Nat. Biotechnol. 2026 | SMRTnet: 在缺乏RNA三级结构信息的情况下预测小分子–RNA相互作用

获取详情及资源:
0 摘要
小分子可以与RNA结合,从而调控其命运和功能,为治疗人类疾病提供了重要机遇。然而,现有用于预测小分子–RNA相互作用(SRIs)的工具通常依赖RNA三级结构的先验信息。相关研究提出了一种名为SMRTnet的深度学习方法,通过多模态数据融合,将两个大语言模型与卷积神经网络和图注意力网络相结合,在RNA二级结构基础上实现SRIs的预测。
SMRTnet在多个实验基准测试中表现出较高的预测性能,显著优于现有工具。针对十个与疾病相关的RNA靶标,SMRTnet的预测结果筛选出40个命中分子,这些RNA靶向小分子的解离常数处于纳摩尔至微摩尔范围。以MYC内部核糖体进入位点为例,SMRTnet预测的小分子结合评分与实验验证命中率高度相关。其中一个预测的小分子能够下调MYC表达,在三种癌细胞系中抑制细胞增殖并促进细胞凋亡。
通过摆脱对RNA三级结构的依赖,SMRTnet拓展了可行RNA靶标的范围,并加速了RNA靶向治疗药物的发现进程。
1 引言
RNA近年来逐渐成为药物发现领域中极具吸引力的靶标。其复杂结构能够被小分子选择性调控,从而影响多种生物学过程,包括pre-mRNA剪接、mRNA翻译、RNA–蛋白质相互作用、非编码RNA加工以及RNA病毒复制等。RNA作为靶点的开发拓展了小分子药物的治疗范围,使针对与疾病相关但传统上被认为“不可成药”的蛋白编码基因成为可能。一个典型例子是Evrysdi(risdiplam),这是首个RNA靶向药物,可在剪接过程中促进SMN2外显子7的包含,用于治疗脊髓性肌萎缩症。
尽管前景广阔,RNA相较于蛋白质仍研究不足,原因在于若干关键挑战,其中最突出的是RNA三级结构难以解析,这限制了RNA靶向小分子的发现。近年来,为绕过这一难题并促进小分子–RNA相互作用(SRIs)的大规模筛选,研究者开发了多种高通量实验方法,例如自动化配体鉴定系统和小分子微阵列。然而,潜在RNA结合分子的化学空间极为庞大,这对实验筛选构成了严峻挑战。
除直接实验测定方法外,也有计算方法被用于预测SRIs。例如,分子对接工具如AutoDock Vina、RLDOCK、NLDock和rDock已被改造或开发,用于小分子与核酸三级结构之间的对接。近年来,深度学习方法也被应用于SRIs预测。其中,RNAmigos2利用变分自编码器和图神经网络预测RNA的潜在结合分子,而RLaffinity引入三维卷积神经网络(CNN)来预测小分子与RNA靶标之间的结合亲和力。尽管取得了进展,这些计算方法仍依赖已知的RNA三级结构,因而在实际应用中受到限制,因为大多数与疾病相关的RNA尚未解析其三级结构,且只有少数具有明确的活性位点。
为此,相关研究开发了SMRTnet(一种利用深度神经网络预测小分子与RNA靶标相互作用的方法),在无需RNA三级结构先验信息的情况下实现SRIs预测。SMRTnet整合了两个大语言模型(LLMs)、卷积神经网络(CNNs)和图注意力网络(GATs),以捕获小分子和RNA的序列与结构特征,并通过多模态数据融合(MDF)对这些特征进行整合,从而实现准确的SRI预测。SMRTnet在多个基于实验验证的SRIs基准测试中表现出优异性能,显著优于现有方法。
此外,该方法还被用于对包含7350种化合物的文库进行筛选,以识别十个与疾病相关RNA靶标的潜在结合分子。微尺度热泳动(MST)实验验证了其中40个命中分子,其解离常数
总体而言,SMRTnet为SRI筛选提供了一种有效途径,展示了人工智能方法在RNA靶向小分子治疗药物开发中的应用潜力。
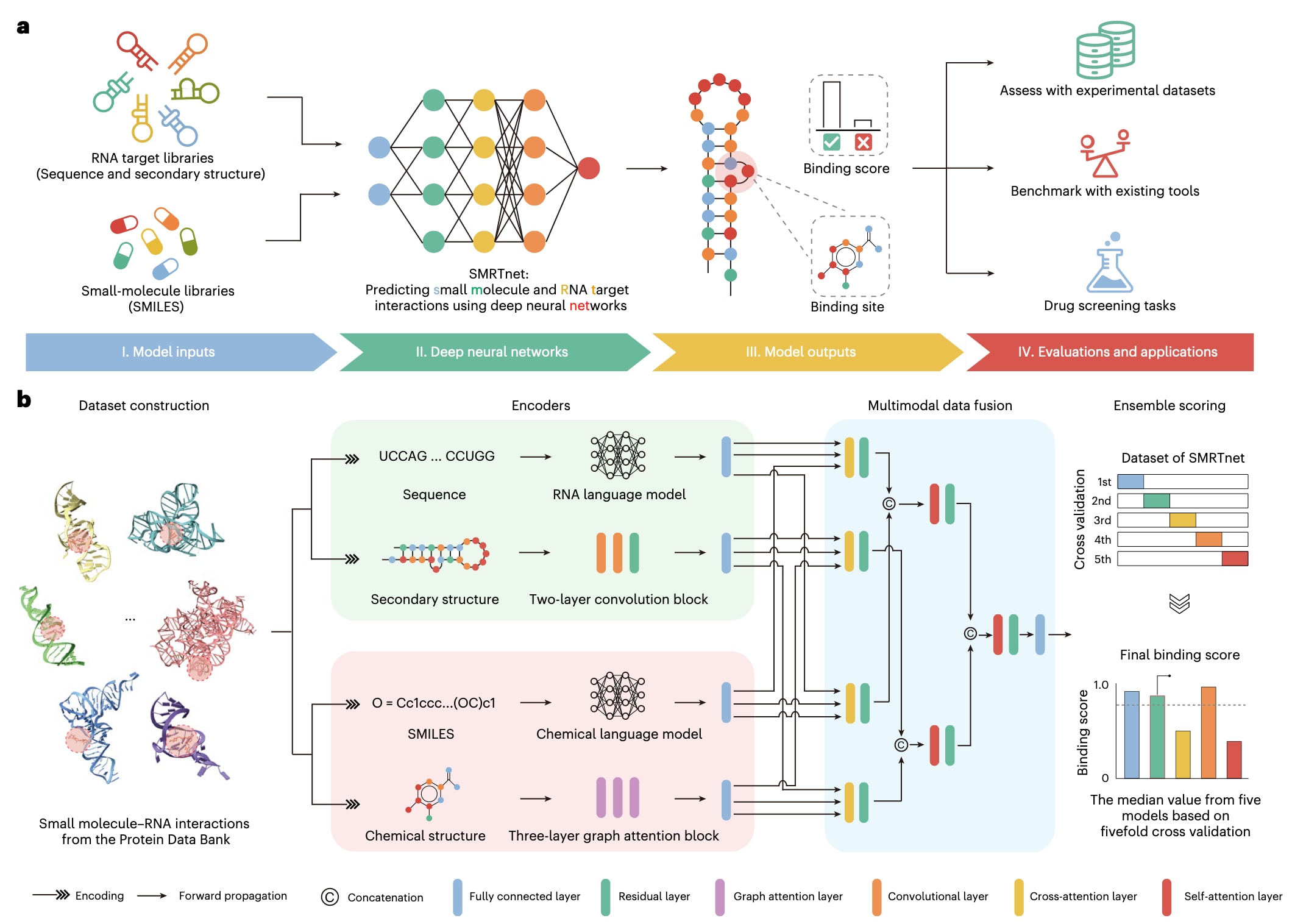
图1|SMRTnet概述。 a,SMRTnet示意图。SMRTnet是一种用于预测小分子–RNA相互作用(SRIs)的深度学习方法。该方法以RNA序列及其二级结构信息,以及以SMILES表示的小分子作为输入,输出结合评分及潜在结合位点。b,SMRTnet的整体架构。训练数据来源于蛋白质数据银行(PDB)。SMRTnet主要由三个核心模块组成:RNA编码器、小分子编码器以及多模态数据融合(MDF)模块。RNA编码器利用RNA语言模型RNASwan-seq对RNA序列进行建模,并通过结合残差神经网络(ResNets)的卷积神经网络(CNNs)处理RNA结构信息。小分子编码器则使用化学语言模型MoLFormer处理小分子的SMILES表示,并通过图注意力网络(GATs)提取其化学结构特征。MDF模块基于注意力机制神经网络逐步整合成对结合信息,随后由全连接神经网络进行解码,预测输入RNA与小分子之间的结合评分。最终,通过集成评分策略生成最终的结合评分结果。
2 结果
2.1 SMRTnet概述
SMRTnet是一种深度学习方法,以带有二级结构信息的RNA序列以及小分子的简化分子线性输入规范(SMILES)作为输入,输出两者之间的结合评分(图1a)。其整体架构包括RNA编码器、小分子编码器、多模态数据融合(MDF)模块以及解码器(图1b和方法部分)。
RNA编码器整合了自研的RNA语言模型RNASwan-seq(扩展数据图1a)以及一个包含残差神经网络(ResNets)的两层卷积神经网络(CNN)(扩展数据图1b),用于同时提取核苷酸序列信息和碱基配对信息,并将其作为输入RNA的表示。小分子编码器则结合了已发表的化学语言模型MoLFormer以及一个三层图注意力网络(GAT)(扩展数据图1c),用于捕获原子组成与化学结构特征,并形成小分子的表示。
为刻画RNA与小分子表示之间在SRIs形成过程中的复杂相互作用,研究构建了一个MDF模块,通过协同注意力机制和自注意力神经网络逐步整合成对结合信息(扩展数据图1d),生成交互表示。该表示随后被输入至一个全连接神经网络解码器,用于预测最终的结合评分。
在模型训练方面,构建了SMRTnet数据集,从蛋白质数据银行(PDB)中收集了1061个高质量三维结构,这些结构至少包含一个RNA和一个小分子(扩展数据图2a和补充表1)。由于每个相互作用位点通常涉及多个RNA片段,提取了这些片段的二级结构信息,最终生成8672个RNA片段与小分子的相互作用样本(扩展数据图2b和方法部分)。这些相互作用作为正样本用于训练和测试。同时,在排除已知相互作用对后,随机配对RNA片段与小分子以构建非相互作用对,作为负样本。
为评估模型的稳健性,按照1:1、1:2、1:3、1:4、1:5和1:10的不同比例采样负样本,与既往研究采用的比例范围保持一致。数据集按照8:1:1的比例划分为训练集、验证集和测试集,并采用基于配体的数据划分策略,以确保测试集中的小分子不出现在训练集和验证集中。此外,还采用五折交叉验证(CV)评估模型稳定性,并基于五个交叉验证模型采用集成评分策略,计算中位结合评分作为最终结果,从而降低单一模型的随机预测误差(扩展数据图2c和方法部分)。
在分类阈值的确定方面,采用
2.2 基于PDB中SRI数据的SMRTnet性能评估
在SMRTnet数据集上进行五折交叉验证时,SMRTnet在不同正负样本比例(1:1、1:2、1:3、1:4、1:5和1:10)条件下均表现出稳健性能,平均受试者工作特征曲线下面积(auROC)达到0.830–0.844(图2a和扩展数据图2e)。在使用不同随机种子生成负样本的情况下,模型性能保持一致,说明负样本随机化过程对整体性能影响较小(扩展数据图2g)。作为对比,目前唯一能够应用于SMRTnet测试集的其他工具RNAmigos2,其auROC仅为0.567–0.596(图2a和扩展数据图2e),明显低于SMRTnet。
为评估小分子结构相似性可能带来的数据泄漏问题,计算了训练集与测试集分子之间的Tanimoto相似度,结果显示在五折交叉验证中平均值始终低于0.75(扩展数据图2h)。进一步设置最大Tanimoto相似度阈值为0.7–1.0后,模型平均auROC仍保持在0.844–0.855之间,表明即使不对小分子相似性进行严格限制,也未出现明显数据泄漏(图2a、扩展数据图2i和方法部分)。
在RNA结合位点相似性方面,排除了测试集中与训练集RNA具有相同多链结合位点的样本。此举使平均auROC从0.844下降至0.798(图2a和扩展数据图2j)。此外,采用基于结构的数据划分策略重新训练SMRTnet,将结构相似的结合口袋(RMscore≥0.75)聚类至同一训练或测试集合,得到平均auROC为0.806(扩展数据图2k)。这些结果表明,尽管RNA侧可能存在轻微数据泄漏,SMRTnet在排除相同或相似RNA结合位点后仍保持较强预测能力。
进一步地,在RNAmigos2的数据集上对SMRTnet进行基准测试,包括在该数据集上重新训练并评估SMRTnet,或在过滤掉具有相似配体和结合位点的数据后,用原始SMRTnet模型进行评估。两种测试中,SMRTnet均展现出与RNAmigos2高度竞争的性能,尽管其训练数据规模更小,且评估时涉及新的化学与结构空间(图2b和扩展数据图2g)。这些结果表明SMRTnet在RNA靶向药物发现中具有良好的稳健性与泛化能力。
为验证SMRTnet是否真正学习到特定RNA–配体相互作用,而非仅依赖配体特征,构建了一个错配测试集:将测试集中每个正样本的配体随机替换为来自其他样本的配体,同时保留原RNA及“正样本”标签。结果显示模型性能显著下降,auROC降至0.572。随后将这些错配样本的标签从“正”改为“负”,模型性能恢复至auROC为0.830(扩展数据图2l)。这些结果表明,SMRTnet确实学习到了SRIs的内在规律,而非简单依赖配体中心特征进行预测。
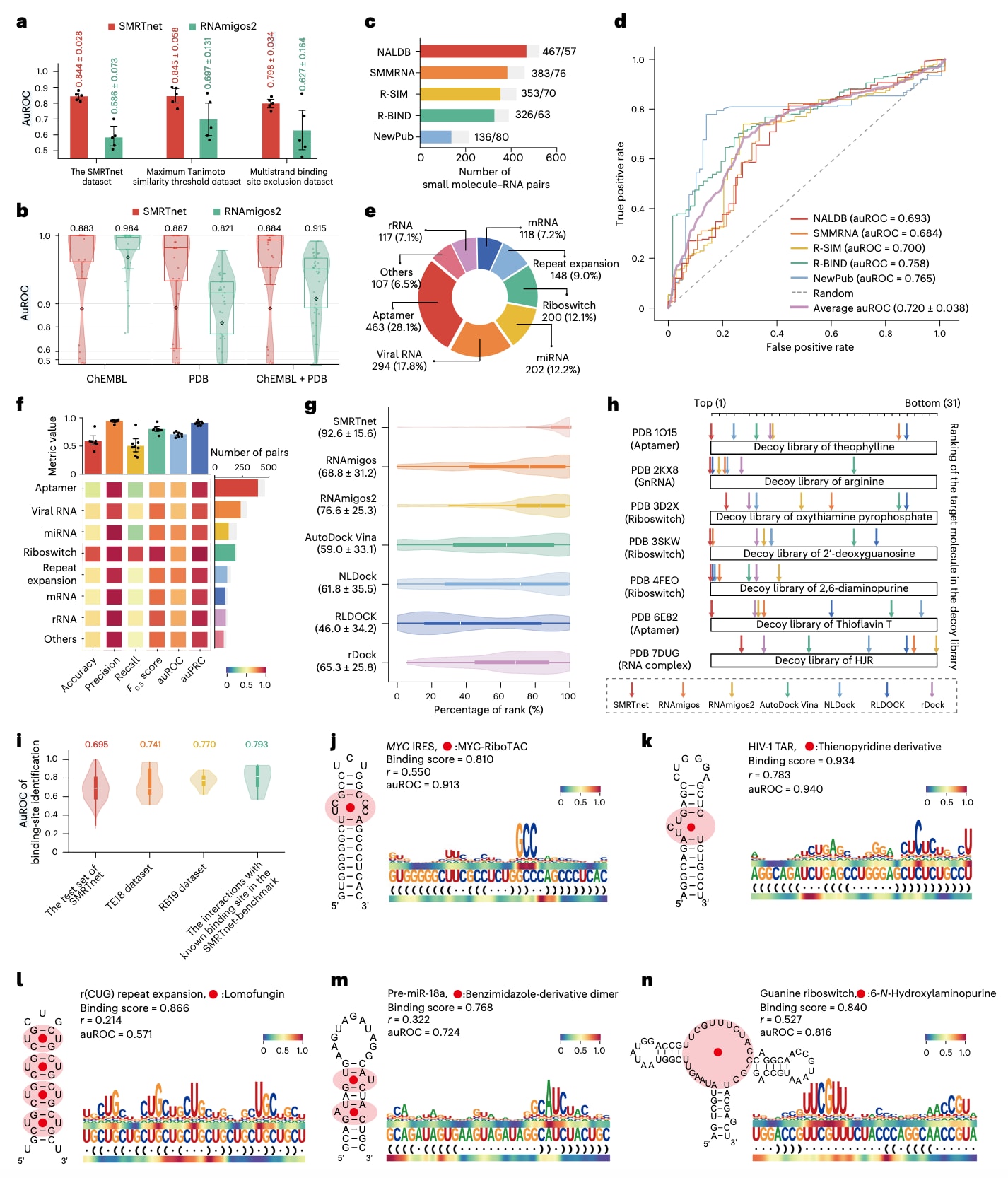
图2|SMRTnet的性能表现。 a,SMRTnet与RNAmigos2在不同测试集上的性能比较,包括SMRTnet原始测试集、施加最大Tanimoto相似度阈值0.7后的修订测试集,以及排除相同多链结合位点后的修订测试集。柱状图叠加散点图展示了在正负样本比例为1:2及其变体条件下,基于五折交叉验证(n=5)得到的auROC结果。粗实线表示五折交叉验证的标准差,数据以均值±标准差形式呈现。b,在RNAmigos2完整测试集上,比较在RNAmigos2子集上重新训练的SMRTnet与RNAmigos2的性能。测试中使用来自不同分子库的诱饵,包括ChEMBL(n=500)、PDB(n=264)以及ChEMBL+PDB(n=764)。数据以箱线图和小提琴图叠加散点图形式展示(n=37),中线表示中位数,箱体上下边界分别为第一和第三四分位数,须线表示IQR×1.5,各分布的均值以数字标注。c,五个实验验证基准数据集中小分子–RNA配对的统计情况。柱状图展示各基准中相互作用(彩色)与非相互作用(灰色)的小分子–RNA配对数量,这些数据统称为SMRTnet-benchmark数据集。d,SMRTnet在SMRTnet-benchmark数据集上的ROC曲线及auROC值,包括NALDB(n=524)、SMMRNA(n=459)、R-SIM(n=423)、R-BIND(n=389)和NewPub(n=216)。e,环形图展示SMRTnet-benchmark数据集中八种RNA类型的比例分布。f,SMRTnet在八种RNA类型上的性能表现,包括aptamer(n=463)、viral RNA(n=294)、miRNA(n=202)、riboswitch(n=200)、repeat expansion(n=148)、mRNA(n=118)、rRNA(n=117)以及others(n=107)。右侧柱状图表示各RNA类型中相互作用(彩色)与非相互作用(灰色)配对数量。上方柱状图表示不同RNA类型的整体性能(n=8),粗实线表示八种RNA类型性能的标准差。g,在诱饵评估任务中,SMRTnet与当前主流工具在SMRTnet测试集(n=684)上的整体性能比较,包括RNAmigos(n=605)、RNAmigos2(n=547)、AutoDock Vina(n=659)、NLDock(n=351)、RLDOCK(n=212)和rDock(n=391)。数据以小提琴图叠加箱线图形式呈现,中线为中位数,箱体上下边界为第一和第三四分位数,小提琴图的上下须线表示IQR×1.5。h,不同计算工具在给定诱饵分子库中对目标分子排名的比较。重叠箭头表示排名并列。箭头越靠左表示目标分子排名越高,越靠右表示排名越低。每个目标分子对应30个诱饵分子。i,SMRTnet在不同数据集上识别结合位点的性能。小提琴图表示SMRTnet在SMRTnet测试集(n=2508)、TE18(n=9)、RB19(n=11)以及SMRTnet-benchmark数据集中具有实验确定结合位点的相互作用(n=5)上的auROC结果。中线表示中位数,箱体上下边界为第一和第三四分位数,小提琴图须线表示IQR×1.5。各数据集上方数字表示结合位点识别的平均auROC值。j–n,SMRTnet对不同RNA的结合位点预测结果,包括MYC IRES(j)、HIV-1 TAR元件(k)、HTT基因中的r(CUG)重复扩增(l)、pre-miR-18a(m)以及鸟嘌呤核糖开关(n)。两条热图轨迹表示SMRTnet在每个核苷酸位置的响应信号,红色表示高注意力区域(HARs)(上轨为序列响应,下轨为结构响应)。RNA结构图中红色圆圈与阴影表示对应小分子的已知结合位点,结合位点周围标注其名称。结合评分在每个小分子–RNA配对之间计算,同时在梯度信号与实验确定结合位点邻近性之间计算Pearson相关系数
2.3 基于已发表研究中SRI数据的SMRTnet性能评估
除基于PDB构建的SMRTnet数据集外,还进一步整理了来自四个数据库(R-BIND、R-SIM、SMMRNA和NALDB)以及22篇最新文献(NewPub)中经实验验证的SRIs数据,并包含相应的非相互作用小分子–RNA对作为负样本。在剔除已出现在SMRTnet数据集中的相互作用后,共获得1665个SRIs和346个负样本(图2c、补充表2和方法部分),该数据集统称为SMRTnet-benchmark数据集。
从相关文献中提取RNA序列及其二级结构信息以运行SMRTnet。结果显示,在SMRTnet-benchmark数据集上,SMRTnet的平均auROC为0.720,其中在SMMRNA子集上的表现为0.684,在NewPub子集上达到0.765(图2d)。进一步根据RNA类型将数据划分为八类,结果表明SMRTnet对大多数RNA类型均无明显偏好,仅在核糖开关(riboswitch)类别中表现出一定差异(图2e、f)。这些结果表明SMRTnet适用于多种RNA类型,并在预测未见过的SRIs方面具有良好能力。
2.4 在诱饵评估任务中SMRTnet优于其他计算方法
采用诱饵评估(decoy evaluation)任务对SMRTnet与现有方法进行基准比较。该任务常用于评估分子对接工具的判别能力。在此任务中,对于每个RNA靶标及其真实结合分子,构建一个与真实分子在物理性质上相似但在化学结构上不同的诱饵分子库,随后由各工具对真实结合分子在诱饵集合中的排名进行排序。
具体而言,对于SMRTnet测试集中每一个SRI,利用DecoyFinder从ZINC15数据库中检索最多30个诱饵分子,并将SMRTnet与四种主流对接工具(AutoDock Vina、NLDOCK、RLDOCK和rDock)以及两种深度学习工具(RNAmigos和RNAmigos2)进行比较。SMRTnet在测试集上的平均排名达到92.6%,显著优于四种对接工具(27.3%–46.6%),也超过两种深度学习方法(16.0%–23.8%)(图2g)。
在七个代表性测试案例中,SMRTnet均将真实结合分子稳定排名于前五位(图2h)。例如,在茶碱结合适配体(PDB 1O15)的案例中,SMRTnet将茶碱本身排名第一,其后为具有相同官能团的诱饵分子,而缺乏该官能团的诱饵则被排在末尾(扩展数据图3a–h)。这些结果表明SMRTnet在从结构高度相似的小分子中识别真实结合分子方面具有更强能力。
此外,还通过在不同数量的SRI预测任务中测量推理时间,对SMRTnet的计算效率进行了评估。结果显示,相较于GPU加速的对接工具Vina-GPU 2.0,SMRTnet具有显著更高的计算效率(扩展数据图2m和方法部分)。
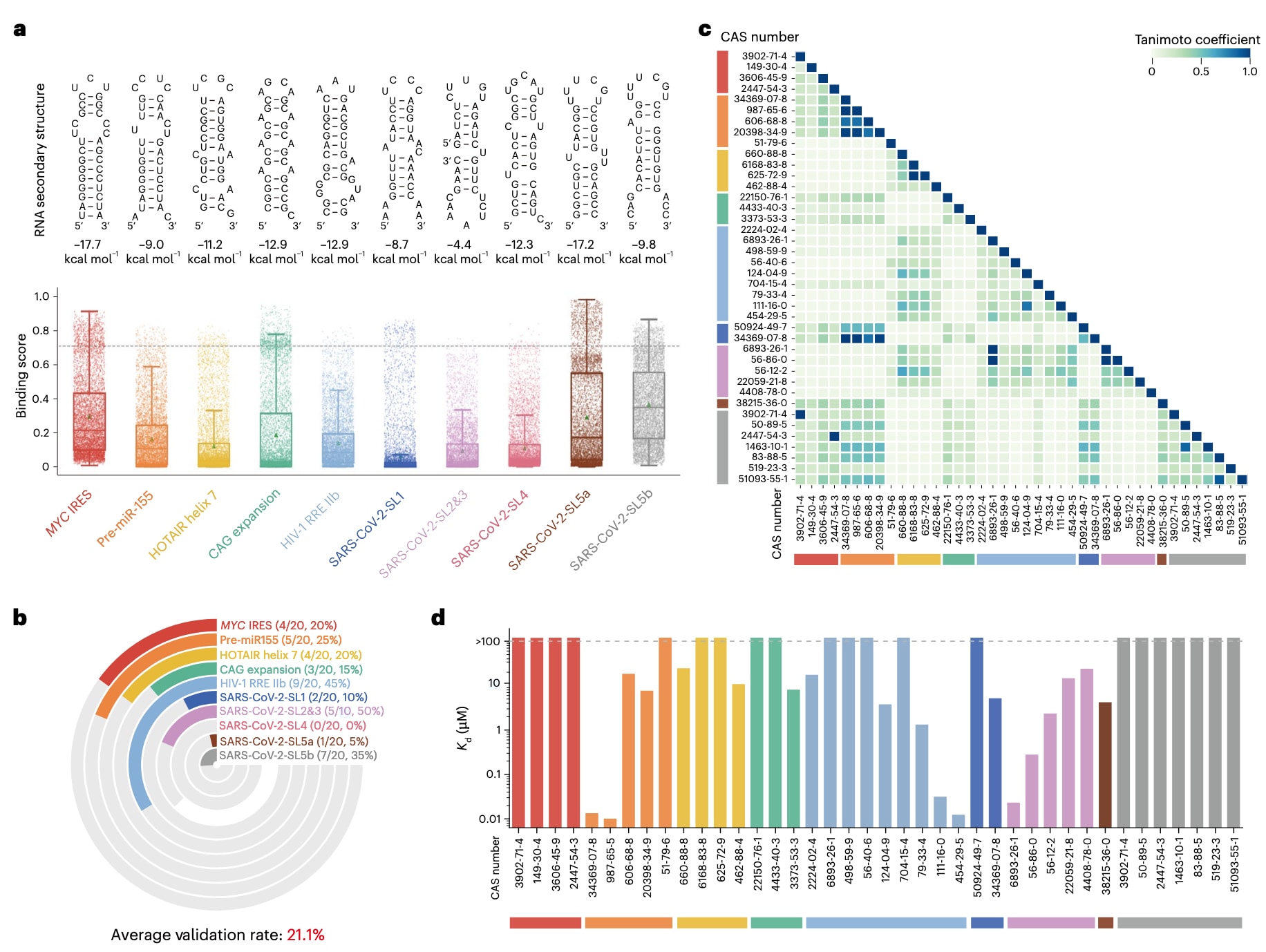
图3|疾病相关RNA靶标的实验验证结果。 a,上图为十个疾病相关RNA靶标的二级结构,其最小自由能由RNAstructure计算得到;下图为SMRTnet在天然产物文库上对这些RNA靶标预测的结合评分分布。数据以箱线图叠加散点图形式呈现,中线表示中位数,箱体上下边界分别为第一和第三四分位数,须线表示IQR×1.5,绿色三角形表示平均值。每个点代表一个化合物与对应RNA靶标之间的预测结合评分(n=7350),灰色虚线表示SMRTnet的分类阈值(0.704)。b,SMRTnet预测结果经微尺度热泳动(MST)实验验证的命中率。彩色区域表示被验证为具有结合能力的化合物,灰色区域表示被验证为不结合的化合物。分子式中的分子表示实验验证为结合分子的数量,分母表示选取进行实验验证的高排名化合物总数,百分比表示各RNA靶标的验证命中率。c,与十个疾病相关RNA靶标发生相互作用的40个化合物的结构相似性分析,采用Tanimoto相似度进行度量。化合物按照其对应RNA靶标进行分组。d,柱状图展示十个疾病相关RNA靶标上40个经实验验证的化合物–RNA相互作用的解离常数
2.5 RNA编码器及实验获得的RNA二级结构数据对准确预测SRI至关重要
通过消融实验系统评估了SMRTnet各组成模块对模型性能的贡献。构建了七种变体模型:(1)仅使用RNA结构编码器和小分子结构编码器;(2)仅使用RNA序列编码器和小分子SMILES编码器;(3)双小分子编码器结合RNA结构编码器;(4)双小分子编码器结合RNA序列编码器;(5)双RNA编码器结合小分子SMILES编码器;(6)双RNA编码器结合小分子结构编码器;(7)完整SMRTnet模型但移除MDF模块。结果显示,完整模型的auROC为0.844,移除MDF模块后(变体7)下降至0.812,在变体5和6中进一步小幅降至0.808和0.802;而在变体1–4中则显著下降至0.561、0.552、0.571和0.591(扩展数据图2n)。这些结果表明,RNA序列与结构信息是模型性能的核心因素,而MDF模块也对实现高预测性能具有重要作用。
进一步聚焦RNA二级结构的作用,构建了仅基于序列的模型SMRTnet-seq,即移除RNA结构特征。该模型在SMRTnet数据集上的平均auROC由0.844下降至0.760,在SMRTnet-benchmark数据集上由0.720降至0.578(扩展数据图2o–p)。此外,将SMRTnet-benchmark数据集中超过80%来源于实验测定的结构数据替换为RNAstructure预测结果后,auROC降至0.664(扩展数据图2p)。这些结果强调RNA二级结构,尤其是实验测定的结构信息,对于获得高预测精度具有关键意义。
同时还比较了不同RNA大语言模型对SMRTnet性能的影响,将RNASwan-seq与RNA-FM和RNAErine进行对比。结果显示,采用RNASwan-seq时模型性能略优于另外两种实现方式(扩展数据图2q)。
2.6 SMRTnet识别RNA上的小分子结合位点
已有研究表明,模型可解释性分析可用于揭示分子相互作用中的结合位点。沿用这一思路,将RNA上对预测结合评分贡献较高的区域定义为高注意力区域(HARs),用于标识小分子结合位点。具体方法为采用Grad-CAM算法量化每个核苷酸对SRI预测结合评分的贡献。
通过与四个数据集中的实验确定结合位点进行比较,评估该方法的准确性。首先,在具有完整结合位点信息的SMRTnet数据集中,五折交叉验证下平均auROC为0.695。其次,在此前提出RNAsite工具的两个基准数据集中,排除无效条目后,SMRTnet的auROC分别达到0.741和0.770,与RNAsite性能相当(图2i和方法部分)。
进一步在SMRTnet-benchmark数据集中选取五个具有已知结合位点的代表性SRI进行分析,包括MYC-RiboTAC与MYC IRES内部环(5′ UUCG/3′ ACCC)的结合、噻吩并吡啶衍生物与HIV-1 TAR RNA中5′ AUCUG/3′ U_C鼓包区域的结合、lomofungin与dystrophia myotonica protein kinase mRNA 3′ UTR中高度结构化的r(CUG)重复扩增内部环(5′ CUG/3′ GUC)的结合、苯并咪唑衍生物二聚体与pre-miR-18a中5′ GAUAG_U/3′ C_AUCUA鼓包的结合,以及6-N-羟胺嘌呤与鸟嘌呤核糖开关三向连接区(5′ AUAAGUU/UUCGUUUCUACC/3′ UCG)的结合。SMRTnet在这些相互作用上的平均auROC为0.793(图2i)。
进一步通过计算梯度信号与实验确定结合位点邻近性之间的Pearson相关系数
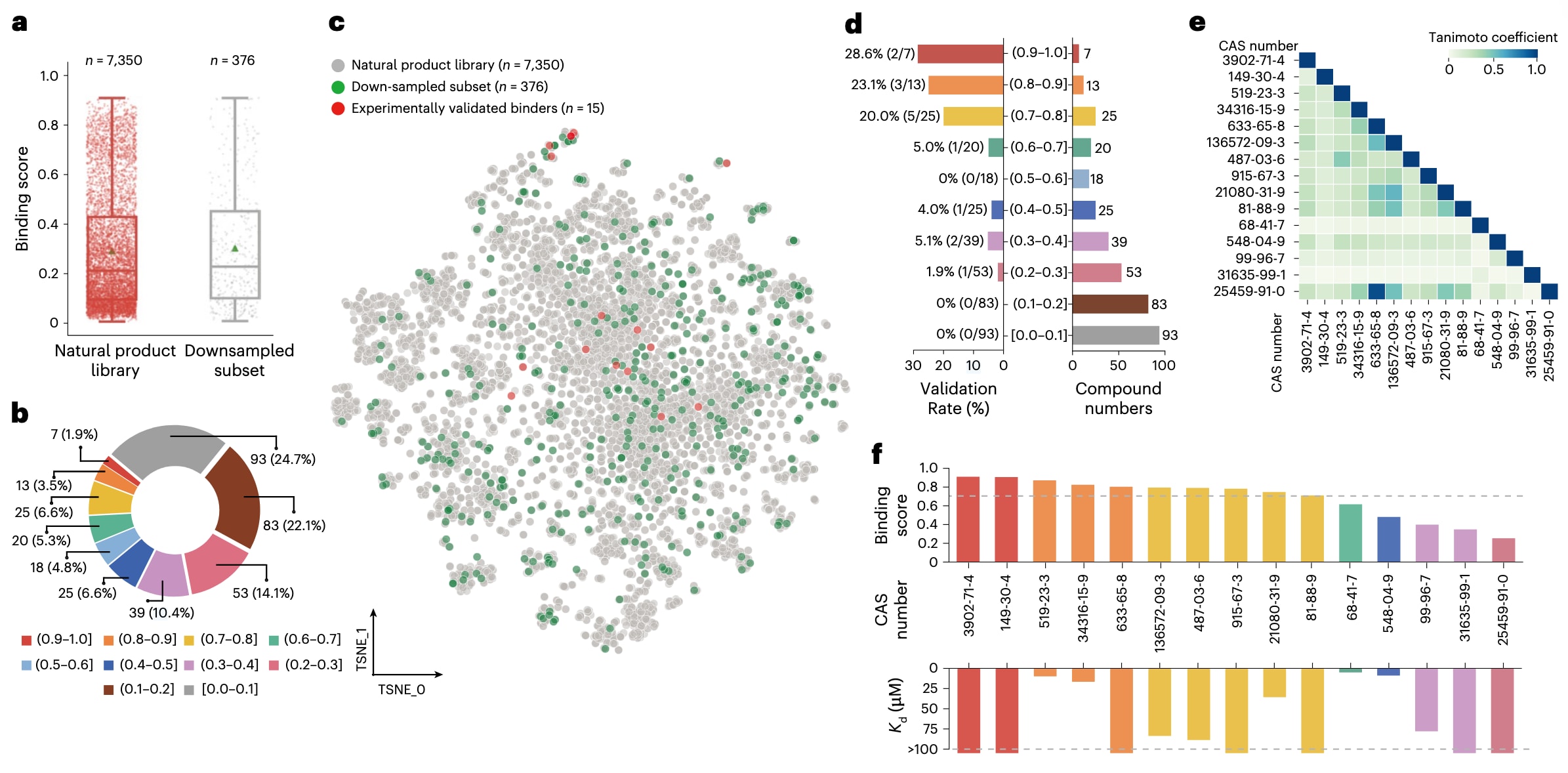
图4|针对不同预测区间的MYC IRES靶向化合物的实验验证结果。 a,SMRTnet对天然产物文库(左)及其下采样子集(右)中靶向MYC IRES的化合物所预测的结合评分分布。数据以箱线图叠加散点图形式呈现,中线表示中位数,箱体上下边界分别为第一和第三四分位数,须线表示IQR×1.5,绿色三角形表示平均值。每个点代表一个化合物与MYC IRES之间的预测结合评分。b,环形图展示在十个预测区间内抽样化合物的数量及比例分布。c,不同化合物数据集的Morgan指纹t-SNE分析结果,包括天然产物文库(n=7350)、下采样子集(n=376)以及从下采样子集中经MST实验验证的结合分子(n=15)。d,下采样子集中被选中化合物在十个预测区间内的分布(右)及其对应的实验验证率(左)。分子式中的分子表示实验验证为结合分子的数量,分母表示随机选取用于实验验证的化合物总数,百分比表示各区间内的验证率。e,与MYC IRES发生相互作用的15个经实验验证化合物的结构相似性分析,采用Tanimoto相似度进行度量。f,15个经实验验证的MYC IRES靶向化合物的预测结合评分与
2.7 SMRTnet预测与疾病相关RNA靶标结合的化合物
利用SMRTnet对包含7350种天然产物和代谢物的文库进行筛选,以寻找能够与十个疾病相关RNA靶标结合的化合物(补充表3和方法部分)。除MYC IRES外,这些RNA靶标还包括在多种癌症中过表达的pre-miR-155、可抑制肿瘤及转移抑制因子的HOTAIR第7螺旋、对病毒复制至关重要的HIV-1 Rev反应元件(RRE)IIB区域、导致亨廷顿病的HTT基因CAG重复扩增。此外,还纳入SARS-CoV-2 5′UTR中的五个RNA结构元件:茎环1(SL1)、参与病毒复制的SL2/3、与亚基因组RNA合成相关的SL4,以及与病毒包装有关的SL5a和SL5b。获取这些RNA靶标的序列和二级结构作为SMRTnet输入。这些RNA具有多样的二级结构特征,可用于评估SMRTnet在不同RNA结构背景下的预测能力(图3a和补充表4)。
针对每个RNA靶标,利用SMRTnet对全部7350种化合物进行结合评分预测。根据结合评分选取每个靶标排名前20且高于分类阈值0.704的化合物,共获得190个预测的SRI用于实验验证(图3a)。实验主要采用微尺度热泳动(MST)的binding check模式,并结合SDS变性测试(SD-test)以排除可能干扰MST测量的内源性荧光化合物(扩展数据图4和方法部分)。在190个预测相互作用中,共有40个获得实验验证,平均验证率为21.1%(图3b)。各RNA靶标呈现出不同的已验证结合分子谱(图3c),表明SMRTnet能够识别RNA结构与配体特异性的细微差异。
随后利用MST的结合亲和力模式测定这40个相互作用的解离常数
2.8 SMRTnet预测的结合评分与MYC IRES的实验验证结果相关
致癌转录因子MYC是多种人类癌症的标志分子,但通常被认为“不可成药”。近期研究提出,靶向位于MYC mRNA 5′UTR中、负责无帽依赖翻译的MYC IRES,可能是一种调控该致癌基因的可行策略。为进一步验证这一思路,从上述化合物文库中随机抽取376种具有不同预测结合评分且结构多样性较高的化合物进行大规模实验验证(图4a–c、补充表5和方法部分)。
在该子文库中,共有15种化合物通过MST被验证为MYC IRES的结合分子,且预测结合评分与实验验证率之间呈明显正相关关系。具体而言,在结合评分为0.9–1.0区间内的7个预测中,有2个(28.6%)被验证为阳性;随着结合评分降低,验证率逐步下降,在0.0–0.1区间内93个预测中无一验证为阳性(图4d、扩展数据图6a和方法部分)。这一结果表明,SMRTnet能够有效优先筛选高预测评分的真实结合分子。
对这15个命中分子进行Tanimoto相似性分析,结果显示其整体结构相似性较低(图4e)。进一步测定这15个命中分子的
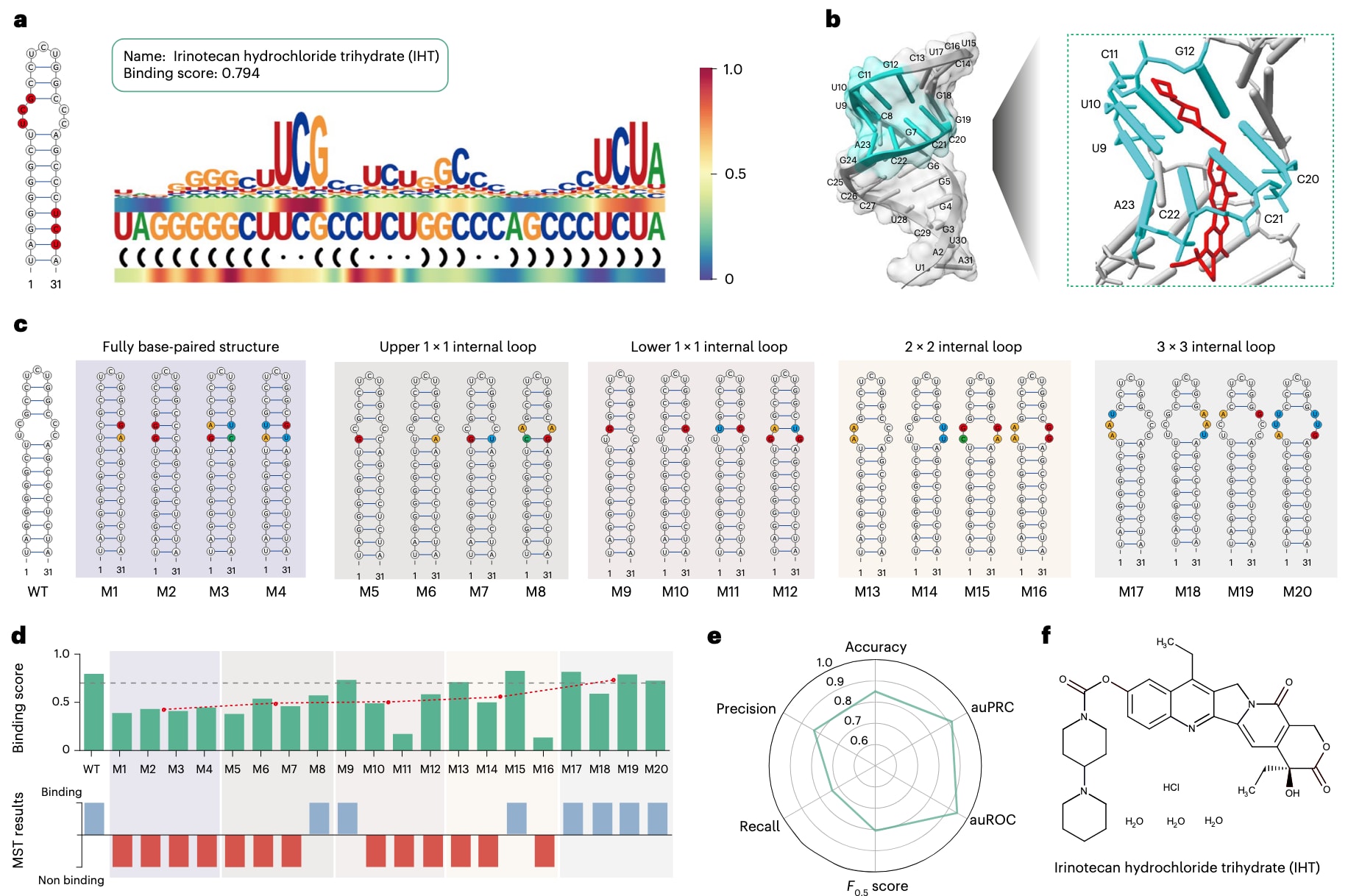
图5|MYC IRES预测结合位点的实验验证。 a,IHT在MYC IRES上的潜在结合位点。RNA二级结构图中以红色标注高注意力区域(HARs),两条热图轨迹显示模型在各核苷酸位置的响应信号,上轨为序列响应,下轨为结构响应,红色表示高注意力。b,利用SimRNA预测的MYC IRES三级结构,其中青色区域表示SMRTnet预测的结合位点;IHT在该位点的分子对接结果通过ChimeraX进行可视化展示。c,20种MYC IRES突变体的二级结构,由RNAstructure预测得到。不同背景颜色表示不同类型的结合位点(上方标注),彩色核苷酸标示突变位置及类型。淡紫色背景表示完全碱基配对结构,亚麻色背景表示上部1×1内部环,浅玫瑰色背景表示下部1×1内部环,木瓜色背景表示2×2内部环,浅灰色背景表示3×3内部环。WT表示野生型。d,不同MYC IRES突变体与IHT之间的预测结合评分分布。下方柱状图为MST实验验证结果。红点表示相同结合位点类型下的平均结合评分,红色虚线表示随着结合位点类型变化的结合评分趋势,灰色虚线表示SMRTnet的分类阈值(0.704)。e,雷达图展示IHT与20种MYC IRES突变体之间计算预测结果与实验验证结果的一致性。f,IHT的化学结构及其全称。
2.9 SMRTnet识别盐酸伊立替康三水合物(IHT)在MYC IRES上的结合位点
在15个被验证可与MYC IRES结合的化合物中,仅盐酸伊立替康三水合物(IHT,CAS:136572-09-3)在理化性质和药物化学特征方面满足药物开发潜力标准,经ADMETlab 3.0评估符合相关指标(扩展数据图7a–j)。为进一步阐明其与MYC IRES相互作用的分子基础,利用SMRTnet预测IHT的结合位点。高注意力区域(HAR)分析将其定位于MYC IRES的内部环(5′ UUCG/3′ ACCC)(图5a和方法部分)。分子对接结果进一步支持这一预测,显示IHT精确定位于相同的内部环区域(图5b)。
为验证该预测位点,设计了20种MYC IRES突变体,并分为五类:(1)保留2×2内部环但改变序列的突变体;(2–4)将内部环分别转变为3×3或1×1构型并伴随序列改变的突变体;(5)完全去除内部环、形成完全碱基配对结构的突变体(图5c和补充表6)。分析结果显示,随着构型由2×2转变为1×1,预测结合评分逐步下降,在完全碱基配对突变体中达到最低;而从2×2转变为3×3构型时,预测结合评分反而升高(图5d和扩展数据图8a–e)。这些预测趋势与实验验证率高度一致,证实了IHT在MYC IRES上的结合位点,并表明SMRTnet在结合位点预测方面具有较高可靠性(图5e、f)。
2.10 靶向MYC IRES的化合物抑制MYC表达和细胞增殖
进一步评估IHT与MYC IRES结合的生物学效应。在HeLa细胞中,IHT处理使MYC mRNA水平降低约56.9%,MYC蛋白水平降低约71.6%,其效果优于MYC-RiboTAC(图6a、b和补充表7、8)。MYC-RiboTAC是一种嵌合分子,通过结合MYC IRES并招募RNaseL诱导MYC mRNA降解。
在三种依赖MYC维持最佳增殖状态的癌细胞系(HeLa、Jurkat和Raji)中,IHT使细胞增殖下降19.6%–48.4%,细胞凋亡增加56.6%–124.2%。值得注意的是,IHT对细胞增殖和存活的影响均强于MYC-RiboTAC,这与其对MYC mRNA和蛋白水平更显著的抑制作用一致(图6c–h和补充表9、10)。
此外,在HEK293T细胞中构建MYC IRES荧光素酶报告系统,并以完全碱基配对的MYC IRES作为对照。结果显示,IHT与MYC-RiboTAC类似,使MYC IRES报告系统(5′ UUCG/3′ ACCC)的荧光素酶活性降低约14.2%,而对对照报告系统(5′ UAUG/3′ AUGC)无显著影响(图6i–k和补充表11)。这些结果表明,SMRTnet能够辅助识别具有生物活性和治疗潜力的疾病相关RNA靶向小分子。
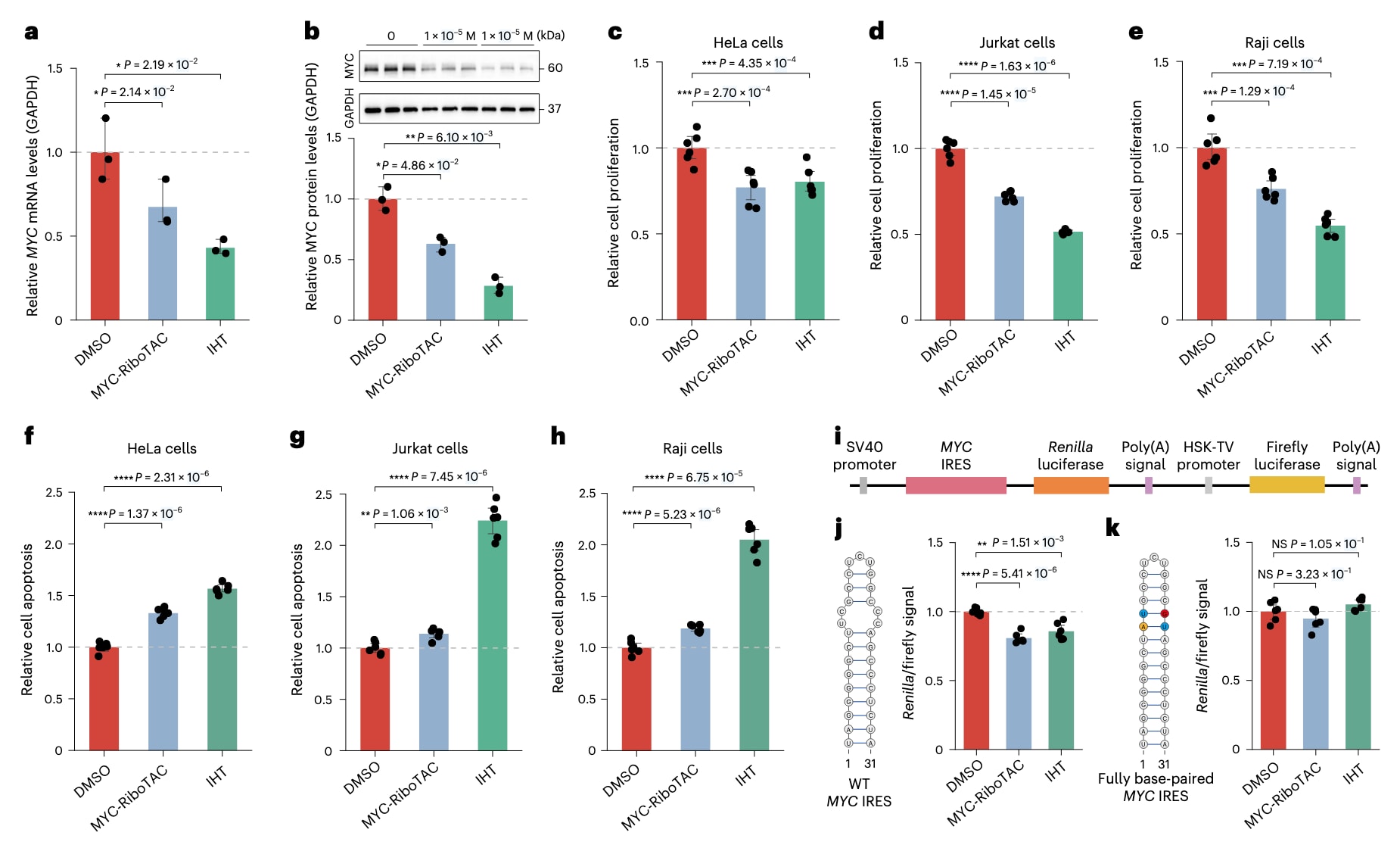
图6|靶向MYC IRES的IHT抑制MYC表达和细胞增殖。 a,b,在HeLa细胞中,以10μM剂量处理48小时后,采用qPCR和western blot分别检测MYC-RiboTAC与IHT对MYC mRNA水平(a)和MYC蛋白水平(b)的影响。数据以
3 讨论
提出的SMRTnet是一种基于RNA序列和二级结构以及小分子SMILES信息预测SRIs的深度学习方法。与现有学习型工具相比,SMRTnet在三个方面具有显著优势。首先,通过整合RNA二级结构信息——这些信息可通过icSHAPE和SHAPE-MaP等实验方法获得——能够捕获仅基于序列方法难以识别的关键决定因素,从而提升预测精度。这种基于二级结构的设计也拓展了对缺乏明确三级结构的疾病相关RNA靶标的适用范围。
其次,SMRTnet整合了两个在数十亿规模RNA或小分子数据上训练的大语言模型,提高了两种模态的表示能力,有助于实现更准确的预测。第三,MDF模块对RNA和小分子编码特征进行融合,生成具有相互作用感知能力的表示,从而稳健刻画SRI发生的概率。
多项实验基准测试及大规模实验验证均表明SMRTnet具有较高预测性能,展示了其在加速RNA靶向治疗药物发现方面的潜力。
目前,经实验验证的SRIs在RNA类型和小分子多样性方面仍然有限。因此,未来需要优先发展高通量筛选平台,以多重化形式高效刻画大规模相互作用数据,例如在多样RNA靶标与小分子文库之间构建系统性筛选数据。这类数据将成为人工智能驱动药物发现的重要训练资源,类似于大量小分子–蛋白质相互作用数据对小分子–蛋白质识别与结合预测能力的提升所起到的推动作用。
需要强调的是,仅有结合并不意味着小分子必然能够调控RNA表达或产生生物学效应。随着化学-RNA互作组学、转录组学及功能筛选等多组学数据不断积累,未来研究可整合这些多模态信息,特别是基因表达谱与表型读出数据,构建能够同时预测结合相互作用及其下游生物效应的人工智能模型,从而弥合靶标结合与功能影响之间的差距,推动RNA靶向治疗的发展。