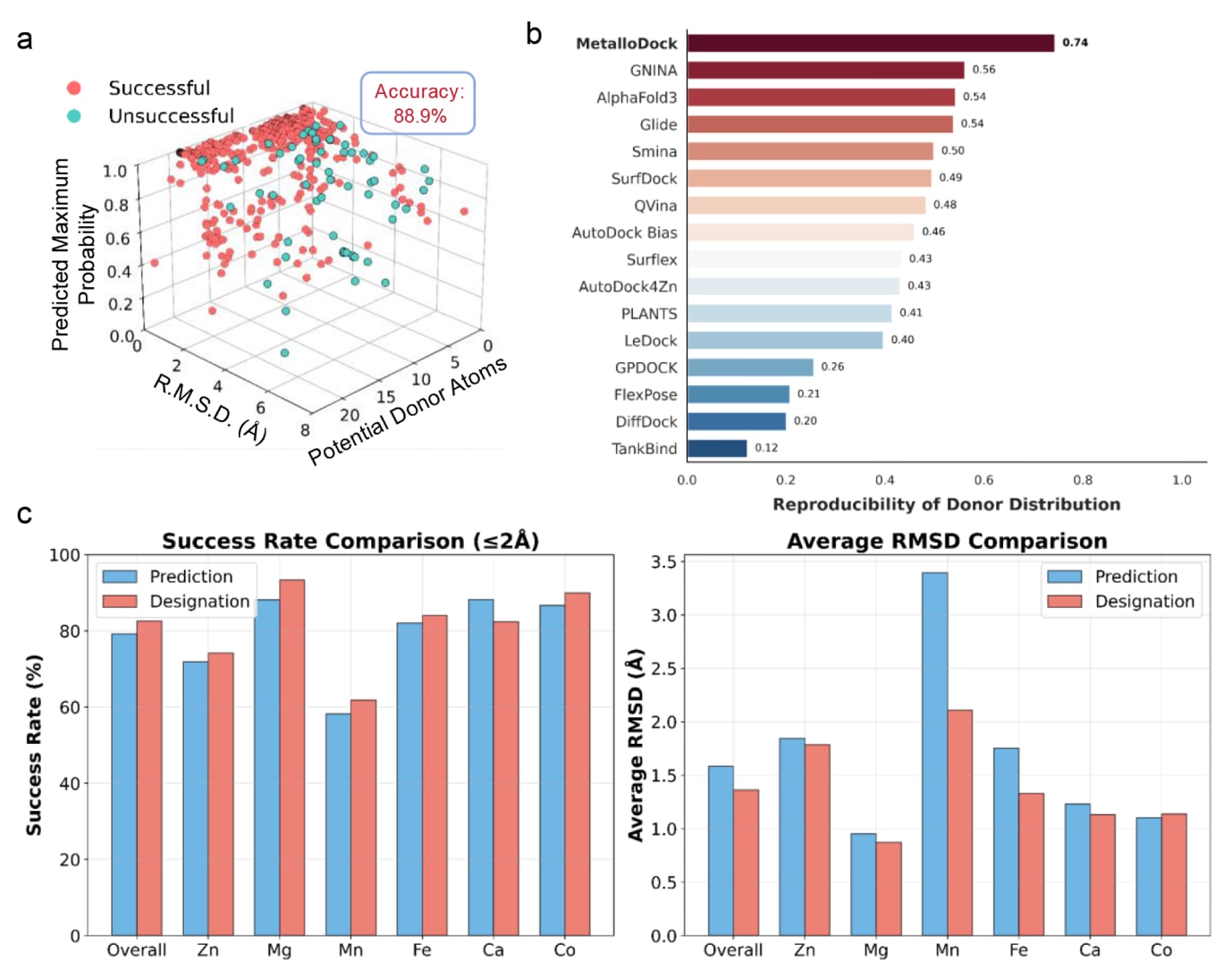
图3|MetalloDock对金属蛋白配体供体原子的预测结果。 (a) 三维散点图展示了数据点在三个变量维度上的分布关系,分别为配体中潜在配位原子的数量(x轴)、对接成功率(y轴)以及模型对所预测配位原子的置信度(z轴)。红色数据点表示模型预测的主要配位原子与真实标注一致的情况,蓝色数据点表示预测不一致的情况。(b) 不同对接模型在金属配位环境中对供体原子分布的再现能力比较,供体原子的预测结果基于CMM在线平台获得。(c) 基于对接成功率和平均RMSD,对比采用MetalloDock自动预测的供体原子与采用用户指定供体原子时的对接结果差异。
2.5 金属配位几何重建能力的评估
在金属蛋白-配体对接任务中,准确预测金属-配体相互作用始终是最具挑战性的关键问题之一。金属离子通常对其配位方式具有明确的几何偏好,但具体的配位模式又会受到周围蛋白环境与配体结构的显著影响,从而表现出高度的复杂性与多样性。因此,对接模型不仅需要正确识别金属中心所处的配位环境,还必须准确预测供体原子与金属中心之间的配位距离和配位角度,才能实现对金属配位几何结构的合理重建。
为系统评估不同模型在金属-配体相互作用预测方面的能力,研究通过对测试集对接结果进行统计分析,开展了以金属配位几何重建为核心的定量评测。具体而言,测量了金属离子与最近潜在配位原子之间的距离,并计算其相对于实验晶体结构中配位距离的偏差。同时,引入配位角偏差作为评价指标,其定义为预测得到的最近供体原子与实验观测的最近供体原子相对于金属中心所形成角度之间的差异。
结果表明,在配位距离预测方面,MetalloDock与晶体结构之间的偏差最小,其中位数偏差为0.123Å(图4a)。在配位角预测方面,该方法的表现仅次于AlphaFold3,中位偏差为18.98°,整体优于大多数传统对接工具和深度学习模型(图4b)。为进一步评估预测配位几何的保真度,研究利用在线平台对不同模型的配位几何重建准确性进行了综合评估。结果显示,MetalloDock在与实验结构的一致性方面取得了67%的再现率,在所有评估方法中排名最高。此外,针对不同金属离子常见配位几何类型的分析表明,该方法在多种配位环境下均保持了具有竞争力的预测性能。这些结果充分说明,MetalloDock在准确捕获金属-配体相互作用并重建金属配位几何结构方面具有显著优势。相关示例进一步展示了该方法在单一金属蛋白结合口袋内区分不同供体原子及配体特异性配位模式的能力(图4c)。不同模型预测的金属-配体配位距离分布与晶体结构分布的对比结果也表明,MetalloDock与实验分布高度重合,突显了其在再现真实配位模式方面的优越性。
为考察金属类型对模型性能的影响,研究选取了金属蛋白中常见的五种代表性金属离子,系统分析了不同金属体系下预测配位距离与配位角的分布及其偏差情况。结果显示,在多种金属离子体系中,MetalloDock在配位几何重建方面整体优于大多数对比方法,尽管在锰离子体系中其配位角预测精度在统计意义上略低于部分模型。这表明,MetalloDock能够有效区分不同金属类型下的金属-配体相互作用特征,并在多种金属离子体系中保持稳健的预测能力。
此外,研究还分析了三种后处理策略对最终金属配位几何结构的影响。结果发现,所有后处理方法在不同程度上均导致配位精度略有下降。其中,基于构象对齐和力场优化的策略主要关注配体几何构象本身,而未充分考虑整体金属蛋白环境,从而破坏了原本精细平衡的配位键关系。基于能量最小化的策略由于缺乏针对金属配位的专用参数,主要依赖通用静电相互作用,难以有效引导配体形成合理的金属配位模式,最终导致供体原子偏离理想结合位置。这些结果进一步强调了在金属蛋白体系中引入金属感知建模策略的重要性。
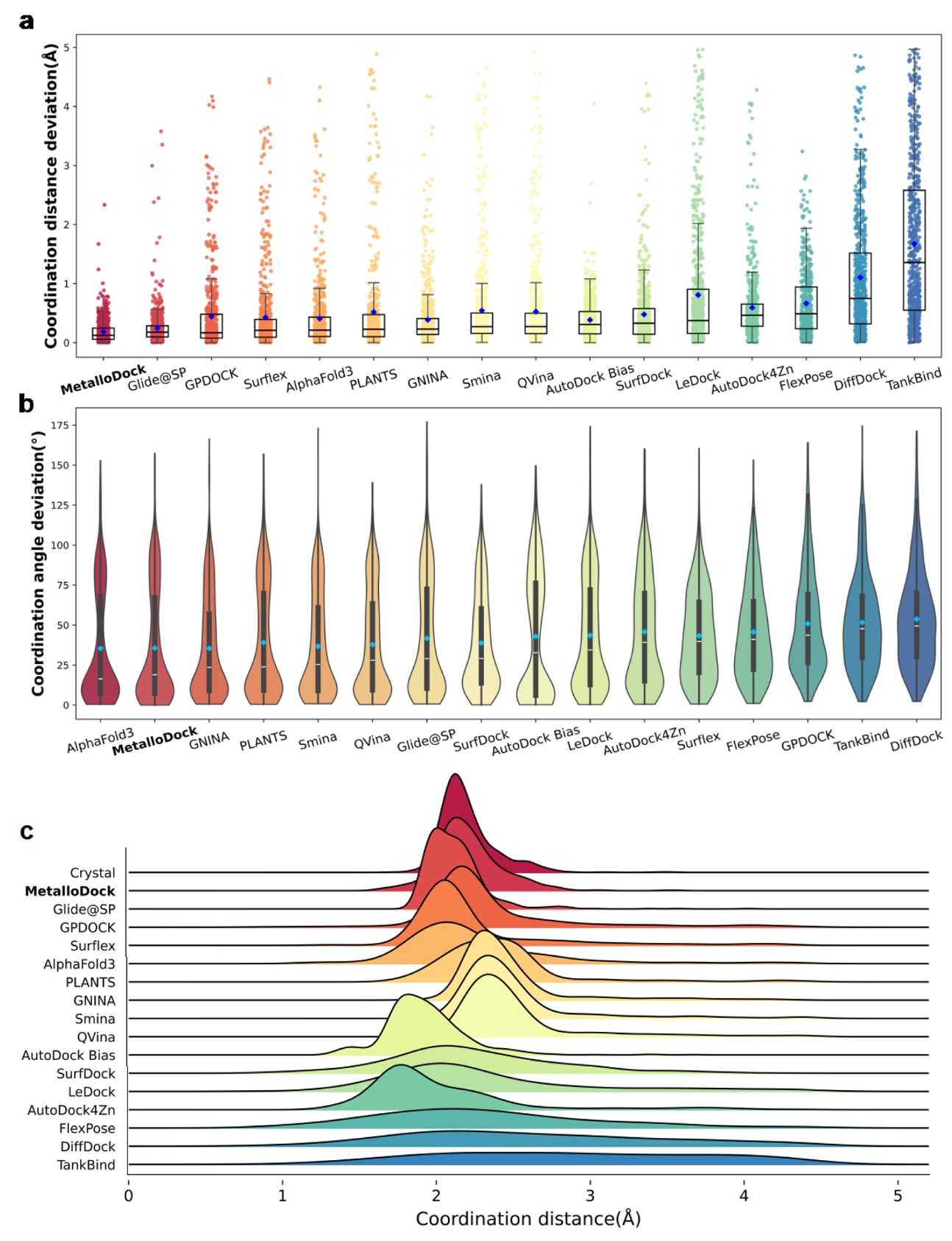
图4|不同分子对接方法在金属配位几何建模中的性能表现。 (a) 各对接模型预测的金属配位距离偏差比较。(b) 各对接模型预测的金属配位角度偏差比较。(c) 不同模型预测的金属-配体配位距离分布与晶体结构中对应分布的岭形图对比。
2.6 金属蛋白靶点的虚拟筛选能力
该研究基于前文构建的金属蛋白虚拟筛选数据集,系统评估了MetalloDock在虚拟筛选任务中的表现,并与四种基于深度学习的分子对接模型以及七种传统对接工具进行了对比分析 (图5)。评估过程中采用了多种常用指标,包括Boltzmann增强受试者工作特征曲线判别能力、精确率-召回率曲线下面积、受试者工作特征曲线下面积以及富集因子等,以全面刻画不同方法在金属蛋白靶点上的筛选性能。对于MetalProGNet,评测流程遵循既有方案,即先使用传统对接工具生成构象,再通过MetalProGNet进行重新打分。
结果表明(图5a, b),不同后处理策略在不同程度上提升了MetalloDock的筛选性能,其整体表现顺序为MetalloDock(FF)、MetalloDock(Minimized)、MetalloDock(Aligned)和未后处理的MetalloDock。其中,在蛋白环境中对配体构象进行全局力场优化的效果反而弱于局部构象优化。该现象可能源于常规力场在能量最小化过程中难以准确描述金属配位相互作用,从而导致金属结合药效团偏离其天然结合位置,最终降低虚拟筛选性能。基于上述结果,研究选择筛选性能最优的MetalloDock(FF)作为代表方法,用于后续与其他对接模型的系统比较。
在综合比较中(图5c),不同方法在金属蛋白靶点上的虚拟筛选总体性能呈现出明显差异,其中MetalloDock(FF)整体排名最高,其后依次为Glide@SP、SurfDock、GNINA、MetalProGNet以及多种传统对接工具。该结果清楚表明,MetalloDock在金属靶向虚拟筛选任务中显著优于所有参与评估的传统方法和深度学习模型,反映出其在准确刻画金属蛋白-配体相互作用方面的优势。相比之下,传统对接工具在金属蛋白体系中的筛选能力普遍较弱,这与其在金属配位建模和活性分子区分方面的固有限制密切相关。除常规基于靶点的评估方式外,研究还引入了基于结构域层面的分析,以降低结构相似靶点之间潜在冗余性的影响。在该评估设置下,MetalloDock同样在各项指标上保持了最佳整体表现。
需要强调的是,虚拟筛选的核心目标在于尽可能富集活性化合物,从而减少后续生化实验验证所需的候选数量,并有效降低实验成本。在这一关键指标上,MetalloDock(FF)在排名前0.5%的化合物中实现了21.8的富集因子,充分体现了其在大规模化合物库筛选中的实际应用价值。这些结果表明,MetalloDock在提升金属蛋白靶向药物发现的效率和准确性方面具有显著潜力。
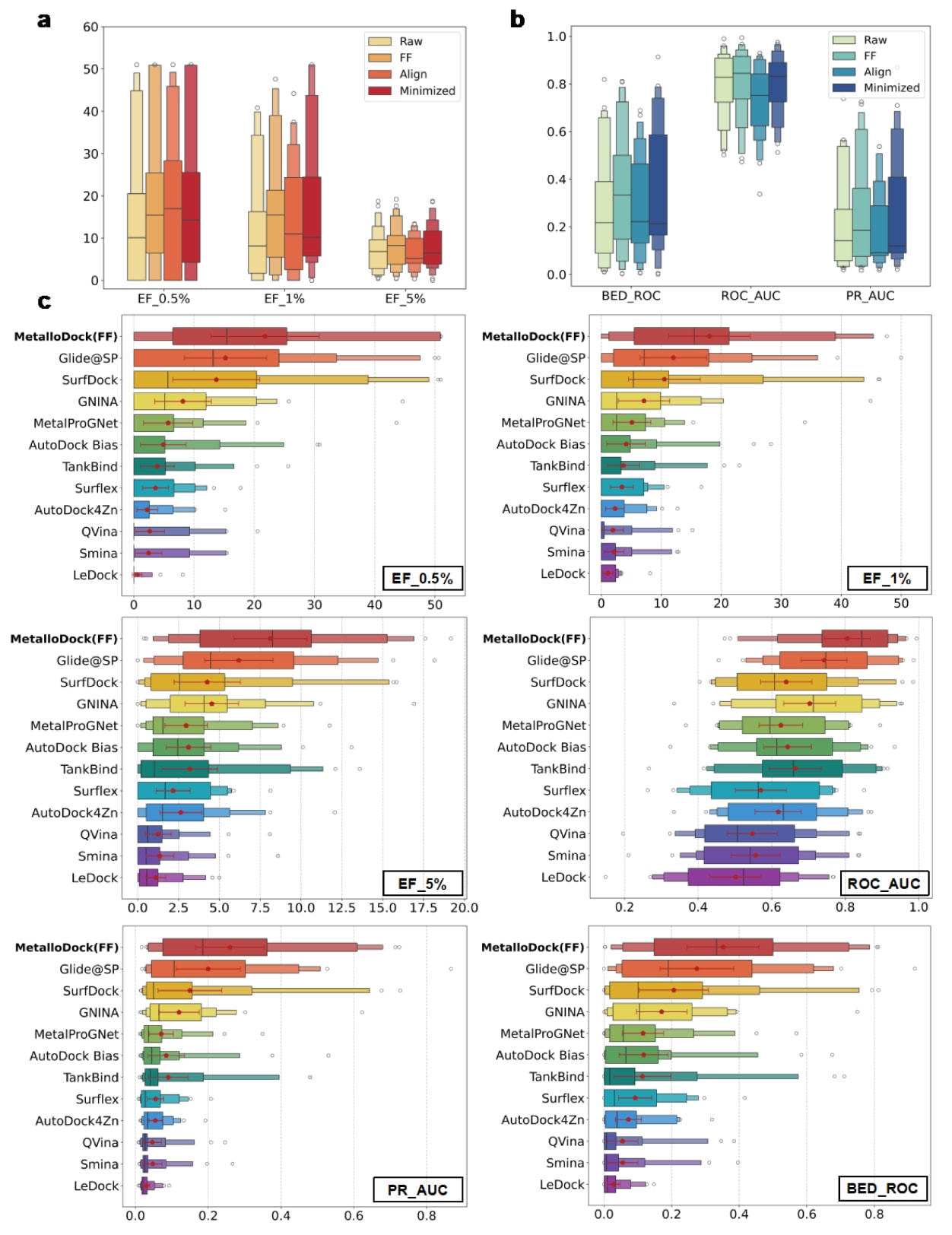
图5|不同分子对接方法在金属蛋白虚拟筛选数据集上的性能表现。 (a,b) 在不同后处理策略下,MetalloDock在六项评价指标上的性能分布情况,包括BED_ROC(α=80.5)、ROC_AUC、PR_AUC、EF_0.5%、EF_1%和EF_5%。(c) 不同对接模型在上述六项指标上的分布情况及其平均值对比,其中包括MetalloDock (FF)、Glide@SP、SurfDock、GNINA、MetalProGNet、AutoDock Bias、AutoDock4Zn、TankBind、Surflex、Smina、QVina和LeDock。
2.7 作为深度学习打分函数的打分能力与排序能力
MetalloDock的打分模块采用混合密度网络架构,该架构在该研究测试以及既有工作中已被证明具有较强的对接与筛选能力。然而,其在纯打分与排序任务中的表现相对有限。为进一步挖掘MetalloDock在打分与排序方面的潜力,研究借鉴了相关框架的思路,在模型训练过程中引入实验测定的结合亲和力信息,以增强模型对能量趋势的相关性。通过加入亲和力引导的损失项对模型进行微调后,在标准基准数据集上对改进模型的打分和排序性能进行了评估。结果显示,经微调后的MetalloDock在打分与排序任务中均取得了具有竞争力的表现,相关结果在补充材料中进行了系统总结。
2.8 MetalloDock通过虚拟筛选鉴定PSMA抑制剂
前列腺特异性膜抗原是一种Ⅱ型跨膜蛋白,在前列腺癌细胞表面,尤其是在晚期及转移性病例中高度表达,而在正常组织中的表达水平极低。这种显著的组织特异性使其成为前列腺癌诊断与治疗中的关键靶点。然而,目前用于PSMA的临床成像试剂和治疗性放射性配体多依赖Glu-urea-Lys这一结构骨架,该类分子易在肾脏、泪腺和唾液腺等组织中发生非特异性富集,从而带来潜在副作用。因此,开发新型PSMA靶向配体仍具有迫切需求。
在该研究中,按照图6a所示流程,基于PSMA晶体结构(PDB ID:3D7H)开展了虚拟筛选研究。筛选所用化合物库为常用商业数据库,共约200168个分子。为保证候选分子的类药性,首先剔除了违反不少于三条Oprea规则或不少于两条Lipinski五规则的化合物。随后,采用MetalloDock对筛选库进行初步对接,并对排名前1%的化合物按ECFP4分子指纹相似性聚类为30组,从中选取27个结构多样性的代表化合物用于后续体外实验验证。
在初步实验中,通过PSMA酶抑制实验评估了上述化合物在10μM浓度下的抑制活性,并以2-(Phosphonomethyl)pentanedioic acid作为阳性对照。结果显示,在27个候选分子中,Z21和Z26表现出超过50%的抑制活性(图6b)。进一步的剂量-反应实验表明,这两种化合物均具有较强的PSMA抑制能力,其IC50分别为0.375μM和0.219μM(图6c)。结构分析发现,两种分子均含有可与
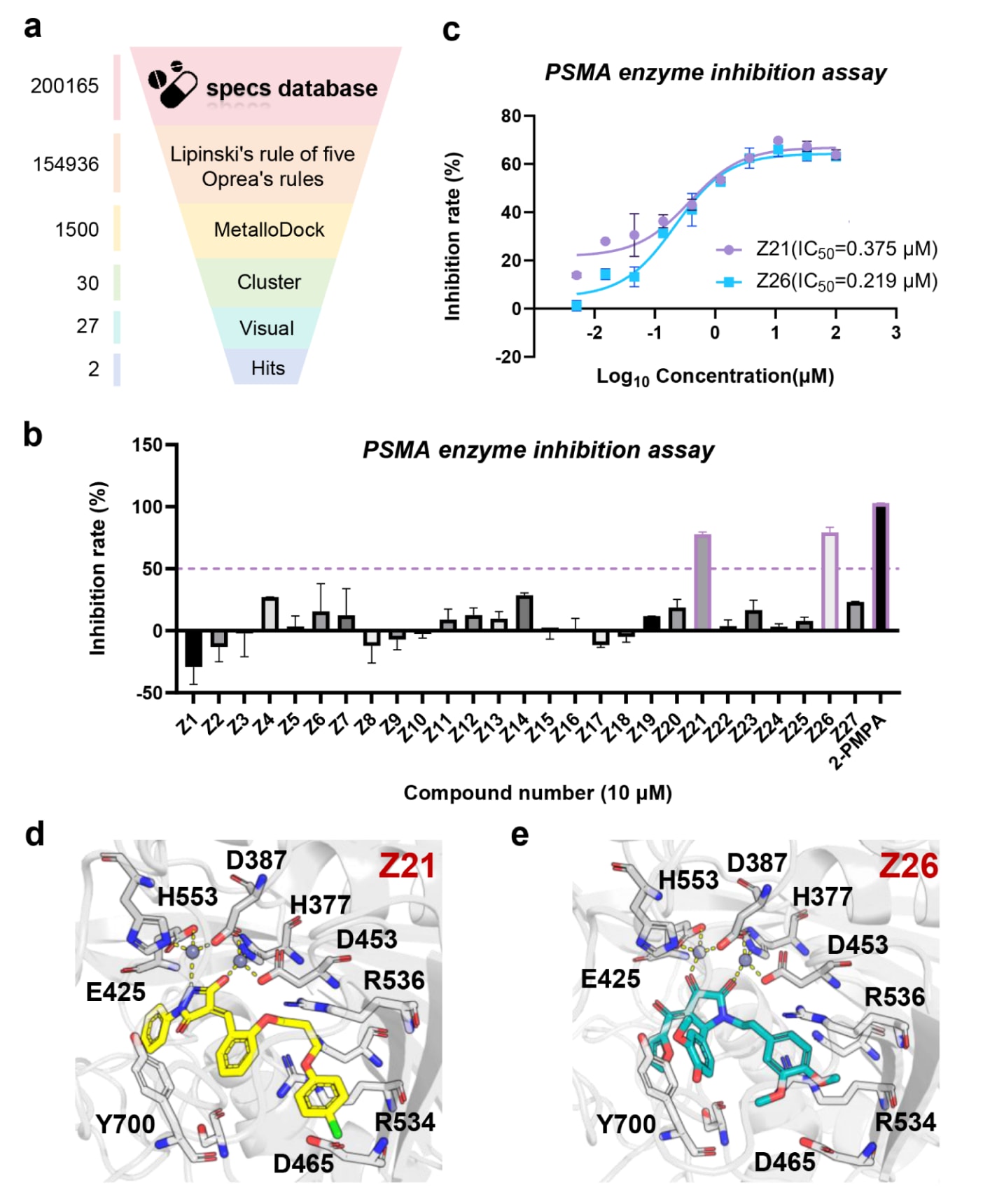
图6|基于MetalloDock鉴定PSMA抑制剂的整体流程。 (a) 以PDB ID:3D7H晶体结构为基础的PSMA靶向虚拟筛选流程示意。(b)通过酶抑制实验评估27个筛选化合物在10μM浓度下对PSMA的抑制活性。(c) 对化合物Z21和Z26在不同浓度条件下的抑制效果进行评估。(d) PSMA与化合物Z21的结合构象及其相互作用模式。(e) PSMA与化合物Z26的结合构象及其相互作用模式。
2.9 MetalloDock辅助酸性聚合酶内切酶抑制剂的理性设计
由于金属配位键的特殊性质以及金属配位体系本身的高度复杂性,金属蛋白抑制剂的理性设计始终面临较大挑战。通常情况下,药物化学家倾向于以金属结合药效团作为核心骨架对分子进行扩展,但这一过程往往需要大量实验资源支持。为评估MetalloDock在金属蛋白理性分子设计中的实际应用价值,该研究构建了一套结合计算设计与实验验证的一体化工作流程(图7a)。
研究以流感病毒酸性聚合酶内切酶为靶点,该酶属于双核锰金属酶。首先选取了三种单金属依赖型金属结合药效团和三种双金属依赖型金属结合药效团作为核心骨架,前者可螯合单个金属离子,后者可同时与两个金属离子发生配位(图7b和图7c)。在此基础上,借助分子生成工具对这些骨架进行结构扩展,在保留金属结合能力的同时引入不同官能团和侧链修饰,以拓展化学空间覆盖范围。随后,利用MetalloDock对生成的候选分子进行对接和打分,并从排名前10%的分子中筛选出合成可行且具有潜在活性的化合物用于实验验证。
最终共合成并测试了22个化合物,其中包括7个单金属依赖型抑制剂和15个双金属依赖型抑制剂。在单金属依赖型分子中,有5个表现出中等抑制活性,显示出进一步优化的潜力。而在双金属依赖型分子中,有4个化合物展现出亚微摩尔级抑制活性,其中优化程度最高的化合物IC50达到191nM(图7d),表现出稳定而显著的抑制效果。由于双核金属酶活性位点中存在两个协同作用的金属离子,通常需要至少三个配位原子才能实现稳定螯合,这使得相应抑制剂的设计难度显著增加。该研究基于金属结合药效团的理性设计策略在保持双金属协同配位结构基础的同时成功获得了高效抑制剂,充分展示了其在双核金属酶抑制剂开发中的应用价值。
综合上述结果可以看出,MetalloDock在不同金属蛋白体系中均展现出良好的泛化能力和实际应用潜力,为新型金属蛋白抑制剂的理性设计提供了有力的计算支持。该工作流程系统体现了人工智能辅助药物设计的优势,为以金属为核心的药物发现提供了一种可扩展且具有普适性的研究范式。
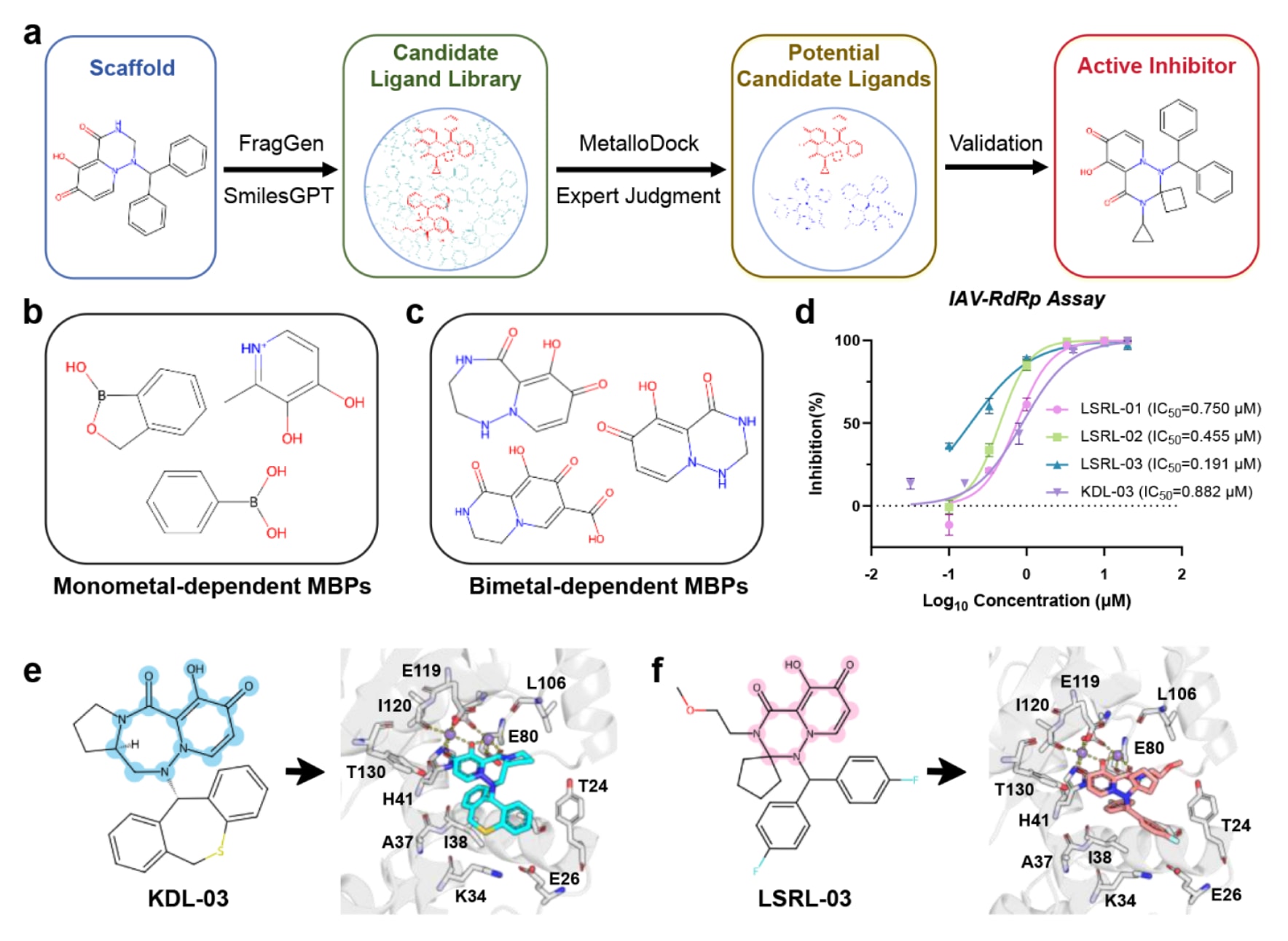
图7|基于MetalloDock的酸性聚合酶内切酶理性药物设计流程。 (a) 针对酸性聚合酶内切酶的理性药物设计整体流程示意。(b) 工作流程中采用的单金属依赖型金属结合药效团的结构。(c) 工作流程中采用的双金属依赖型金属结合药效团的结构。(d) 在不同浓度条件下对LSRL-01、LSRL-02、LSRL-03和KDL-03抑制活性的评估结果。(e) 酸性聚合酶内切酶与化合物KDL-03的结合构象及其相互作用模式。(f) 酸性聚合酶内切酶与化合物LSRL-03的结合构象及其相互作用模式。
3 结论
传统分子对接工具和现有的深度学习模型在准确刻画金属蛋白-配体相互作用以及开展高效金属蛋白靶向虚拟筛选方面均存在明显局限。为应对这些挑战,提出了MetalloDock这一全新的分子对接框架,其通过引入自回归几何生成机制,显著提升了金属蛋白-配体对接的精度。该方法能够有效捕获金属中心多样化的配位几何特征,并在金属蛋白-配体相互作用预测方面展现出前所未有的准确性。为对金属蛋白-配体对接模型进行严格评估,研究构建了一套完整的基准评测体系,包括金属蛋白-配体复合物数据集和金属蛋白虚拟筛选数据集。其中,金属蛋白-配体复合物数据集为训练深度学习模型准确学习金属-配体相互作用提供了基础资源,有效应对了金属配位几何多样性带来的建模挑战;而金属蛋白虚拟筛选数据集则为系统评估对接模型在金属蛋白靶向药物发现中的筛选能力提供了标准化平台。二者相互配合,旨在缩小金属蛋白计算建模与实验验证之间的差距,从而提升人工智能驱动的金属蛋白靶向药物研发流程的可靠性。
在系统的基准测试中,MetalloDock在对接精度方面整体优于传统方法和其他深度学习对接模型,并在金属-配体相互作用建模方面表现出卓越能力,能够精确重建复杂的金属配位几何结构。此外,回顾性虚拟筛选实验表明,该方法在大规模化合物库中高精度识别金属蛋白抑制剂方面具有显著潜力。综合来看,MetalloDock是一种稳健且具有实际应用价值的金属配位对接工具,适用于真实药物研发场景。展望未来,其模块化设计有望与QM/MM精修流程实现无缝整合,从而更准确地刻画传统经典力场难以充分描述的极化效应、电荷转移效应以及金属中心的电子结构特征,进一步提升复杂配位环境下的结构保真度和能量排序能力。
对金属蛋白-配体相互作用的深入理解是推动金属蛋白抑制剂发现和治疗创新的重要基础。随着金属蛋白靶向药物研发领域的不断发展,MetalloDock有望成为药物研究中的关键工具。其对金属-配体相互作用复杂性的精准刻画能力,不仅能够加速药物发现的早期阶段,也为新一代金属蛋白抑制剂的理性设计提供了有力支持。通过有效连接计算建模与实验验证,MetalloDock具备推动金属蛋白抑制剂研发取得实质性进展的潜力,并有望为多种人类疾病的治疗策略优化以及精准医学的发展带来重要推动作用。