JMC 2026 | PLMCA:一种用于口袋识别与结合亲和力预测的通用多模态蛋白–配体交叉注意力框架
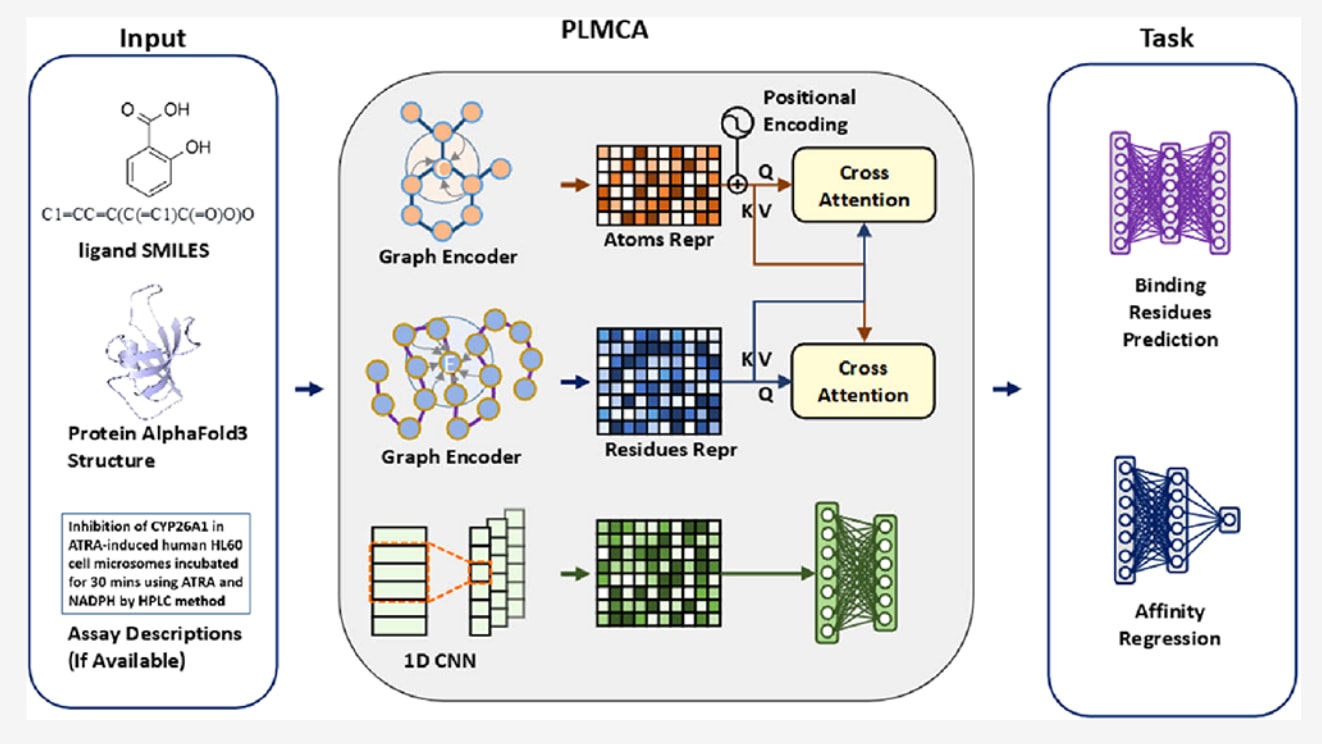
药物–靶标相互作用的精准建模是计算机辅助药物设计中的核心问题,而同时实现结合口袋识别与结合亲和力的可靠预测仍然极具挑战性。近年来,蛋白语言模型和图神经网络的发展显著提升了对序列与结构信息的建模能力,但如何在统一框架中高效融合多源异构信息、并在未见蛋白或未见配体场景下保持稳健泛化,仍缺乏成熟解决方案。发表在JMC的这项工作提出PLMCA,一种通用的多模态蛋白–配体交叉注意力框架,通过整合蛋白语言模型嵌入、三维几何特征、理化性质描述以及配体分子图表示,实现了对结合位点与结合亲和力的协同建模。该方法在PDBbind21和ChEMBL等基准数据集上系统验证了其准确性与泛化能力,并以开源代码和在线服务器形式提供,展示了面向真实药物研发流程的实用潜力。

获取详情及资源:
- 📄 论文: https://doi.org/10.1021/acs.jmedchem.5c03431
- 💻 代码: https://github.com/heyigacu/plmca
- 🛜 网站: https://hwwlab.com/Webserver/plmca
0 摘要
药物–靶标结合亲和力的准确预测仍然是药物发现中的核心挑战之一,其根本原因在于需要同时整合来源异构的序列信息、结构信息以及理化性质信息。该研究提出PLMCA,一种多模态蛋白–配体交叉注意力框架,在统一的模型架构中融合了来自两种蛋白语言模型的蛋白序列嵌入、三维几何特征、理化描述符以及配体的分子图表示。PLMCA还将来自ChEMBL数据库的实验测定条件作为辅助输入引入模型,以减轻批次效应并降低测量噪声的影响。在PDBbind21数据集上,PLMCA在随机划分、未见配体以及未见蛋白三种设置下,在Kd和Ki预测任务中表现出与当前最先进方法相当或更优的性能。在ChEMBL_mini数据集上,PLMCA在IC50、Kd和Ki预测任务中分别取得了0.531、0.635和0.519的R2值。此外,PLMCA在蛋白结合口袋预测任务中同样表现稳健,在未见蛋白设置下AUPR最高可达0.655。
1 引言
药物-靶标相互作用预测是现代药物发现的核心组成部分,尤其在药效学研究中,理解小分子如何与蛋白质或核酸等生物靶标相互作用,对于评估药物的疗效与效力至关重要。DTI预测支撑了虚拟筛选、药物再利用以及潜在不良反应识别等关键应用,为治疗候选物的优化提供指导。传统的高通量筛选和生化实验方法通常成本高昂且耗时较长,因此,结合机器学习与人工智能的计算方法逐渐成为提升DTI预测效率与可及性的关键工具。尽管近年来取得了显著进展,计算型DTI预测在药效学应用中仍面临若干限制。
其中一个主要问题在于,能够预测定量疗效指标的模型数量有限,例如半数抑制浓度IC50或半数有效浓度EC50。这类指标对于量化药物效力、筛选和排序候选分子具有决定性作用,并直接影响药效学特征的刻画。然而,现有多数模型仍停留在二分类层面,仅判断药物与靶标是否发生相互作用,而未能刻画相互作用强度,从而在计算疗效评估中留下明显空白。另一个关键挑战来自负样本数据质量不足,这直接削弱了模型的可靠性。常用数据库往往缺乏系统整理且经实验验证的非相互作用药物-靶标对,研究中常依赖随机配对或计算推导得到的负样本,这类数据并未经过实验确认,容易包含潜在的真实相互作用,从而抬高假阳性率,造成过于乐观的性能评估,并显著降低模型的泛化能力。在药效学研究中,准确区分不相互作用与相互作用同样重要,这一缺陷尤为突出。
此外,面向蛋白-配体结合亲和力预测的开源工具仍然匮乏,尤其是直接针对IC50和EC50等药效学相关指标的工具。部分方法侧重于结合位点识别而非亲和力本身,而许多深度学习模型缺乏易用的平台支持,限制了其在实际研究中的推广。尽管如此,近期的机器学习进展为DTI预测提供了新的思路。一些基于序列的深度模型能够在无需三维结构信息的情况下预测配体结合残基,为后续亲和力预测奠定基础;融合知识图谱嵌入与蛋白序列表示的方法在多类数据集上取得了更高的预测精度,并有利于下游的定量分析;基于图结构的框架通过归纳式链接预测和领域自适应机制,提高了对新化合物和新靶标的泛化能力;还有统一建模DTI、结合亲和力以及激活或抑制机制的模型,借助自监督预训练在冷启动场景下表现突出,并能够直接服务于IC50和EC50等关键指标的预测。然而,这些方法大多未以集成化、用户友好的形式开放,限制了研究人员的使用。
针对上述问题,提出了一个新的开源预测框架PLMCA,用于蛋白-配体结合亲和力估计与结合口袋预测。该模型利用多模态蛋白特征嵌入,并通过图神经网络学习分子与蛋白的结构表示,同时引入交叉注意力机制实现蛋白与配体之间的特征互学习。在高质量数据集和创新的相互作用建模架构支持下,PLMCA在多项亲和力预测任务中展现出优异的准确性与泛化能力。通过以开源平台形式提供这一模型,不仅提升了先进机器学习方法的可获得性,也为更精确的候选药物筛选提供了可靠工具,从而加速早期药物研发进程。
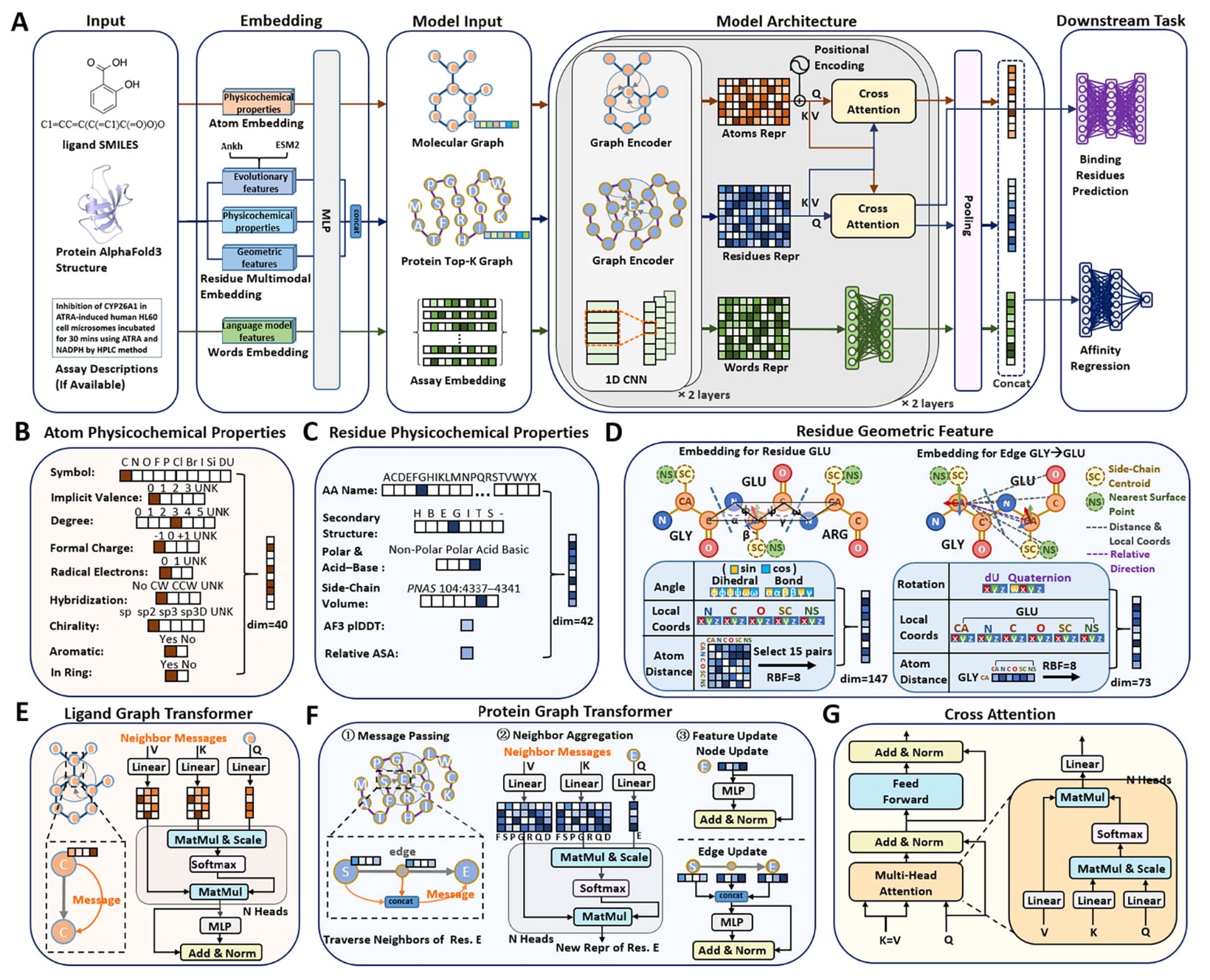
图1|PLМCA的模型架构。 (A)PLMCA的整体流程,以配体SMILES、蛋白质结构和实验测定描述作为输入。(B)用于配体原子嵌入的原子级理化性质特征。(C)用于蛋白残基嵌入的残基级理化性质特征。(D)基于局部原子坐标以及距离和角度信息构建的残基几何特征。(E)用于分子表示学习的配体图Transformer。(F)用于残基表示学习的蛋白图Transformer,通过消息传递和特征更新实现表示建模。(G)用于刻画配体−残基相互作用和特征融合的跨注意力模块,随后接入结合残基预测和亲和力回归等下游任务。
2 方法
2.1 数据集
第一个数据集PDBbind21用于训练蛋白配体结合位点预测模型。由于PDBbind21中大量晶体结构存在残基缺失的问题,这些结构被替换为来自AlphaFold蛋白结构数据库的对应预测结构,以保证蛋白结构与序列之间的一致性,并避免后续蛋白嵌入过程中出现语义不一致。同时,包含字符“X”的蛋白序列以及长度超过1500个残基的序列被移除,以降低超长序列带来的建模与计算负担。对于配体,统计其重原子数量,仅保留重原子数不超过225的分子。经过上述筛选后,最终保留了7135个蛋白-配体复合物。
在数据划分阶段,采用MMseqs2对蛋白序列进行聚类,序列一致性阈值设为70%,比对覆盖度阈值设为80%,以确保未见蛋白划分中不存在高度相似的序列。在未见配体设置下,首先计算Morgan指纹,并利用Tanimoto系数衡量分子相似性,设置70%的相似性阈值,以保证训练集与测试集之间配体的低相似性。针对未见蛋白和未见配体两种划分方式,其训练集与测试集的相似性分布如图S1所示。此外,该数据集中还包含2462条Kd和1624条Ki注释样本,这部分数据被进一步用于评估PLMCA的亲和力学习能力。
第二个数据集ChEMBL用于蛋白-配体亲和力预测任务的训练。从ChEMBL数据库中收集了三类实验测量值:IC50,Kd和Ki。仅保留具有明确数值且小于10^12的记录,以剔除无效或极端测量值。所有单位统一转换为μMol/L,并对亲和力数值进行−log10变换,分别对应IC50,Kd和Ki三种亲和力指标。最终得到由配体-蛋白-实验描述-亲和力标签构成的四元组数据。对蛋白和小分子同时进行长度筛选后,获得857102条IC50样本,54033条Kd样本以及387339条Ki样本。此外,通过从IC50,Kd和Ki数据集中各随机抽取10000条记录,构建了ChEMBL_mini数据集。对于同一蛋白-配体对对应的多条实验测量值,采用其中位数作为最终标签,从而得到三个规模较小的数据子集,用于高效评估。
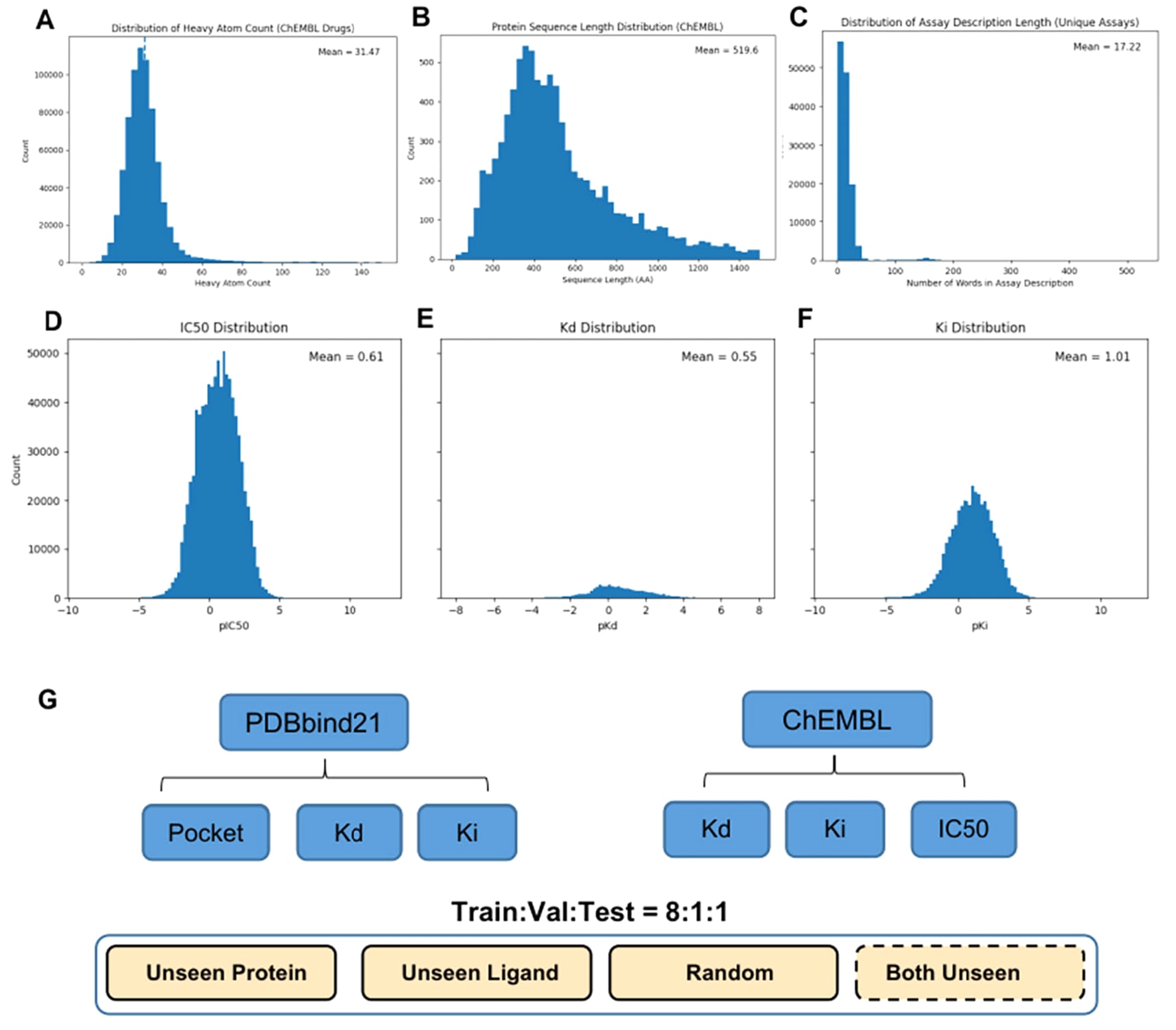
图2|数据集统计与数据划分策略。 (A)ChEMBL药物数据集中化合物重原子数的分布。(B)ChEMBL靶点数据集中蛋白序列长度的分布。(C)在去除重复实验测定后,以词数衡量的实验测定描述长度分布。(D−F)ChEMBL数据集中结合亲和力数值的分布情况,分别对应pIC50(D)、pKd(E)和pKi(F),虚线表示各分布的平均值。(G)该研究所使用的数据集及评估划分方式概览,其中PDBbind21用于带有Kd和Ki标注的口袋级结合位点预测,ChEMBL用于基于Kd、Ki和IC50测量值的亲和力预测,并在随机划分、未见蛋白、未见配体以及蛋白和配体同时未见四种评估设置下进行分析。
2.2 特征工程
该研究提出了一种用于药物-靶标结合亲和力预测的深度学习模型,将图神经网络与双向跨模态注意力机制相结合,实现对异构信息的有效融合。这些信息来源于药物分子图、蛋白图以及实验检测条件的文本描述。在特征工程方面,蛋白质采用多模态表示方式,包括ESM2嵌入、Ankh嵌入、理化性质特征以及几何结构特征。蛋白三维结构来源于AlphaFold蛋白结构数据库。为捕获蛋白序列中的上下文与进化信息,使用预训练的蛋白语言模型ESM2对氨基酸序列进行编码。同时,引入Ankh-large蛋白语言模型作为补充,以增强蛋白表示的多样性与鲁棒性。
除序列嵌入外,还进一步引入显式的理化与结构先验特征。基于处理后的蛋白三维结构文件,使用DSSP程序计算每个残基的二级结构类型和溶剂可及表面积。溶剂可及表面积通过残基特异的最大值进行归一化,得到相对溶剂可及性。每个氨基酸残基被编码为42维特征向量,包含氨基酸类型、化学类别和理化性质分组的独热编码,二级结构状态编码,以及从Cα原子提取并归一化的相对溶剂可及性和pLDDT值。为准确刻画蛋白的三维几何构象,参考LABind框架在残基层面构建局部坐标系。每个残基的局部参考系由主链原子N,Cα,C和O定义,几何信息被映射到以残基为中心的参考框架中,从而实现旋转和平移不变的表示,并生成残基及残基-残基边的几何特征。所有蛋白图均基于Deep Graph Library实现。
配体分子以SMILES字符串形式提供,并通过RDKit工具解析为分子图结构。每个配体被表示为无向图,其中节点对应原子,边对应原子之间的化学键。针对每个原子节点,构建基于其化学环境的离散特征向量,包含原子类型,隐式化合价,自由基电子数,原子度数,形式电荷,杂化状态,手性信息以及芳香性和成环性的指示特征。所有类别型特征均采用独热编码,从而使每个原子节点具有固定长度的数值特征表示。
为了充分利用ChEMBL数据库中实验层面的语义信息,还引入了基于自然语言的实验描述特征。每条实验记录对应的文本来源于ChEMBL的Assay Description字段,涵盖实验类型、测量条件及生物学背景等信息。这些实验描述通过预训练的BioBERT模型生成嵌入表示,用于辅助亲和力预测。
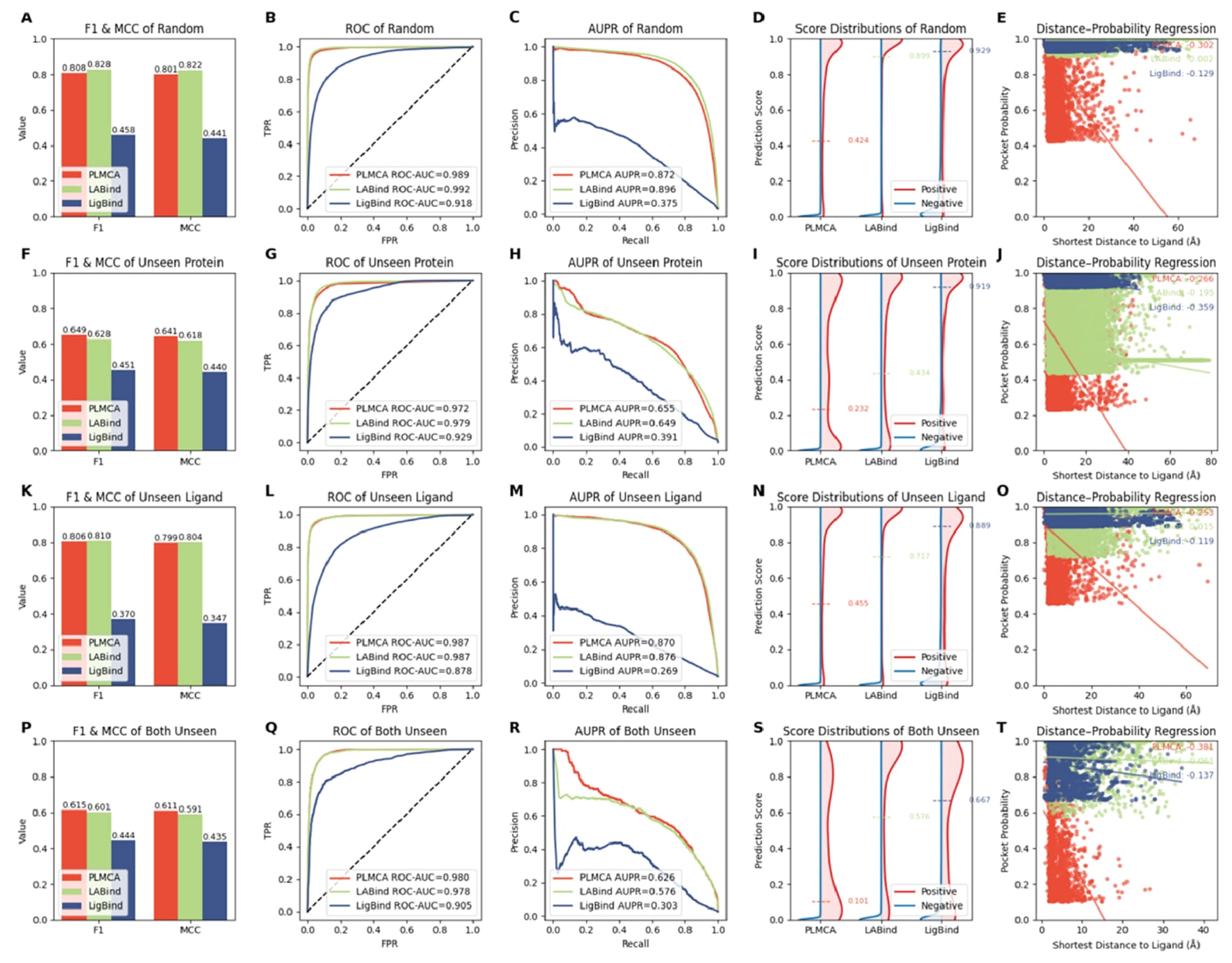
图3|PLMCA、LABind和LigBind在四种评估设置下的性能对比。 (A−E)随机划分条件下的结果,包括F1/MCC、ROC、AUPR、得分分布以及距离−概率回归。(F−J)未见蛋白设置。(K−O)未见配体设置。(P−T)蛋白和配体同时未见设置。在所有评估条件下,PLMCA相较于基线方法表现出更高的预测准确性、更强的区分能力以及更一致的距离−概率关系。
2.3 深度学习架构
在蛋白质表示学习中,采用多模态特征融合策略。来自不同来源的蛋白质特征首先通过各自独立的多层感知机映射到统一的潜在空间,以实现尺度对齐和非线性特征变换。随后,将来自ESM和Ankh蛋白语言模型的隐藏表示与几何结构特征和理化性质特征在特征维度上进行拼接,并通过一个融合多层感知机进行进一步整合。该过程可形式化表示为
其中,
整体模型采用统一的架构设计,将多通道图编码与双向跨模态注意力机制相结合,从而在同一框架下同时建模配体−蛋白结合位点与结合亲和力。如图1所示,首先将配体分子图和蛋白Top-K残基图分别输入各自的图编码器,通过多层图Transformer进行消息传递和上下文聚合,得到高维的原子级和残基级表示。与此同时,实验测定条件的文本描述通过一维卷积网络编码为语义向量。随后,引入双向跨模态注意力作为模型的核心模块,原子与残基表示分别作为Query、Key和Value,实现对配体−蛋白相互作用的精细建模,同时融合实验语义信息以增强上下文感知能力。
标准的跨注意力形式为,当Query来自模态A而Key和Value来自模态B时,其定义如下
其中,
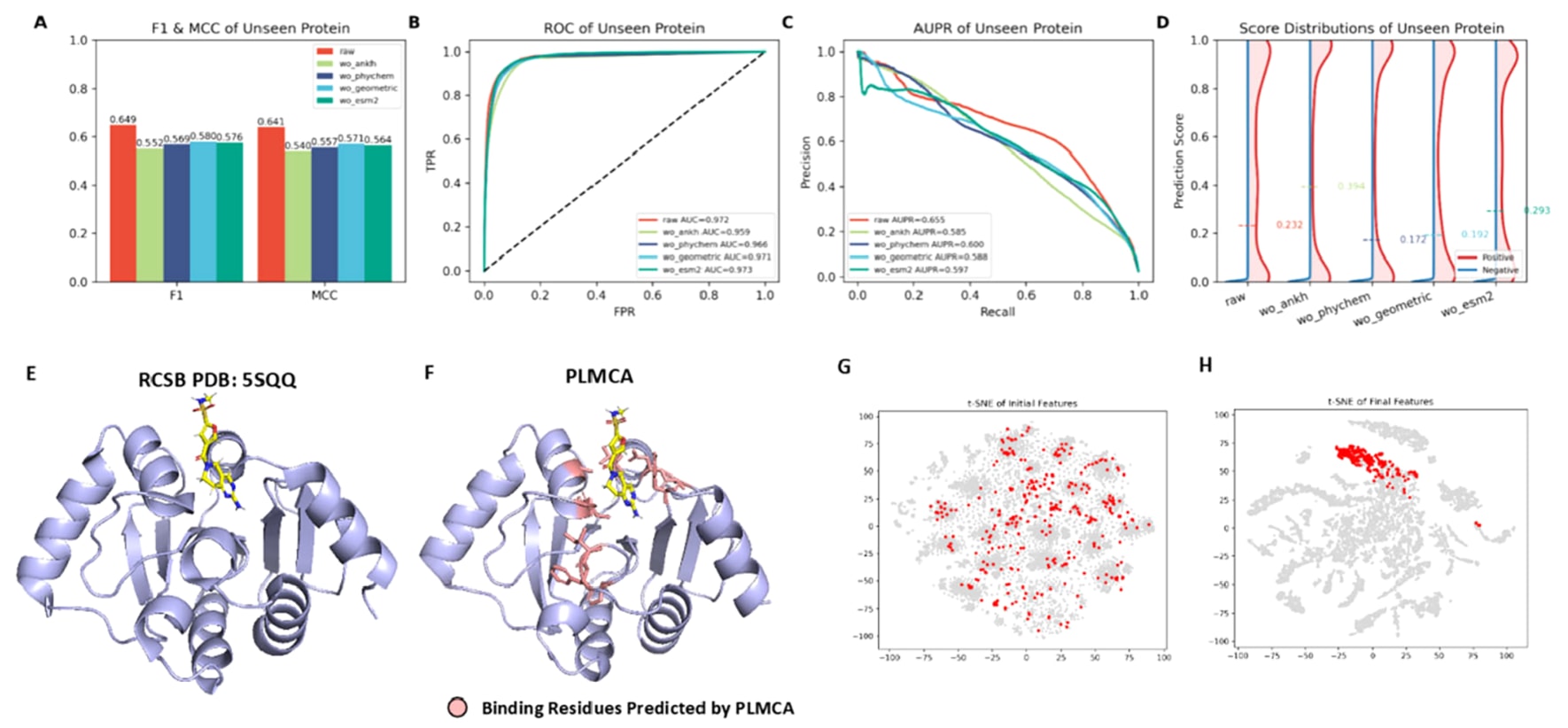
图4|(A)在不同消融设置下,未见蛋白条件中的F1和MCC指标。 (B)特征移除变体对应的ROC曲线。(C)未见蛋白条件下的AUPR曲线。(D)正负样本的得分分布情况。(E)参考蛋白−配体复合物的晶体结构,PDB编号为5SQQ。(F)PLMCA预测的结合残基映射到蛋白结构上的结果,与实验观测到的结合位点表现出较强的一致性。(G)初始残基特征的t-SNE可视化。(H)学习后特征的t-SNE可视化,显示出结合残基在特征空间中的聚类效果得到明显提升。
2.4 训练策略
为构建稳健的配体−蛋白结合位点与结合亲和力预测模型,采用了标准的深度学习训练策略。原始数据集按照8:1:1的比例划分为训练集、验证集和测试集。在训练过程中,使用AdamW优化器,其中结合位点预测任务采用交叉熵损失函数,结合亲和力预测任务采用均方误差损失函数。学习率在结合位点预测任务中设置为0.0005,在结合亲和力预测任务中设置为0.001。为缓解过拟合问题,引入了早停机制,当验证集损失在连续10个epoch内未得到改善时即终止训练。每个epoch结束后,模型都会在验证集上进行评估,并保存性能最优的模型参数及其对应的预测结果,用于后续分析和测试。在配体−蛋白结合位点预测任务中,训练通常在一块显存为24 GB的NVIDIA RTX 4090 GPU上耗时1至2小时,系统内存占用约为32 GB。相比之下,在ChEMBL数据集上进行蛋白−配体结合亲和力预测时,训练在同样的24 GB RTX 4090 GPU上大约需要2天时间,系统内存消耗约为64 GB。
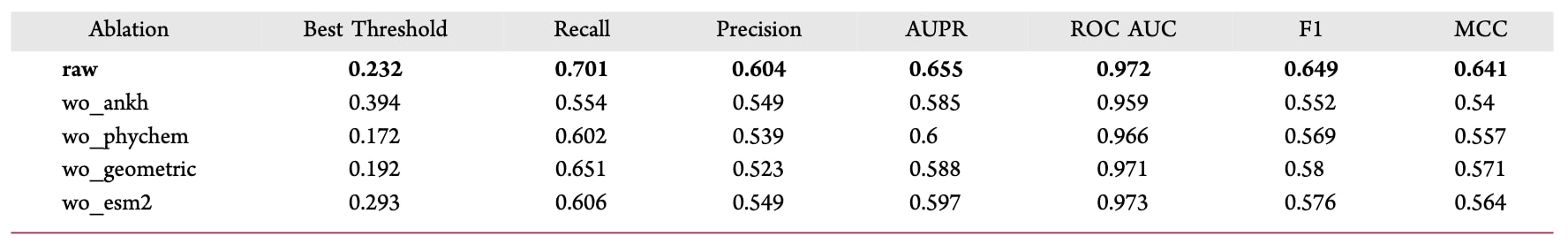
表1|PDBbind21口袋数据集的性能对比结果。
2.5 Webserver
PLMCA Web服务器旨在实现高效且易用的分子与蛋白亲和力预测。前端基于React构建,并采用Ant Design进行界面设计,从而提供响应式且直观的用户体验。后端采用基于Django实现的模型−视图−控制器架构,部署了预训练的机器学习模型以支持实时预测。服务器支持SMILES分子结构字符串和UniProt蛋白标识符等输入格式。该平台在保证预测精度的同时兼顾可解释性,为药物研发流程提供了有力支持。
3 结果
3.1 数据集统计
为全面刻画该研究中使用的数据集,对来自ChEMBL的分子、蛋白以及实验测定相关属性分布进行了分析,如图2所示。图2(A)展示了化合物中重原子数的分布,大多数分子包含少于40个重原子,平均值约为31,表明该数据集主要覆盖典型的类药化学空间。图2(B)给出了蛋白序列长度的分布范围,从短肽到长度超过1000个氨基酸的蛋白不等,平均长度约为520个残基。图2(C)显示了实验测定描述长度的分布情况,大多数描述少于40个单词,平均长度约为17个单词。图2(D−F)分别展示了以pIC50、pKd和pKi表示的结合亲和力数值分布,这些数值通过对原始亲和力测量结果进行
3.2 PDBbind21口袋数据集上的比较
在随机划分、未见蛋白、未见配体以及蛋白和配体同时未见这四种评估设置下,PLMCA整体表现与LABind具有竞争力,并在未见蛋白场景中显著优于LABind(图3)。在随机划分条件下,PLMCA取得了0.808的F1值、0.828的MCC、0.989的AUC以及0.872的AUPR(图3A−D)。在该设置中,PLMCA优于LigBind,但性能略低于LABind。在未见蛋白设置下(图3F−J),PLMCA的F1值为0.649,MCC为0.628,明显优于LABind和LigBind。对应的ROC曲线(AUC=0.972)和AUPR值(0.655)表明,即使测试集中所有蛋白在训练阶段均未出现,模型仍保持了较强的泛化能力(图3G−H)。在未见配体设置中,即测试集中所有配体均不同于训练集,PLMCA依然取得了与LABind相当的性能(图3K−O),具体表现为F1为0.806、MCC为0.799、AUC为0.987、AUPR为0.870。在最具挑战性的蛋白和配体同时未见设置下,PLMCA仍取得了最高的整体性能,F1值为0.615、MCC为0.604(图3P),略高于LABind(F1=0.601,MCC=0.591)。对应的ROC和AUPR曲线(图3Q−R)进一步表明,在严重分布偏移条件下,PLMCA依然能够保持有效的预测能力。此外,还评估了Top-K召回率和精确率指标,结果见表S1,结论与上述分析一致。总体来看,尽管PLMCA在预测得分上对正负残基的可分性略低,但距离−概率回归分析(图3E、J、O、T)验证了预测的结合概率与真实空间距离之间具有较强相关性,使模型能够对更靠近配体的残基赋予更高的置信度。综合而言,PLMCA在未见蛋白场景中表现出尤为突出的优势,说明多模态蛋白表示的引入有效提升了模型对新蛋白结构的泛化能力。
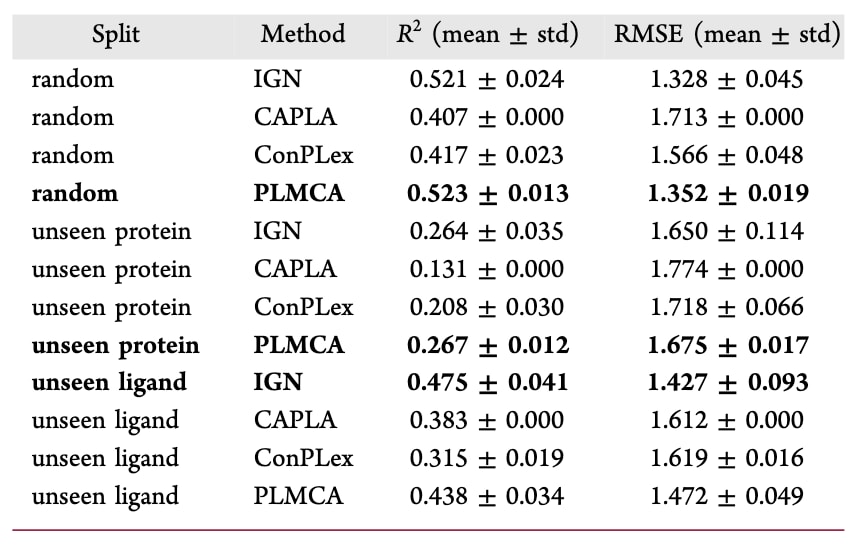
表2|不同模型在Kd数据集上的性能对比结果
3.3 消融实验
为评估PLMCA在未见蛋白条件下的泛化性能,在多种消融设置下进行了系统比较(图4A−D和表1)。如图4A所示,完整模型(raw)取得了最高的F1值0.649,明显优于去除ANKH(0.552)、去除理化性质特征(0.569)、去除几何特征(0.580)以及去除ESM2嵌入(0.576)的消融版本。MCC指标上也呈现出相同趋势,完整模型达到0.641,而各消融模型的MCC范围为0.540至0.571。这些结果表明,各类特征均对模型性能具有正向贡献,完整的特征表示能够带来最强的预测能力。ROC曲线(图4B)进一步验证了方法的稳健性,完整模型的AUC为0.972,略高于不含ANKH(0.959)、不含理化性质特征(0.966)、不含几何特征(0.971)以及不含ESM2嵌入(0.973)的变体。图4C中的精确率−召回率分析显示,完整模型的AUPR为0.655,高于各消融设置下的结果(AUPR介于0.588至0.608之间)。ROC和AUPR曲线的整体提升表明,PLMCA能够有效捕获来源于结构和序列的关键信息,用于结合残基的识别。以参考结构5SQQ为例(图4E),PLMCA预测的关键结合残基与晶体结构中观测到的结合位点高度一致,预测得分最高的前10个残基如图4F所示。最后,t-SNE可视化结果展示了模型学习表示的有效性。初始特征(图4G)中,结合残基的分布较为分散,而经PLMCA优化后的特征(图4H)形成了更加紧凑且分离良好的结合簇,说明模型能够有效重塑特征空间,从而增强结合残基与非结合残基之间的区分度。
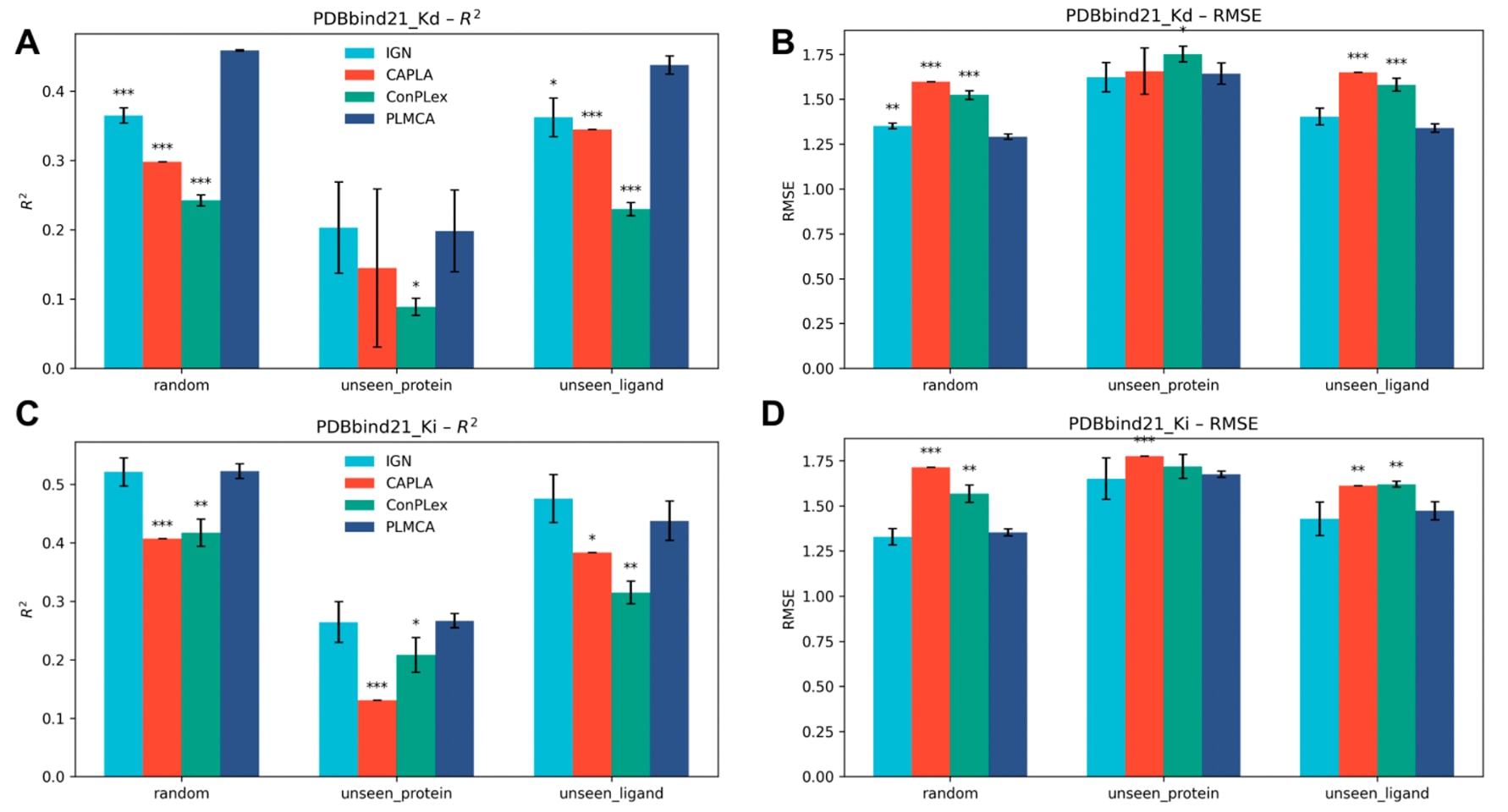
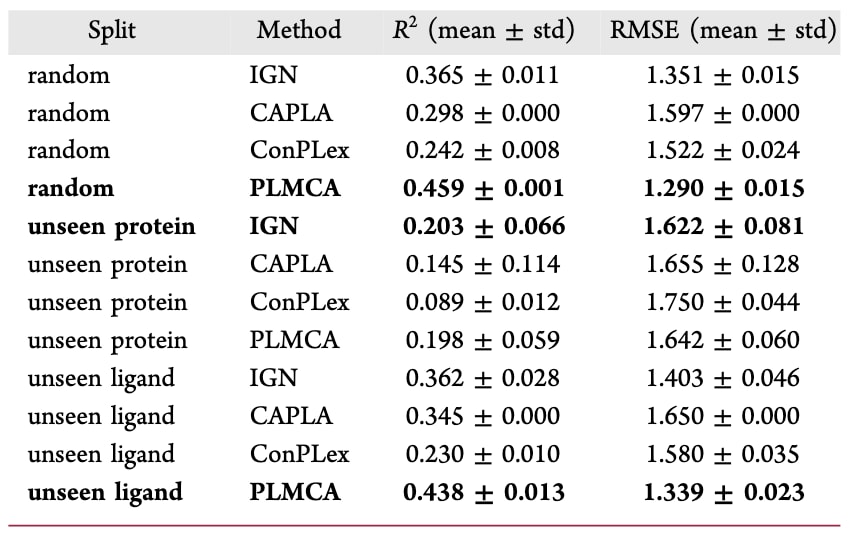
3.4 PDBbind21亲和力数据集上的比较
在表S2中,对多种常用基线模型的输入特征、模型架构及相关信息进行了对比。图5汇总了在PDBbind21亲和力基准上、采用随机划分、未见蛋白和未见配体三种数据划分策略时,五次独立运行的平均回归性能。性能指标包括Kd和Ki数据集上的
表3|不同模型在Ki数据集上的性能对比结果
3.5 ChEMBL亲和力数据集上的性能
在ChEMBL_mini数据集上,对PLMCA在IC50、Kd和Ki三种亲和力预测任务中的回归性能进行了评估(图6)。在IC50任务上,PLMCA取得了0.531的
3.6 Webserver
为提升用户可访问性,已将PLMCA部署为Web服务器,并免费开放,地址为https://hwwlab.com/Webserver/plmca。PLMCA是一款面向药物−靶点相互作用预测的快速、准确且具备可解释性的工具。平台提供两类预测任务,包括针对特定靶点的虚拟筛选以及针对特定化合物的靶点挖掘。用户可通过PLMCA主页进入预测页面(图S2)。在虚拟筛选任务中,用户首先选择支持的靶点,然后输入候选化合物的SMILES字符串,系统提供“Example”按钮用于快速填充示例。输入格式要求每行一个SMILES字符串,每次请求最多支持500个分子。平台通常在数分钟内返回预测结果,包括每个化合物对应的IC50、Ki和Kd数值。
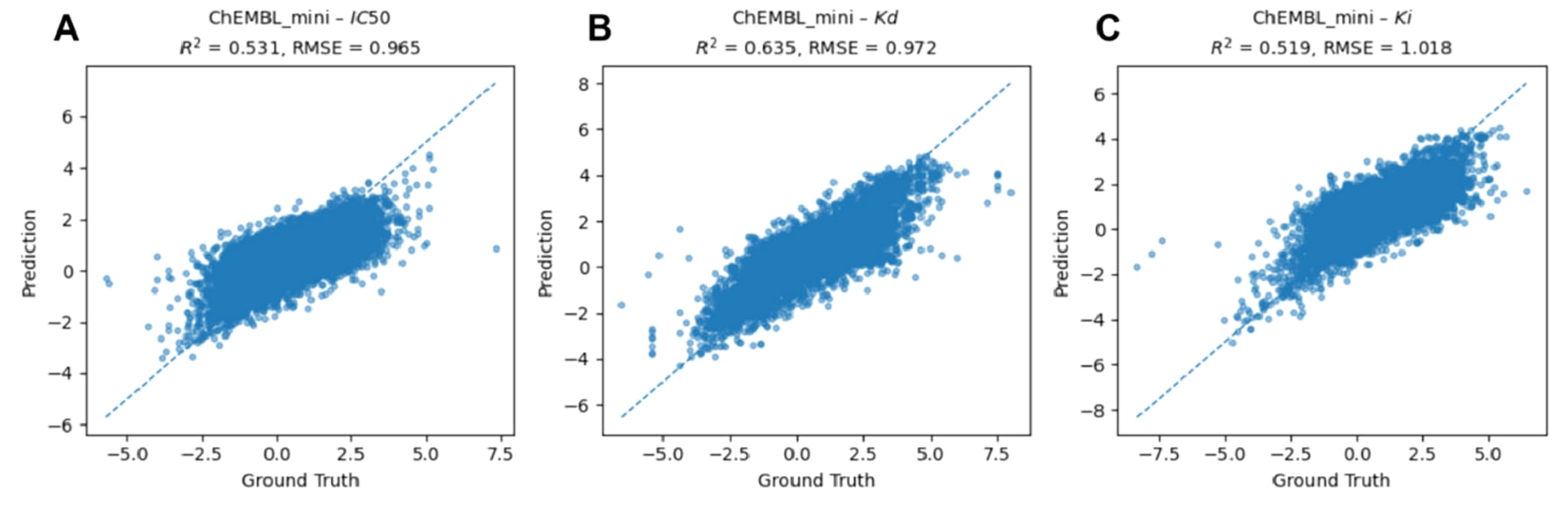
图6|ChEMBL_mini亲和力基准上的回归性能。 ChEMBL_mini数据集中IC50(A)、Kd(B)和Ki(C)任务的预测值与真实值对比,亲和力数值以
4 讨论
PLMCA框架的创新性主要体现在其多模态融合策略上,该策略有效应对了药物−靶点结合亲和力预测中的关键瓶颈。该模型首次在同一体系中同时整合了蛋白序列的语义信息(由两个蛋白语言模型编码)、三维几何特征、理化性质描述符以及配体分子图表示,并通过蛋白−配体跨注意力机制对这些异构模态进行统一建模。这种设计突破了传统方法通常仅关注精细结构表征或全局上下文信息之一的局限。此外,通过引入来自ChEMBL数据库的实验测定条件作为辅助输入,模型有效缓解了批次效应和测量噪声,从而显著提升了整体鲁棒性。值得注意的是,在未见蛋白等具有挑战性的场景下,PLMCA仍保持了较强的泛化能力,在ChEMBL_mini的IC50数据集上取得了0.531的
尽管如此,该模型仍存在一些局限性。首先,对多模态信息的依赖可能显著增加计算复杂度,从而在高通量虚拟筛选应用中限制其效率。目前,该框架尚未引入显式的稀疏化或近似策略,例如局部感知注意力、残基剪枝或低秩注意力变体,以降低计算开销。未来若能引入此类技术,有望在保持相互作用建模精度的同时显著提升计算效率,从而拓展PLMCA在大规模蛋白体系和真实药物研发流程中的适用性。其次,模型性能在很大程度上依赖于各模态数据的质量和完整性,任一模态中存在缺失或噪声信息,都可能对预测稳定性产生不利影响。此外,当前工作尚未系统评估模型对不同结合机制(例如变构调控或共价结合)的适应能力,其在更复杂相互作用类型上的泛化性能仍有待进一步验证。
总体而言,PLMCA通过多模态协同与上下文感知建模,为AI驱动的药物设计提供了一种新的统一框架。未来工作可进一步探索模型轻量化、跨模态表示对齐的优化策略,以及在更广泛结合机制下的泛化能力提升,从而加速精准药物发现的进程。