Cell 2026 | PocketXMol: 基于原子相互作用实现三维分子生成的统一建模

获取详情及资源:
0 摘要
该研究提出了PocketXMol,一种原子级模型,用于统一处理与蛋白口袋相互作用相关的生成式任务。通过将原子提示作为任务规范,该模型无需针对具体任务进行微调,即可支持多种分子建模任务,包括小分子和多肽的结构预测以及从头设计。在13项计算基准测试中,PocketXMol在其中11项上取得了优异表现,在其余两项上也保持竞争力,整体性能超过55个基线模型。在实际应用中,该模型被用于设计抑制caspase-9的小分子,其抑制效果可与商业化的广谱caspase抑制剂相媲美。同时,利用PocketXMol生成了能够结合PD-L1的多肽,其成功率显著高于传统文库筛选方法。选取的三个代表性多肽进一步接受实验验证,结果证实了其在细胞水平上的特异性,并表明其在分子探针开发和治疗应用方面具有潜在价值。总体而言,PocketXMol为人工智能辅助药物发现提供了一个通用平台,并为未来更广泛的应用场景奠定了基础。
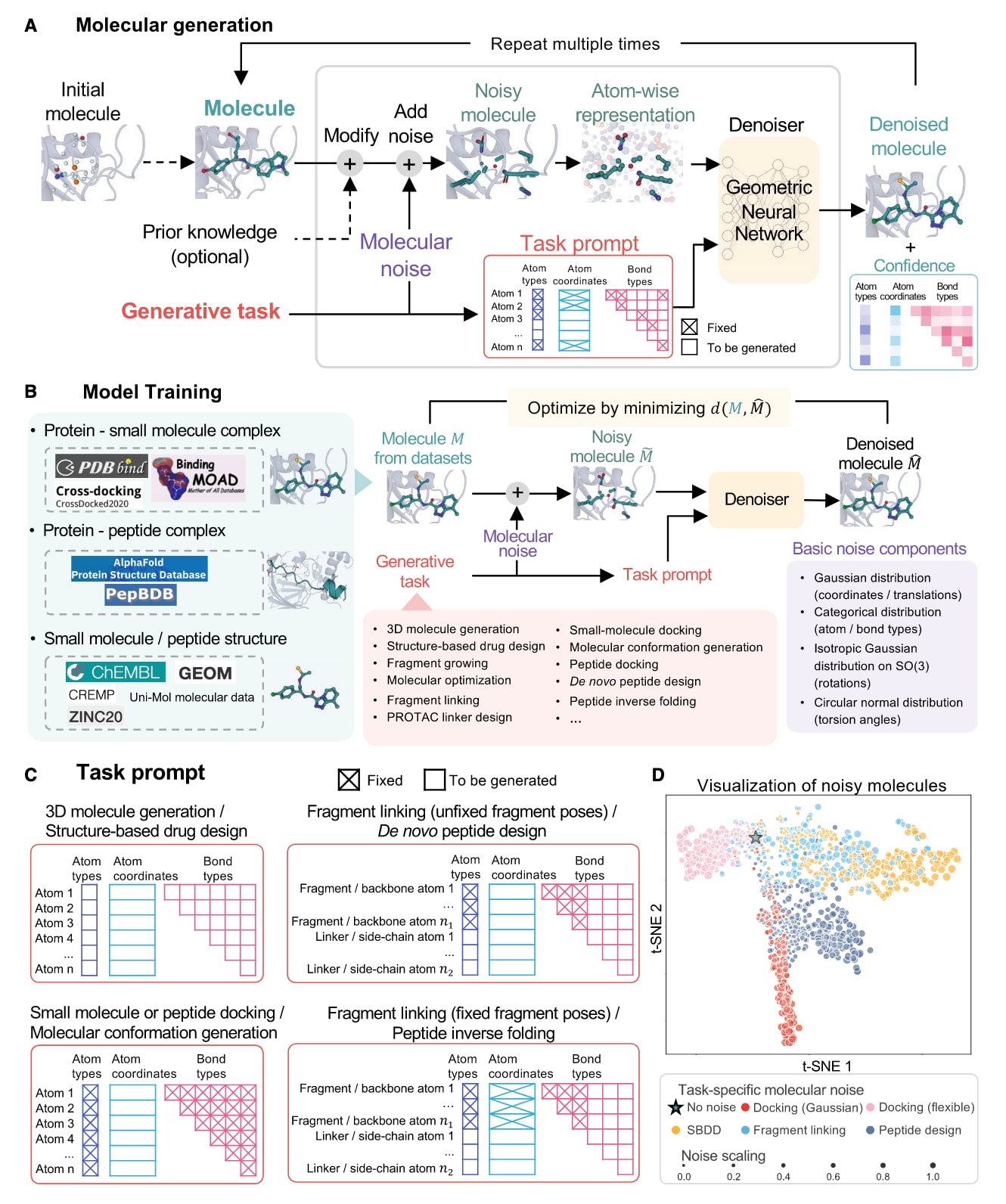
图1|PocketXMol框架。 (A)生成流程的示意图。(B)训练过程示意。PocketXMol采用通用去噪框架,整合来自多种分子类型的结构数据,在统一噪声空间中实现多任务联合建模。
1 引言
人工智能已在分子结构预测和分子设计领域带来深刻变革,但现有模型通常依赖针对特定任务定制的专用算法。一个关键认识在于,所有分子任务本质上都受原子相互作用这一普适物理规律所支配。因此,一个自然的问题是,是否可以借鉴自然语言和计算机视觉等领域的基础模型范式,基于大规模数据构建一个统一的原子级模型,以刻画分子相互作用的基本规律。然而,要实现多任务统一仍面临多重挑战。尽管文本提示在大语言模型中已被证明有效,自然语言在定义分子任务时却缺乏精确性,尤其是在涉及复杂空间关系或多个分子片段时更为明显。现有生成模型的另一局限在于,尽管在单一任务上表现出色,其依赖特定任务的先验分布与采样过程,限制了多任务联合学习的整合能力。此外,当前尚缺乏统一的分子表示方式,不同模型采用类型特异的表示格式,在本质上限制了跨任务与跨分子类型的迁移能力。
为应对上述问题,提出了PocketXMol这一统一的原子级生成模型,其核心由三部分构成:首先,通过原子级任务提示机制,直接在原子层面定义输入与输出,相较文本描述提供更精确的控制能力;其次,构建“通用去噪器”架构,将不同任务分布映射至统一的噪声空间,从而实现无需任务特定微调的多任务联合训练;再次,采用原子级任务表示方式,绕过显式氨基酸建模,提升不同分子类型之间的可迁移性。为训练该模型,构建了包含三维分子结构的大规模数据集,涵盖11985300个小分子,39911个蛋白-多肽复合物,以及来自PDBBind、Binding MOAD、CrossDocked2020、PepBDB、AlphaFold DB、ChEMBL、ZINC、GEOM-Drug、CREMP以及Uni-Mol dataset的85434个蛋白-小分子复合物等数据。
在评估方面,模型在13类生成任务上进行了系统测试,包括小分子对接、线性与环状多肽对接、三维构象预测、基于结构的药物设计、三维分子生成、片段连接、PROTAC设计、片段延伸、分子优化、线性与环状多肽设计以及多肽逆折叠等,并与55种基线方法在51项指标下进行比较。结果显示,PocketXMol在13项任务中的11项取得显著优势,在其余任务上亦保持高度竞争力。同时,模型在多种实际应用场景中展现出实用价值,包括结合先验知识的约束对接、酶-底物筛选、虚拟筛选以及非标准氨基酸设计。
在实验验证方面,利用PocketXMol设计了16种caspase-9抑制剂,其中筛选出1个有效分子,能够抑制caspase-9并在ABT-737处理条件下抑制下游caspase-3及多聚ADP-核糖聚合酶1(PARP1)的裂解。此外,还设计了靶向程序性死亡配体1(PD-L1)的多肽,在382条合成多肽中鉴定出15个结合亲和力达到
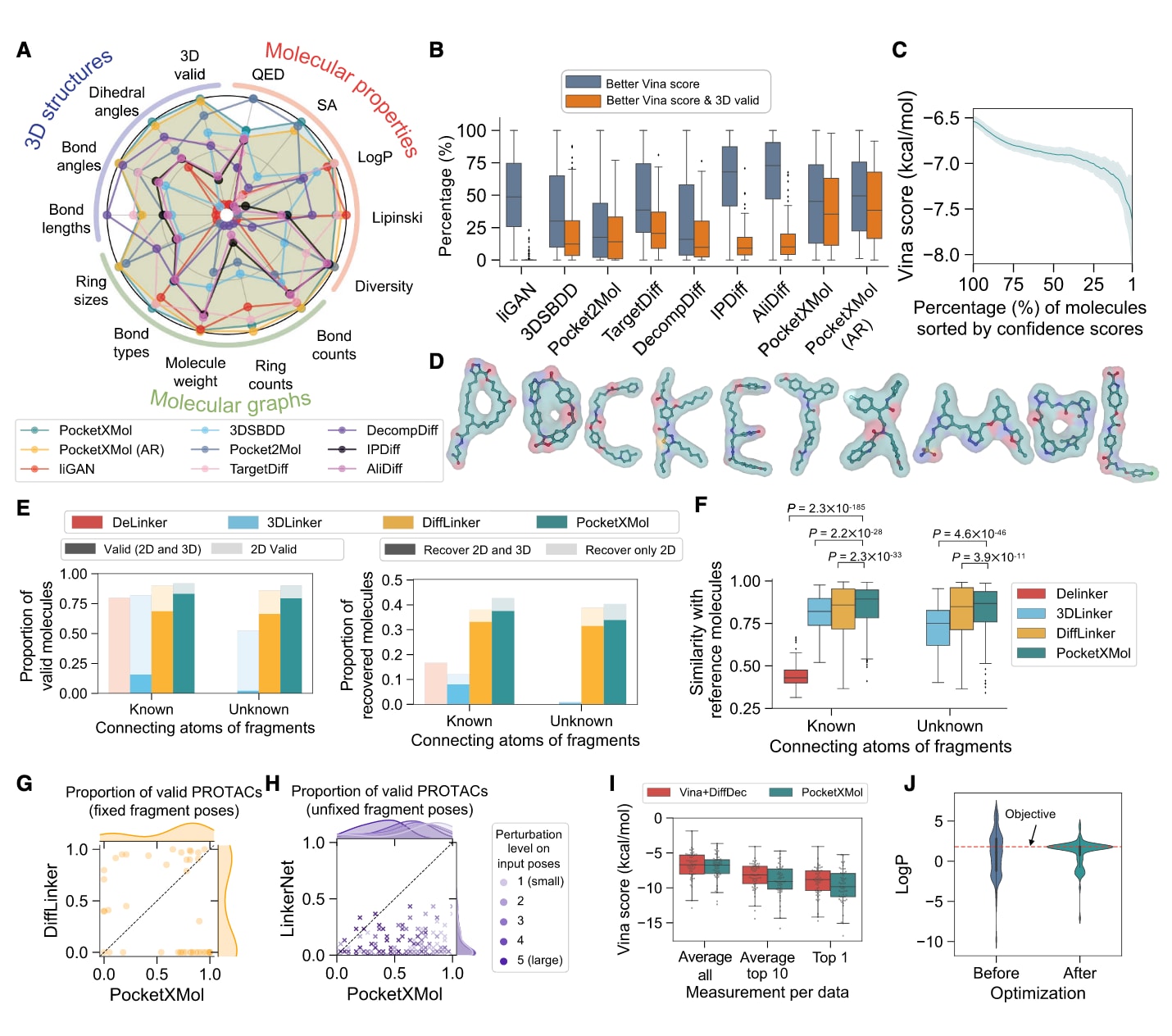
图2|小分子设计任务的性能表现。 (A)基于结构的药物设计(SBDD)任务结果。所有指标均归一化至0到1区间,其中1表示最佳方法,0表示最差方法。(B)在各个蛋白口袋上,同时具有更优Vina评分和三维结构有效性的生成分子比例。与不同基线方法相比,PocketXMol对应的
2 结果
2.1 PocketXMol概述
PocketXMol将输入分子抽象为由原子与化学键构成的集合,以学习原子层面的基本相互作用规律(图1A、1B和S1)。每个任务均通过任务提示进行定义,该提示由一组二值指示变量组成,用于控制哪些分子变量需要被生成(图1C)。模型主体为几何神经网络,通过迭代方式对带噪输入进行加噪与去噪。不同于采用预定义噪声调度的扩散模型,PocketXMol根据任务提示自动确定不同原子组分所需噪声的类型与尺度,从而将多种不同噪声形式映射至统一且可观测的分子表示空间。这一机制使模型能够仅依据受噪声扰动的分子直接识别任务类型,而无需额外的任务特定参数(见STAR Methods)。基于这种统一表示,所有生成任务在综合数据集上进行联合训练,以学习共享的结构模式(图1B和1D)。因此,PocketXMol无需针对不同任务进行微调即可直接应用,相比许多预训练模型具有显著优势。
2.2 小分子设计任务中的性能表现
基于结构的药物设计(SBDD)任务旨在根据蛋白口袋结构设计三维小分子。针对100个基准蛋白口袋,为每个口袋生成100个分子,并与多种SBDD方法进行比较。覆盖分子性质、二维分子图质量及三维结构保真度等14项指标的评估结果显示,PocketXMol在其中11项指标上排名第一(图2A;表S2)。在基于refine策略与自回归采样两种生成方式下,模型表现保持一致,表明其优势主要来源于训练机制本身,而非特定采样策略(图2A)。
对于每个口袋,统计生成分子中Vina评分优于参考分子的比例,并结合PoseBusters三维有效性筛选以控制结构有效性与结合强度之间的潜在权衡。PocketXMol在每个口袋上生成的高质量三维分子比例中位数为35.37%,显著高于第二名基线方法的20.54%(图2B和S2A)。同时,模型给出的置信度评分与Vina评分呈相关性,有助于对候选分子进行有效优先排序(图2C和S2B)。
为评估模型对输入口袋结构变化的鲁棒性,还基于不同来源的口袋结构进行分子生成。考虑了四类口袋结构:(1)由AlphaFold预测的结构;(2)经Rosetta重新打包侧链的结构;(3)来自Protein Data Bank且序列相同的结构;(4)来自Protein Data Bank且序列相同的apo结构(见STAR Methods)。不同来源口袋的结果整体一致,尤其是在AlphaFold预测结构与Protein Data Bank检索结构上表现稳定(图S2D–S2F;表S2)。侧链重排口袋产生较弱Vina评分,可能与口袋部分遮挡有关;而apo结构引起的分子性质变化较小,符合holo与apo构象差异的预期。
2.2.1 三维分子生成
相较于SBDD任务,三维分子生成任务不依赖蛋白口袋约束,在药物发现中可作为计算机虚拟筛选候选分子的替代策略。生成1000个与真实药物分子尺寸相近的分子,并与多种基线模型进行比较。结果表明,PocketXMol生成的分子具有较高有效性,其原子间距离分布与测试集中类药分子的分布高度一致(图S2C和S2G)。
进一步测试了在体积形状约束下引入先验知识的能力。在预设体积形状限制条件下生成分子,所得结果不仅符合给定形状要求,同时在拓扑结构与空间构型上保持合理性(图2D;表S2),显示出模型在受限条件下进行可控生成的能力。
2.2.2 片段连接
围绕已有片段或分子的分子设计策略包括片段连接、片段延伸以及分子优化,这些方法通过对现有分子的特定化学基团进行修饰,以获得期望性质,在药物研发中被广泛应用。片段连接的核心在于设计合适的连接子,将彼此分离的片段整合为完整且稳定的分子结构。这一技术亦可拓展至PROTAC药物开发,其中需要构建分子连接子以连接靶向蛋白的配体与招募E3泛素连接酶的配体。
在DiffLinker dataset上,针对固定片段连接设计模型进行基准测试,在已知或未知连接原子的条件下,为每对片段生成100个三维分子。结果显示,PocketXMol在二维与三维有效性、结构恢复率以及与参考分子的三维相似性方面均优于基线方法(图2E和2F)。在PROTAC连接子设计任务中,基于包含43个PROTAC分子的测试集,为每对warhead-E3配体生成100个连接子。DiffLinker需要事先提供两个三维片段的相对位置,而LinkerNet则无需该信息。因此在两种条件下分别进行比较。给定片段构象时,PocketXMol生成有效分子的比例达到59.60%,显著高于DiffLinker的34.03%,并且在与真实PROTAC分子的结构相似性方面表现更佳(图2G和图S2I)。在未知片段构象条件下,对输入片段施加多级随机扰动,并在每个扰动水平下为每个口袋生成30个分子。PocketXMol的分子有效性达到56.88%,明显优于LinkerNet的6.99%,同时保持与真实PROTAC分子的较高结构相似性(图2H和图S2I)。
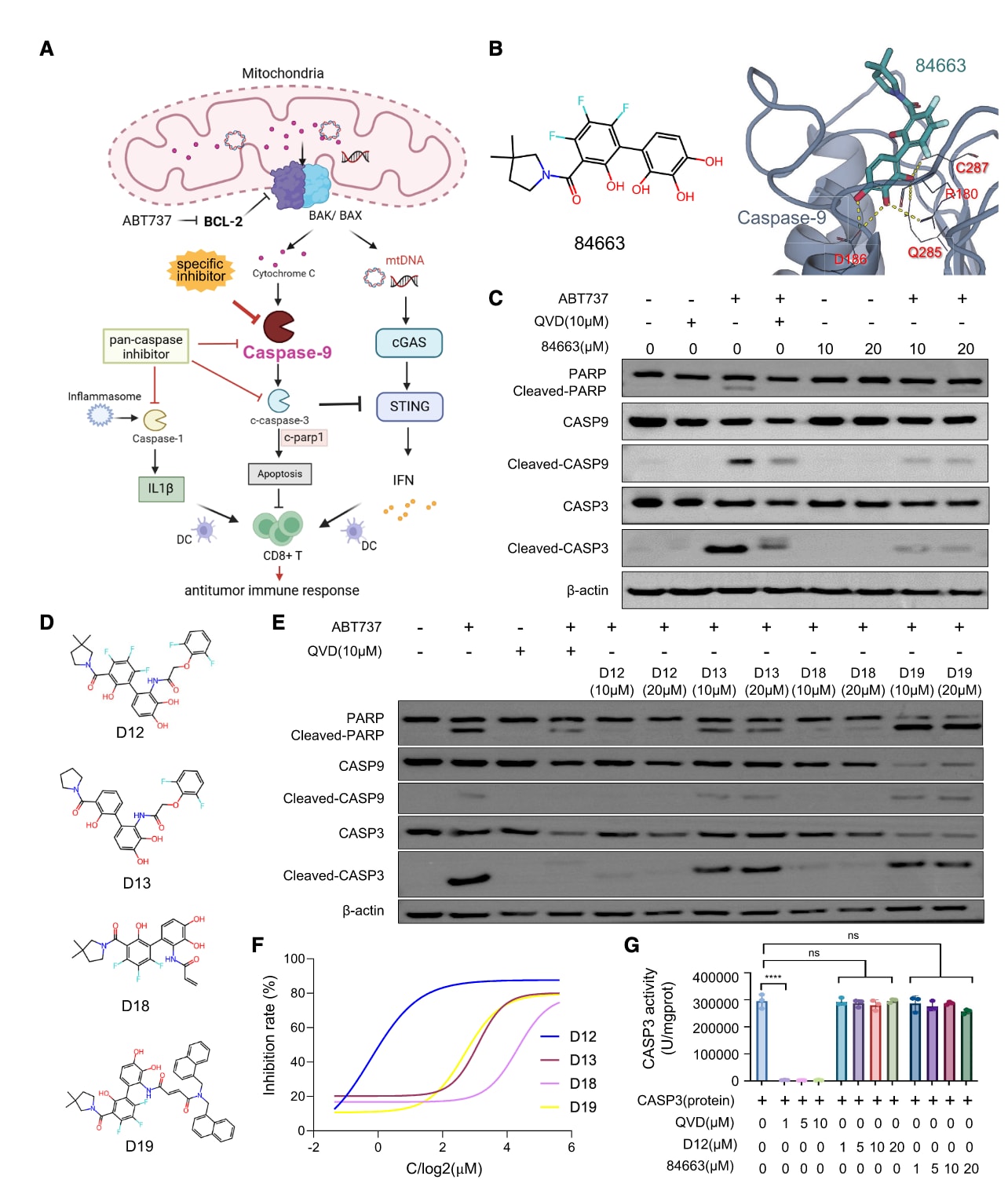
图3|Caspase-9抑制剂设计。 (A)caspase-9相关信号通路示意图。(B)设计分子84663及PocketXMol生成的结合构象。黄色虚线表示由PyMOL识别的极性相互作用,相关氨基酸以残基名称和编号标注。(C)Western blot检测PARP及caspase-9/-3的表达水平。MC38细胞分别接受QVD或84663处理,并与或不与ABT-737(10μM)联合处理。“CASP”为“caspase”的基因名称。(D)第二轮优化并验证可结合caspase-9的分子(D12、D13、D18和D19)。(E)Western blot检测PARP及caspase-9/-3表达。MC38细胞接受不同caspase抑制剂处理,并与或不与ABT-737联合使用。(F)caspase-3活性实验用于测定各分子抑制由caspase-9介导的ABT-737诱导caspase-3活化的半数有效浓度(EC50)。(G)利用caspase-3检测试剂盒,对caspase-3蛋白(0.5ng/mL)与不同caspase抑制剂混合后的酶活性进行分析。另见图S3。
2.2.3 片段延伸
片段延伸策略以较小分子片段为起点,通过化学修饰逐步扩展为性质更优的完整分子(图S2K)。在无片段构象信息条件下,针对每个口袋-片段对生成100个分子,并与采用AutoDock Vina进行预对接、结合DiffDec生成的两步法基线进行比较。结果表明,PocketXMol在分子完整性、与参考分子的相似性以及与靶标的结合亲和力方面均优于DiffDec(图2I、S2K、S2M和S2N)。分子完整性提高26.03%,与参考分子的相似性提升47.64%,并取得更优的Vina评分。此外,生成分子的Vina评分相较测试集中原始分子亦有提升(图S2L)。
2.2.4 分子优化
分子优化旨在在保持结构相似性的同时提升目标性质。PocketXMol在该任务中采用与SBDD相同的任务提示,但以输入分子为初始状态并施加较低噪声水平,从而生成部分保留原结构的变体。结构相似性通过噪声强度进行隐式调控,噪声越低,生成分子与原分子的相似度越高。每轮生成后筛选出性质改进的分子作为下一轮种子分子(见STAR Methods)。
以100个测试分子的LogP值优化为例,目标值设定为1.8这一常见理想区间。优化后的分子迅速向目标值收敛,其误差显著小于初始分子(单侧配对
2.2.5 Caspase-9抑制剂设计
由caspase-9介导的内源性细胞凋亡过程是肿瘤内源性免疫感知通路的重要调控环节(图3A)。然而,作为细胞死亡的核心调节因子,caspase家族蛋白在抗肿瘤治疗中具有高度多样化的功能,而现有首创类caspase抑制剂难以有效区分不同家族成员。基于PocketXMol设计了靶向caspase-9的候选化合物,合成16个设计分子(图S3A),其中编号84663(图3B)能够有效抑制ABT-737诱导的caspase-9和caspase-3活化(图3C和S3B)。该分子与已知caspase抑制剂相似性较低(图S3C),在常见化学数据库中亦无高度相似结构(图S3D和S3E)。
在此基础上对84663进行结构优化,合成15个优化分子(图S3H),其中D12、D13、D18和D19表现出显著活性(图3D)。所设计分子,尤其是D12,在抑制caspase-9和caspase-3活化方面的效果可与QVD-OPh(QVD)及Z-LEHD-FMK TFA相媲美(图3E、3F和S3F)。caspase-3酶活实验显示,D12与84663并不直接抑制caspase-3(图3G),不同于QVD,支持其通过结合caspase-9发挥作用的机制。利用丙氨酸突变体(C287、R180、Q285;D186未表达)进行表面等离子共振实验发现,突变后结合亲和力降低(图S3G),其中C287A突变导致结合能力下降约30倍,进一步验证了预测的结合位点。
2.3 多肽设计性能
在原子级表示框架下,多肽设计在建模层面上不再与小分子设计存在本质区别。PocketXMol将多肽生成视为一种特殊的片段延伸任务:在给定多肽主链原子与三维蛋白口袋结构的条件下,同时生成侧链原子的类型与坐标,以及主链原子的空间坐标(图4A)。这一建模方式将多肽结构与序列的协同设计转化为端到端的分子原子生成过程,不同于显式建模氨基酸单元的方法。此外,模型直接输出全原子坐标,无需后续对接或侧链重排等后处理步骤。
2.3.1 线性多肽设计
为验证该设计策略,基于PepBDB对接基准数据集构建了包含35个蛋白-多肽复合物的测试集,针对每个蛋白口袋进行多肽设计。为评估模型是否学习到氨基酸的形成规律,对生成的侧链原子嵌入进行平均,并对所得氨基酸嵌入进行可视化(图4B)。聚类结果与真实氨基酸类型高度一致,调整兰德指数为0.813,且嵌入空间的排列反映了侧链之间的化学相似性。嵌入还捕捉到结构化的语义关系:例如谷氨酸(E)与谷氨酰胺(Q)之间的嵌入差异,与天冬氨酸(D)与天冬酰胺(N)之间的差异高度相似,两者均对应于
大多数生成的侧链对应标准氨基酸,且在不同口袋与多肽长度条件下均保持稳定分布(图4D)。将PocketXMol与标准RFdiffusion流程进行比较,后者先利用RFdiffusion生成主链结构,再通过ProteinMPNN设计序列,并使用Rosetta进行侧链重排。针对每个测试蛋白,两种方法均最多生成100条由标准氨基酸组成的多肽。结果显示,PocketXMol生成的氨基酸分布更接近测试集真实分布,Jensen-Shannon散度为0.16,低于RFdiffusion的0.23(图4E),平均序列恢复率亦更高(37.2%对32.1%;图S4C)。
在结构质量方面,PocketXMol生成结构的均方根偏差(RMSD)更低,Rosetta能量评分更优(图4F和S4D)。其生成多肽的二级结构比例更接近测试集分布,表明结构偏倚更小(图4G)。同时,满足MolProbity结构质量标准的多肽比例更高,且模型置信度评分与MolProbity评估结果相关(表S3)。
进一步在与PepBDB相似度较低的Q-BioLiP数据集上进行相同分析,结果保持一致(图S4A–S4J)。类似于SBDD任务,还评估了在不同口袋构象下的鲁棒性。整体性能与基于holo结构的结果基本一致(图S4K–S4N)。对于AlphaFold预测结构、Protein Data Bank检索结构或apo结构,主链RMSD略有升高,但该指标本身对口袋对齐高度敏感。基于Rosetta预测的结合能在apo结构中略显不利,可能源于其表面性质与holo构象之间的差异。
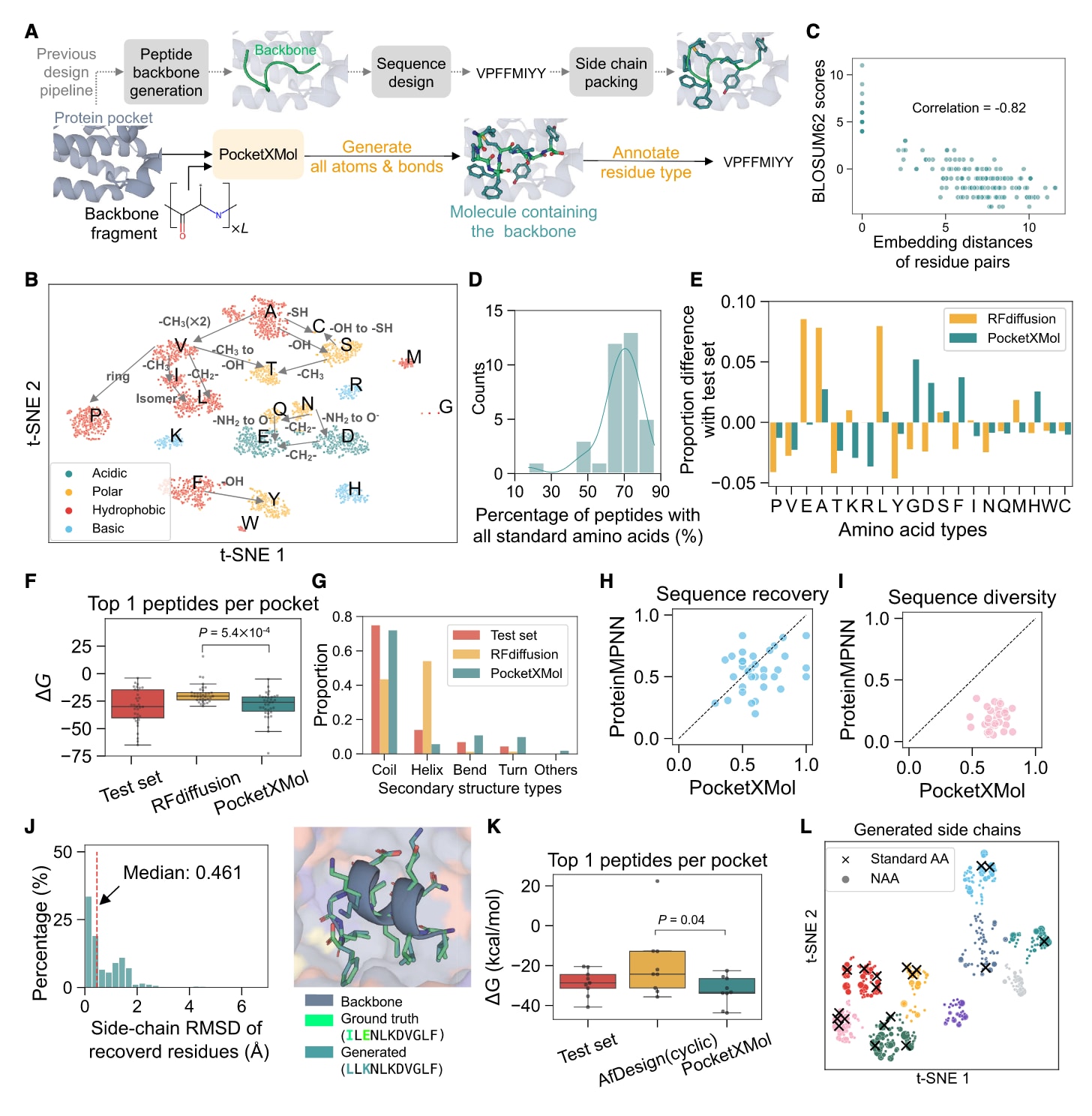
图4|多肽设计性能。 (A)PocketXMol与传统多肽设计流程的差异。(B)BLOSUM62替换得分与氨基酸嵌入距离之间的关系。(C)氨基酸嵌入的t-SNE可视化结果。(D)测试集中各蛋白口袋生成的由标准氨基酸组成的多肽比例。(E)生成多肽与测试集在氨基酸组成上的差异。(F)蛋白-多肽复合物的Rosetta结合能分布。
2.3.2 多肽逆折叠
多肽逆折叠旨在在已知主链结构与蛋白口袋结构的条件下设计多肽序列。与ProteinMPNN相比,PocketXMol在序列恢复率上表现更优(61.7%对54.4%,单侧配对
循环多肽设计方面,构建了包含26个蛋白-循环多肽复合物的数据集,并根据靶标序列一致性将其聚类为9组,每组选取一个代表性口袋。针对每个口袋最多生成100条循环多肽,并与AfDesign进行比较。结果显示,PocketXMol在每个口袋的top-1与top-10Rosetta结合能方面更优,在序列恢复率、主链RMSD及二级结构组成方面表现相当(图4K、S4O和S4P;表S3)。
2.3.3 含非标准氨基酸多肽设计
由于PocketXMol直接生成侧链原子而非预测氨基酸类型,因此天然支持非标准氨基酸(NAA)的设计。在直链多肽设计中,模型同时生成标准与非标准残基。在所有含NAA的多肽中,共识别出454种独特的NAA侧链,在尺寸、出现频率、新颖性与理化性质方面呈现多样性(图4L、S4S和S4T;表S4)。将高频NAA映射至其最接近的标准残基后进行Rosetta能量比较,结果显示含NAA多肽在能量上相当甚至更优(图S4Q和S4R),表明模型利用NAA以优化结合相互作用。
2.3.4 PD-L1结合多肽设计
进一步利用PocketXMol设计靶向PD-L1的10肽,并通过实验验证其结合亲和力与特异性。针对三个PD-L1口袋结构(PDB:3BIK、4ZQK和5IUS),生成多肽后,结合自置信度评分、FoldX、Rosetta以及AlphaFold3等工具进行筛选与排序(见STAR Methods)。最终选取382条多肽,通过表面等离子共振成像实验验证其与PD-L1的结合能力。结果显示,15条多肽达到
为排除排序过程的影响,在未进行后处理排序的情况下再次测试382条多肽,其中9条达到
在采样与筛选流程中获得的15条
体内靶向实验采用FDA批准的吲哚菁绿(ICG)染料进行成像。将与ICG偶联的多肽以50μM经尾静脉注射至PD-L1阳性的非小细胞肺癌模型小鼠体内,注射后0.5小时即在肿瘤部位观察到富集,4小时达到荧光峰值(图5E)。游离ICG未显示肿瘤信号,表明多肽探针具有特异性靶向能力。12小时离体成像显示多肽组肿瘤信号强烈,对照组几乎无信号(图5F和S5F)。肝脏与肾脏信号升高反映循环与代谢过程中的非特异性分布。相比之下,通过文库筛选获得的阳性多肽未能维持持久的肿瘤靶向(图S5G–S5J)。上述结果表明,由PocketXMol设计的多肽在体内具有优异的靶向性与选择性,适用于肿瘤成像与诊断。
进一步测试多肽P282是否能够抑制程序性死亡蛋白1(PD-1)/PD-L1相互作用。流式细胞术结果显示,在P282存在条件下PD-1与PD-L1的结合显著下降,且随多肽浓度升高,PD-1-Cy5信号进一步降低(图5G),提示其在肿瘤免疫治疗中的潜在应用价值。为验证结构预测结果,对PocketXMol识别的关键PD-L1残基进行丙氨酸扫描突变并测定结合能力(表S5)。对于P65与P73,多数突变导致亲和力下降超过10倍,支持预测的结合界面;对于P282,仅一个关键突变产生显著影响,其余影响较小,可能与其α螺旋结构的稳定性有关。
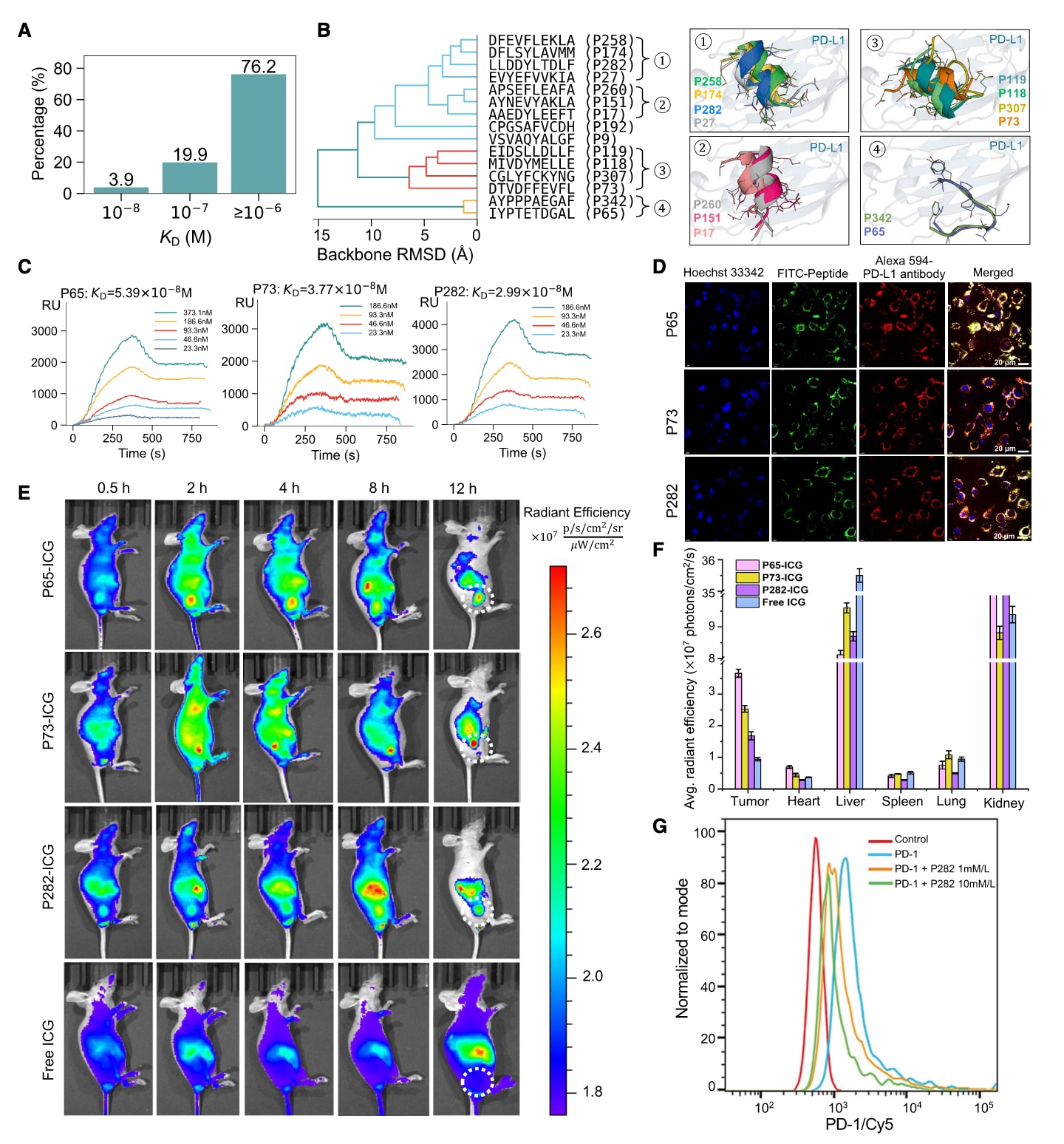
图5|PD-L1结合多肽设计。 (A)不同解离常数范围内设计多肽所占比例。(B)
2.4 分子结构生成性能
在PoseBusters基准数据集上评估了PocketXMol的小分子对接性能,该数据集包含428个蛋白-配体复合物。任务目标是在刚性蛋白口袋中对柔性配体进行对接。按照基准协议,各基线方法在默认设置下为每个蛋白提供一个对接构象。PocketXMol则为每个口袋生成100个构象,并通过自置信度评分(自排序)或经调优的置信度预测器(调优排序)选取最佳构象。计算原子级RMSD,并统计RMSD低于2Å的构象比例。结果显示,PocketXMol在自排序和调优排序下分别达到82.5%和83.4%,仅次于在已定义口袋条件下的AlphaFold 3(图6A)。RMSD与排序得分呈相关性,且随着每个口袋选择构象数量增加,性能进一步提升(图6B–6D)。在“oracle排序”(即每个口袋选择最佳构象)条件下,性能可达96.5%,表明通过更先进排序策略仍有显著提升空间。RMSD与分子性质无显著相关性,说明不存在分子偏倚(图S6A)。
进一步采用PoseBusters标准评估对接有效性,统计同时满足RMSD<2Å且通过PoseBusters有效性筛选(PB-valid)的构象比例。在428个复合物上,PocketXMol自排序与调优排序分别达到78.5%和79.4%(图6F;表S6)。在包含308个复合物的PoseBusters v.2数据集上,分别达到84.7%与84.4%,与AlphaFold 3在已定义口袋条件下报告的84.4%相当(图6G)。
为评估对口袋结构变化的鲁棒性,使用替代口袋结构进行对接。由于PocketXMol在训练中严格遵循输入坐标并避免空间冲突,引入PocketFlexible适配版本(PocketXMol-PF),通过在扰动口袋上微调提升对结构变化的容忍度(见STAR Methods)。对接构象仅依据模型自置信度排序。PocketXMol-PF在实验结构与Rosetta重排口袋上的表现与原始PocketXMol在实验结构上的性能相当,而原始模型在重排口袋上表现较差(图S6C)。对于其他口袋结构,在主链偏差适中的情况下,PocketXMol-PF仍保持较高准确率(图S6D)。随着口袋与原始结构偏差增大,配体RMSD有所升高,但主要源于结构对齐差异而非对接失败。小分子数据集中主链偏差整体较小,SBDD测试集、中对接测试集及下采样训练集的主链RMSD中位数分别为0.42Å、0.51Å和0.46Å(图S6B),表明AlphaFold预测口袋可作为PocketXMol-PF的可靠输入。
先验知识可通过分子编辑或任务提示方式整合。例如在已知片段构象的条件下进行对接。评估了包含已知分子中心位置、键长以及锚定原子近似或精确坐标等先验信息的对接任务。所有先验均降低RMSD,将RMSD<2Å的比例从83.4%提高至84.6%–85.5%,其中精确锚定原子坐标带来最大提升,源于对可行构象空间的大幅约束(图6E和6H)。
2.4.1 酶-底物识别
将PocketXMol应用于酶-底物对接任务,在多个酶-底物数据集中评估其自置信度评分区分活性与非活性底物的能力。结果显示,对接置信度在不同酶家族中均具有判别能力(图6J、6K和S6E),整体优于AlphaFold 3。四类酶家族的平均AUROC分别为0.65、0.62、0.85和0.61,而AlphaFold 3分别为0.51、0.53、0.59和0.60。
为实现直接活性预测,利用PocketXMol预测酶-底物复合物结构,提取神经网络隐藏表示,并在带有活性标注的数据上训练逻辑回归分类器(图S6F)。不同划分下的交叉验证结果显示,分类器优于原始对接置信度,AUROC提升5.9%–51.9%,AUPR提升35.2%–123.5%,表明模型学习到可迁移的相互作用模式,可通过简单模型有效预测酶-底物活性。
此外,通过量化底物及其反应基团与催化残基之间的距离,评估PocketXMol与AlphaFold 3生成催化合理构象的能力(图S6G)。在卤化酶与糖基转移酶中,PocketXMol更频繁地将底物及其反应基团定位于催化残基附近,在5Å范围内的比例显著更高(分别为54.15%对36.88%,95.07%对80.79%)。同时,反应基团更倾向于朝向催化位点而非底物中心,显示出更有利的催化取向。
2.4.2 虚拟筛选
进一步在DEKOIS 2.0基准上评估PocketXMol对接置信度区分真实结合物与诱饵分子的能力。在未进行任务特定微调的情况下,PocketXMol与PocketXMol-PF在六项标准指标上均达到与最佳基线KarmaDock相当的性能。两者的ROC_AUC中位数分别为0.787和0.788,与KarmaDock的0.786相当(图S6H)。PocketXMol-PF表现略优,与其针对缺乏holo结构场景优化的设计相一致。
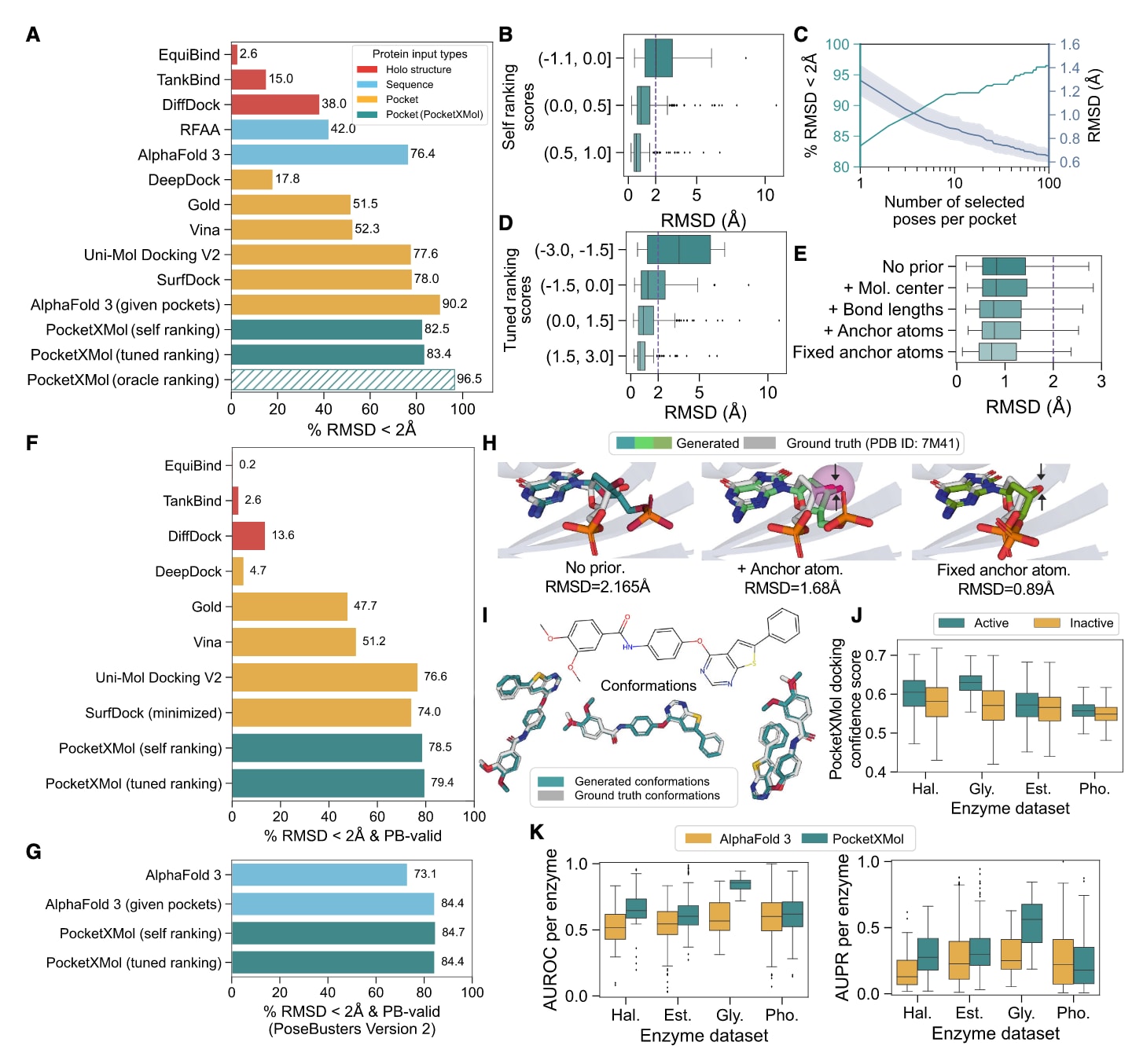
图6|小分子结构生成性能。 (A)小分子对接中RMSD<2Å的构象比例。(B,D)排序得分与对接RMSD之间的关系:(B)自排序,(D)调优排序。箱线图展示四分位数及
2.4.3 分子构象生成
PocketXMol在不依赖蛋白口袋条件下即可为小分子生成有效构象。按照既有研究的评测流程,将其与专门的构象预测工具进行比较。结果显示,PocketXMol的性能与专业方法相当(表S6),并能够恢复与低能量真实构象高度一致的三维结构(图6I),表明其在构象空间建模方面具备良好能力。
2.4.4 线性多肽对接
在建模框架中,多肽对接与小分子对接采用相同处理方式。在包含79个蛋白-多肽复合物的数据集上,将PocketXMol与多种多肽对接方法进行比较,并额外纳入两种小分子对接工具,将多肽视作小分子进行处理。采用刚性口袋对接策略,对每个口袋生成100个构象并根据排序得分选择一个最佳构象。PocketXMol在自排序与调优排序下分别获得平均DockQ值0.53与0.58(图7A;表S6),表现突出。小分子对接工具在该任务中显著逊色(图S7A),说明小分子与多肽对接工具之间的迁移能力有限。
随着每个口袋选取更多候选构象,对接性能进一步提升;在oracle排序条件下DockQ达到0.73,表明排序策略仍有优化空间(图7B)。对接准确性与多肽性质相关:较短或螺旋/折叠含量较低的多肽更易获得高精度结果,可能源于其更高柔性及与小分子的相似性(图7C)。值得注意的是,长度超过15个残基的多肽未包含在训练集中,但仍能以合理精度完成对接,体现出良好的泛化能力。在Q-BioLiP中更近期的蛋白-多肽复合物基准测试结果亦保持一致(图7D和S7B)。
得益于原子级表示,模型能够自然处理含有修饰氨基酸的多肽。测试集中13条含非标准残基的多肽均实现良好对接(DockQ≥0.49),且非标准侧链原子的RMSD较低(图7G和7H)。相比之下,采用最接近标准氨基酸替代策略的AlphaFold-Multimer仅有6条达到DockQ≥0.49(图7F)。鉴于非标准氨基酸在多肽药物中的广泛应用,PocketXMol提供了更具实用性的对接能力。
在替代口袋结构上评估对接性能时,原始PocketXMol与微调后的PocketXMol-PF均表现稳定(图7E和S7C–S7E),显示出对口袋结构变化的鲁棒性。对于较长或结构更刚性的多肽,PocketXMol-PF略有优势。
此外,演示了约束对接策略,通过固定多肽不同子集原子,包括最接近口袋的原子、N端残基原子、两端残基原子或全部主链原子。随着约束增加,对接精度不同程度提高(图7I)。固定N端或两端原子分别提升准确率27%和50%,显示出在环结构建模任务中的潜力,例如抗体互补决定区(CDR)等柔性片段附着于刚性骨架的场景。在固定整个主链、仅生成侧链位置的特殊情况下,侧链RMSD中位数为1.1Å。
2.4.5 循环多肽对接
在包含26个蛋白-循环多肽复合物的数据集上,按照靶蛋白序列一致性分为9个簇,将PocketXMol与AlphaFold 2、HighFold以及AfCycDesign进行比较。PocketXMol为每个复合物生成100个构象并依据排序得分选取最佳构象,基线方法采用默认设置。计算每个复合物的DockQ并在簇内取平均。结果显示,PocketXMol与专门方法表现相当(图7J;表S6),平均DockQ介于0.82至0.87之间。
2.4.6 从头设计分子的对接精度
为评估从头设计分子的对接精度,设计配体并分析其与参考配体在化学或序列相似性上的关系与预测三维构象准确性之间的联系。对于小分子,基于分子指纹阈值0.9和蛋白序列一致性0.3,从Protein Data Bank中筛选23个非冗余蛋白-配体复合物。对于多肽,基于多肽序列一致性阈值0.6,从PepBDB与Q-BioLiP中筛选46个复合物。训练与测试数据构建过程中已去除蛋白冗余。
在小分子任务中,最大公共子结构(MCS)覆盖率与其RMSD呈负相关(图S6I),且在高相似度配体中,PocketXMol获得的RMSD低于AlphaFold 3(图S6J)。在多肽任务中,更好的序列比对对应更低的Cα与全原子RMSD(图S7F和S7G),当相同残基数≥8时达到亚埃级精度,并持续优于AlphaFold 3。此外,设计构象与重新对接构象之间保持一致(图S6K和S7H–S7K),表明模型对从头设计配体的结合结构预测具有良好的泛化能力与准确性。
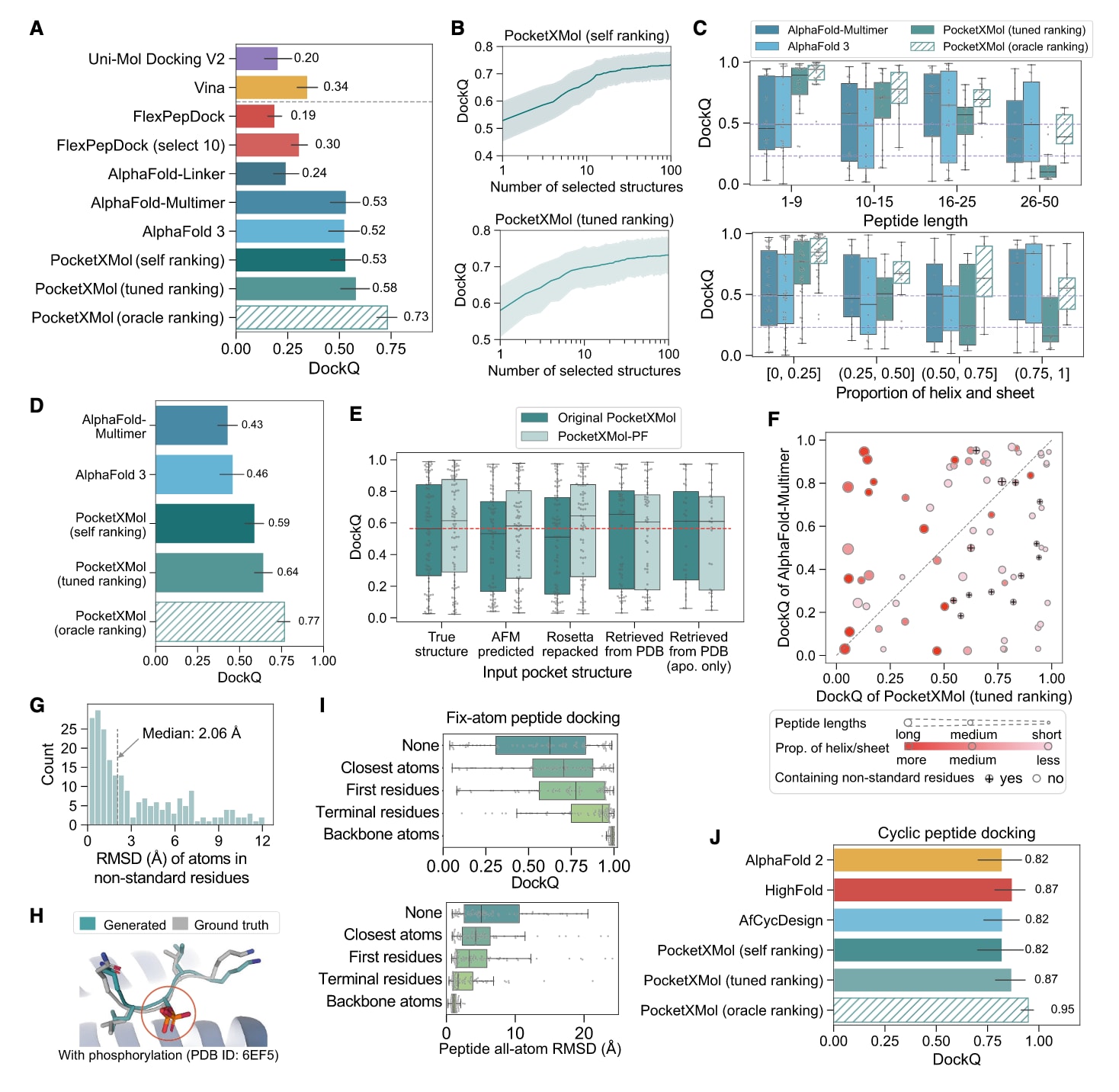
图7|肽分子对接性能评估 (A)在PepBDB测试集上的肽分子对接DockQ评分。柱状图展示平均值及95%置信区间(n=79)。(B)在每个结合口袋从100个生成结构中选择不同数量构象时的对接表现。折线图展示平均值及95%置信区间(n=79)。(C)DockQ评分与肽长度(上)以及螺旋与折叠结构比例(下,n=79)之间的关系。箱线图展示四分位数及1.5×IQR范围。(D)在BioLiP测试集上的肽分子对接DockQ评分。柱状图展示平均值及95%置信区间(n=79)。(E)在PepBDB测试集上,不同PocketXMol变体以及不同输入口袋结构条件下的对接结果。箱线图展示四分位数及1.5×IQR范围。(F)在PepBDB测试集上,PocketXMol与AlphaFold-Multimer之间DockQ评分的直接比较。(G)非天然氨基酸(NAAs)中原子的对接RMSD。(H)一个含有磷酸化修饰的已对接肽结构示例。(I)在引入先验知识条件下,PocketXMol的肽分子对接性能。箱线图展示四分位数及1.5×IQR范围(n=79)。(J)环肽对接的DockQ评分,先在簇内取平均,再在簇间取平均。误差线表示95%置信区间(n=79)。
3 讨论
PocketXMol在原子层面对分子进行建模,无需预先定义氨基酸或特定化学基团等化学实体。尽管已有部分模型能够生成原子级结构,但通常针对不同分子类别采用各自独立的表示方式,且缺乏对跨分子类型迁移能力的系统关注。即便是微小的原子改变,也可能使一个残基被归类为非标准形式,从而形成完全不同的表示体系,阻碍知识迁移。PocketXMol提出统一的任务表示框架,在单一体系中编码多种分子任务,并显式建模任务之间的关系。这种设计在结构预测与分子设计之间建立了桥梁,使类似AlphaFold 3和RoseTTAFold All-Atom等模型的能力得以扩展至分子设计领域。通过将多种噪声形式统一映射至同一表示空间以实现多任务学习,该框架在其他领域亦具有推广潜力。例如在图像生成中,可将高斯噪声、模糊、遮挡与像素化等扰动整合至统一框架中进行建模。
3.1 研究局限性
PocketXMol主要面向以蛋白口袋为核心的任务,尚未扩展至蛋白-核酸对接或相关设计等其他相互作用场景。其计算复杂度呈二次增长,限制了在大型复合物上的应用范围。此外,训练数据覆盖度有限,对含有稀有元素或金属离子的配体处理能力仍受约束。
模型依赖预先定义的口袋结构,无法与配体联合推断或优化口袋构象,因此在口袋边界不明确或来源于低分辨率结构时应用受限。尽管在预测口袋或非天然口袋条件下表现出一定鲁棒性,若能实现口袋生成或柔性口袋建模,将有助于拓展至诱导契合对接及口袋设计等更复杂场景。
此外,针对所设计分子尚缺乏实验解析结构(例如X射线晶体结构)的验证,限制了对生成结合构象准确性的全面评估。虽然功能实验已提供活性证据,但直接的结构确认对于充分验证生成预测仍至关重要。未来若能开展更多实验验证,包括对设计复合物进行晶体学或冷冻电镜(cryo-EM)解析,以及合成并测试更复杂的配体类别如循环多肽,将进一步增强模型能力的证据基础并拓展其应用范围。