Nat. Methods 2024 | DeepMSA2:基于大规模宏基因组数据提升深度学习蛋白单体及复合物结构预测性能
深度学习方法的快速发展正在持续推动蛋白结构预测能力的边界,其中AlphaFold2及其多聚体扩展模型在单体与复合物建模中取得了里程碑式进展。然而,大量预测结果表明,模型性能在很大程度上仍受限于输入多序列比对(MSA)中所蕴含的进化信息质量,尤其是在同源序列稀缺或蛋白参与复杂相互作用的情形下。随着宏基因组测序数据规模的爆炸式增长,如何高效、系统地挖掘这些序列资源,并将其转化为对结构预测真正有用的进化约束,成为当前领域面临的核心问题之一。DeepMSA2正是在这一背景下提出,通过迭代式搜索与多策略融合的方式,统一构建单体与多体MSA,并结合深度学习驱动的筛选机制,显著提升了结构预测中进化信息的有效性。该工作不仅在CASP评测和人类蛋白质组建模中展示了稳定而显著的性能提升,也为重新审视“输入信息优化”在深度学习结构预测中的基础性作用提供了有力证据。

获取详情及资源:
0 摘要
通过在基因组与宏基因组序列数据库中进行迭代式比对搜索,该研究提出了DeepMSA2流程,用于统一构建蛋白单链与多链的多序列比对(MSA)。大规模基准测试表明,与当前最先进的方法相比,DeepMSA2生成的MSA能够显著提升蛋白三级结构与四级结构预测的准确性。集成DeepMSA2的完整流程参与了最新一届CASP15实验,其构建的复合物结构模型质量明显优于AlphaFold2-Multimer服务器(v.2.2.0)。进一步的数据分析显示,DeepMSA2的主要优势在于其平衡的比对搜索策略与有效的模型选择机制,以及对超大规模宏基因组数据库的深度整合能力。这些结果展示了一条通过先进MSA构建来提升深度学习蛋白结构预测性能的新路径,并进一步证明,对于基于深度学习的结构预测方法而言,输入信息的优化与预测模型本身的设计同等重要。
1 引言
近年来,在国际性社区评测Critical Assessment of protein Structure Prediction(CASP)中,蛋白结构预测取得了显著进展。在CASP14中,端到端的深度学习方法AlphaFold2能够为约三分之二的单结构域蛋白序列生成原子级精度的结构预测。随后,AlphaFold2被扩展为用于多链蛋白复合物结构预测的AlphaFold2-Multimer,并在多种情况下展示了生成高质量复合物模型的能力。AlphaFold2框架将从MSA中提取的共进化特征作为自注意力网络的核心输入,用于训练并生成三维蛋白结构模型。因此,输入MSA的质量是决定是否能够获得高精度模型的关键因素。正因如此,开发能够准确识别并比对多样同源序列的方法,被认为是提升结构预测精度的重要方向之一。随着宏基因组序列数据库规模的指数级增长,快速且准确地构建MSA已成为一项极具挑战性的任务。
在这一方向上的一个代表性工作是ColabFold,其通过用更敏感且更高效的MMseqs2替代HHblits,并结合非冗余的宏基因组数据库,加速了AlphaFold2的MSA生成流程。相比之下,另一种更为激进的策略是引入蛋白语言模型,利用深度学习Transformer网络,通过对输入序列进行掩码并训练模型恢复被遮盖部分来学习共进化信息。训练完成后,该类语言模型甚至可以在不依赖MSA的情况下进行结构预测。将语言模型与AlphaFold2结构模块结合的方法,如ESMFold与OmegaFold,在某些缺乏可检测同源序列的孤立蛋白上,能够生成优于AlphaFold2的结构模型。然而,总体而言,这些方法并未在单体蛋白预测上带来显著的整体精度提升,而蛋白复合物结构预测仍然是一个更加严峻的挑战。
例如在CASP14实验中,仅有约7%的测试蛋白复合物能够获得令人满意的模型,即界面接触评分(ICS)>0.8。在CASP15中这一情况得到明显改善,表现最优的方法(包括该研究提出的流程)可在多达47%的案例中生成满意模型。但目前仍缺乏证据表明,新近提出的AlphaFold2改进方法(如ColabFold)在复合物建模性能上能够显著超越原始AlphaFold2。同时值得注意的是,在CASP15实验中,表面上不依赖MSA的语言模型方法如OmegaFold,在同源序列较少的目标上表现不佳,这表明蛋白语言模型中进化信息不足的问题,与基于MSA方法中MSA过浅所带来的问题本质上是相似的。
为了系统性地评估最优MSA在蛋白结构预测中的潜在贡献,该研究提出了DeepMSA2(图1),一种受此前迭代式单体MSA构建方法DeepMSA启发的分层式策略。与DeepMSA相比,DeepMSA2不仅将流程从单体扩展至多聚体,还整合了多种新开发的MSA生成流程,基于包含约400亿条序列的超大规模基因组与宏基因组数据库构建多个候选MSA,并引入一种由深度学习驱动的MSA评分策略,用于选择最优MSA。该研究在来自CASP13–15的大规模单体与多体数据集上,对DeepMSA2进行了系统而严格的基准测试,结果表明,与当代最先进方法相比,该流程在提升蛋白三级结构与四级结构建模精度方面具有显著优势。DeepMSA2及其相关结构数据库已向学界免费开放,该研究的结果对未来MSA构建方法以及基于深度学习的蛋白结构与功能预测技术的发展具有重要启示意义。
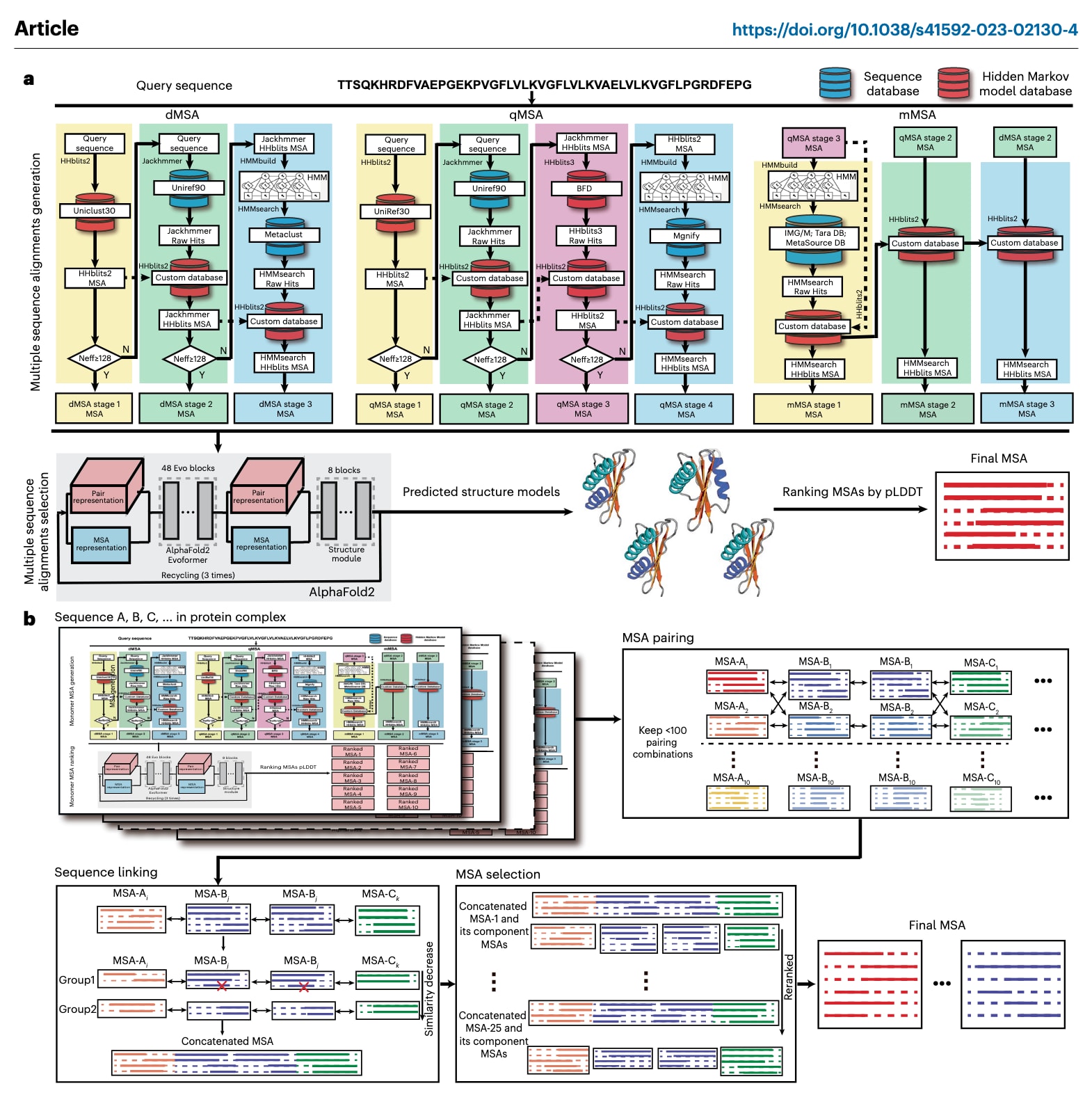
图1|DeepMSA2用于单体与多聚体MSA构建的流程示意图。 a,DeepMSA2-Monomer包含两个主要步骤:首先是迭代式MSA生成步骤,该步骤融合了dMSA、qMSA和mMSA三种算法,随后基于预测结构模型的置信度评分,利用深度学习方法对候选MSA进行排序与筛选。b,DeepMSA2-Multimer包含四个步骤,依次为单体MSA生成、MSA配对、序列连接以及拼接MSA的选择。IMG/M表示来源于Joint Genome Institute的宏基因组数据库,具体说明见Methods中JGIclust部分。
2 结果
DeepMSA2由两条相互独立的流程组成,分别用于单体和多聚体的MSA构建。对于单体MSA构建(图1a),该方法并行采用三种基于不同搜索策略的模块,即dMSA、quadrupole MSA(qMSA)和mMSA,从由全基因组与宏基因组序列库组成的多样化数据库中获取原始MSA。在这三种MSA生成模块中,整体遵循相似的逻辑流程:首先使用初始查询序列在数据库中进行搜索,若未获得足够数量的有效序列,则进一步对规模更大的数据库进行迭代式搜索。最终,来自三个模块的最多十个原始MSA将通过一种快速的深度学习引导预测过程进行排序,以选取最优MSA。对于多聚体MSA构建(图1b),该方法通过连接来源于相同直系同源关系的不同组分链的单体序列,生成多条复合序列。具体而言,每条链中排名前M的单体MSA会与其他链的MSA进行配对,从而生成MN个混合多聚体MSA,其中N为复合物中不同单体链的数量。随后,依据MSA深度与单体链折叠评分即预测的局部距离差异检验分数(pLDDT)的组合评分,按照下文公式(3)所定义的方式选择最优多聚体MSA。两种MSA构建流程的完整细节在Methods部分中给出。
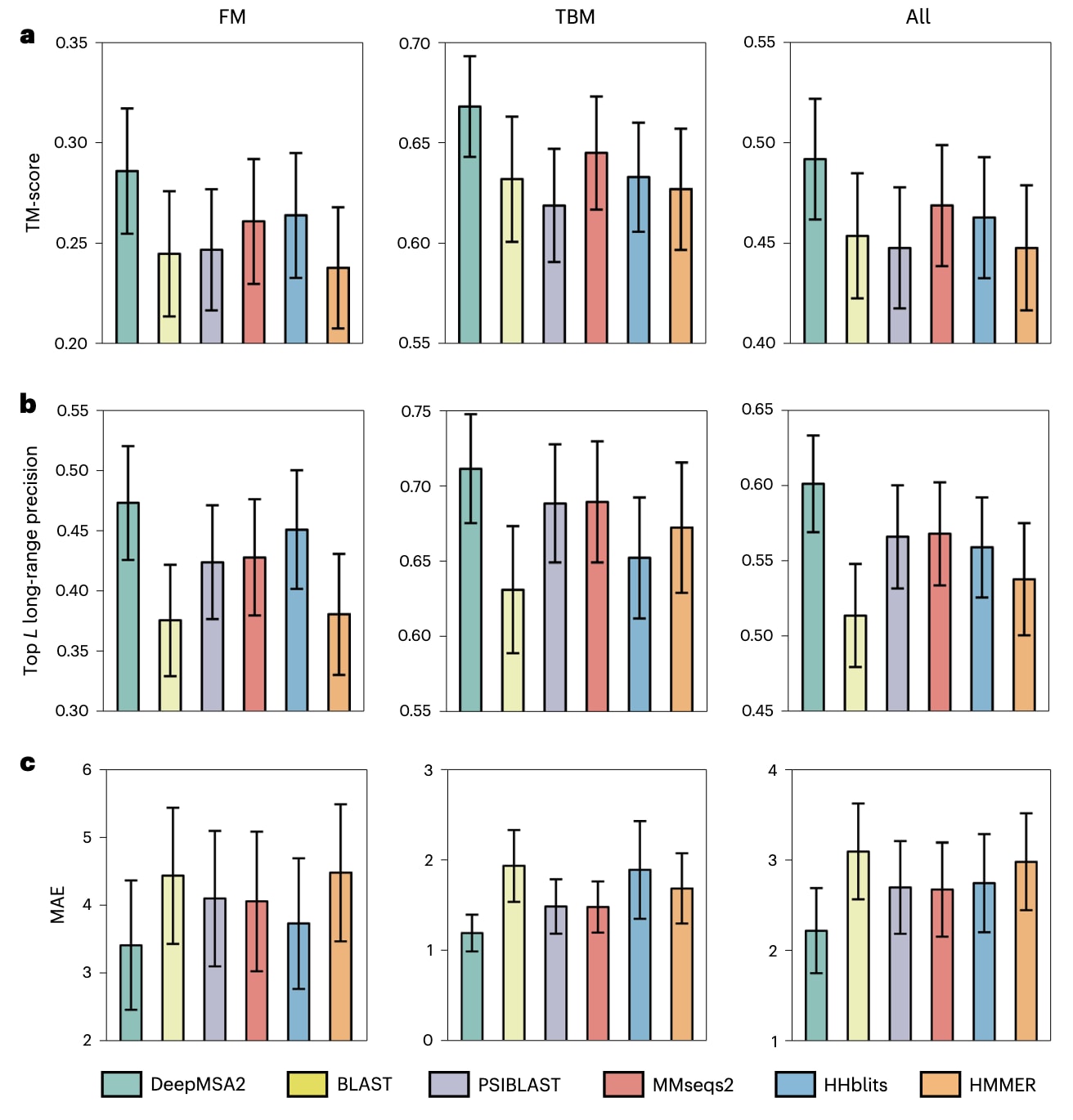
图2 | DeepMSA2与五种对照方法在293个CASP13–15单体结构域上,用于辅助模板识别与深度学习空间约束预测的MSA对比结果。 a,由HHsearch识别到的首个结构模板的平均TM-score。b,前L个长程残基–残基接触预测的精度,其中L为序列长度,长程定义为序列间距|i − j| ≥ 24。c,由DeepPotential预测的前5L个长程残基–残基距离的平均绝对误差MAE。柱状图的高度表示各指标的均值,误差棒表示基于Student t分布计算的95%置信区间。根据评测方的分类,CASP结构域被划分为自由建模(FM)和模板建模(TBM)。在a中,“All”“TBM”和“FM”三列分别对应287、155和132个单体结构域,而在b和c中,对应的样本数分别为271、146和125。
2.1 使用DeepMSA2提升单体结构预测的准确性
首先评估了DeepMSA2生成的单体MSA在模板识别与基于深度学习的空间约束预测中的表现,并与五种常用流程进行了对比,包括BLAST、HHblits、HMMER、MMseqs2和PSIBLAST。总体而言,在三项评估指标上,DeepMSA2均优于上述对照方法。这三项指标分别为通过HHsearch识别的结构模板的平均TM-score(图2a及补充表1),由DeepPotential预测的前L个长程接触的精度(L为序列长度,长程指序列间距|i − j| ≥ 24)(图2b及补充表2),以及前5L个长程距离的平均绝对距离误差MAE(公式(6),图2c、补充图1及补充表3)。此外,还比较了六种MSA在有效序列数Neff(公式(1)),平均序列一致性以及比对覆盖度等方面的差异(补充图2及补充表4),结果显示DeepMSA2在收集同源序列时能够实现更均衡的比对覆盖与多样性。关于单体MSA特征及其对模板识别与空间约束预测潜在影响的更深入讨论,总结于补充讨论文本1中。
作为对DeepMSA2在端到端三维结构预测中作用的直接检验,该研究实现了一个AlphaFold2的改进版本,其中将默认输入MSA替换为DeepMSA2生成的MSA。为简便起见,下文将这一混合流程称为基于DeepMSA2的蛋白折叠方法DMFold。在图3a中,对CASP13–15实验中132个自由建模(FM)单体蛋白目标,比较了DMFold与AlphaFold2预测模型的TM-score。为准确反映FM结构域的特点,在运行程序时,分别排除了CASP13、CASP14和CASP15中2018年5月、2020年5月和2022年5月之后发布的所有模板。结果表明,在63%的案例中即132个目标中的83个,DMFold生成的模型TM-score高于AlphaFold2。DMFold模型的平均TM-score为0.821,相比AlphaFold2的0.781提高了约5%,且单侧Student t检验得到的P值为1.82 × 10−4,表明该差异具有统计显著性。
值得注意的是,这一优势主要体现在较为困难的结构域上。对于86个两种方法均取得TM-score >0.8的结构域,二者的平均TM-score几乎一致,DMFold为0.925,AlphaFold2为0.922。然而,在其余46个至少有一种方法表现不佳的结构域中,二者差异显著,DMFold的平均TM-score为0.626,而AlphaFold2仅为0.517,P值为2.86 × 10−4。在这46个困难结构域中,DMFold在18个目标上的TM-score比AlphaFold2高出0.1以上,而AlphaFold2仅在4个目标中表现出类似优势。
在补充表5中,进一步列出了AlphaFold2与DMFold所使用MSA中的序列统计信息。尽管AlphaFold2收集到的同源序列总数略多于DeepMSA2(2724对2279),但DeepMSA2生成的MSA具有更高的平均有效序列数,Neff为93.7,而AlphaFold2仅为84.5。这表明DeepMSA2能够识别出更加多样化的同源序列,从而构建出在有效意义上更“深”的MSA。造成这一改进的一个重要原因在于,引入了来自Tara数据库(TaraDB)、MetaSource数据库(MetaSourceDB)以及JGIclust数据库的内部宏基因组序列,显著扩展了生物序列空间的覆盖范围。此外,DeepMSA2所采用的多层级迭代式搜索策略,也有助于收集更加多样但仍具相关性的同源序列。
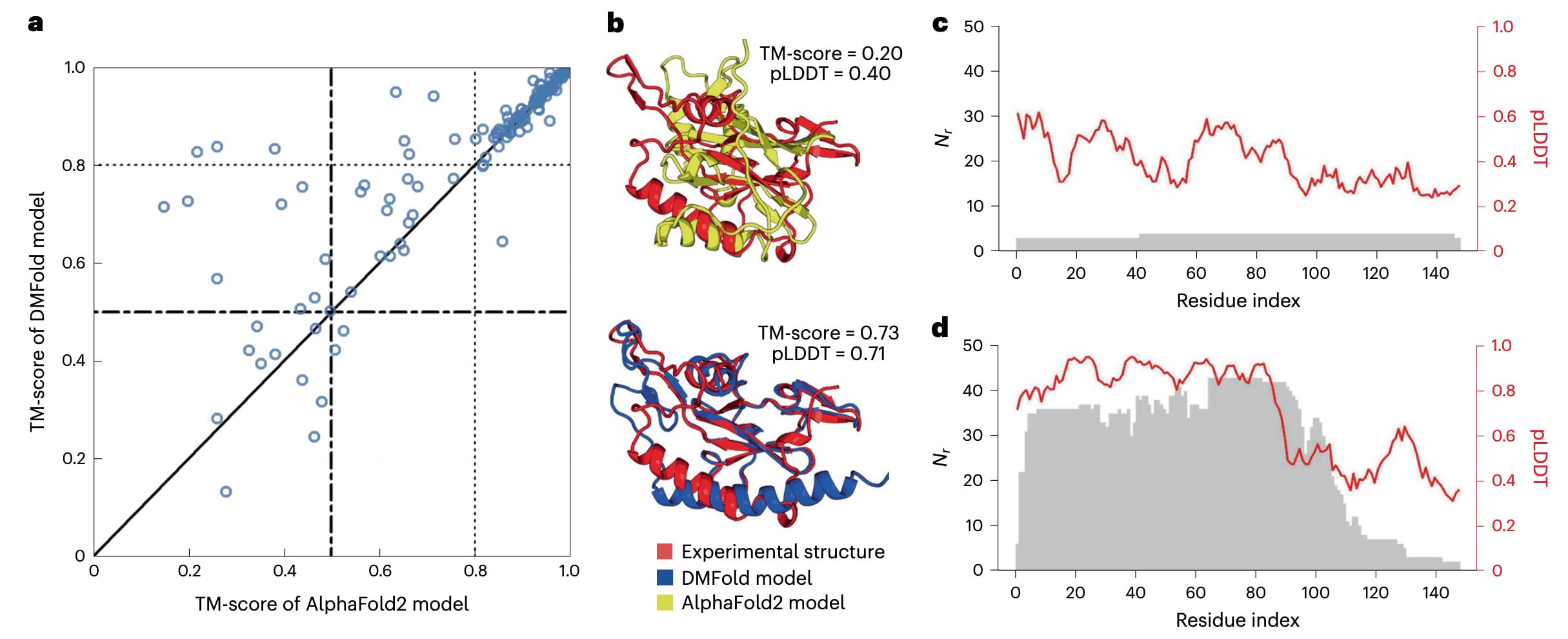
图3 | DMFold在蛋白单体结构域上的预测性能与AlphaFold2的基准对比。 a,在来自CASP13、CASP14和CASP15的132个自由建模(FM)单体结构域上,DMFold与AlphaFold2预测结果的TM-score逐一对比。b–d,以FM结构域T1043-D1为案例,展示AlphaFold2与DMFold生成的结构模型(b),以及基于AlphaFold2所用MSA(c)和DeepMSA2所用MSA(d)时,沿蛋白序列分布的每个位点比对残基数Nr(灰色)与pLDDT评分(红色)。
2.2 利用DeepMSA2对困难蛋白的人类蛋白质组建模
为进一步评估该方法在大规模结构建模中的实际应用价值,该研究将DeepMSA2/DMFold流程应用于人类蛋白质组。鉴于DeepMind团队近期发布了AlphaFold2 Structure Database(DB),分析重点放在其中5,042条被判定为困难序列的蛋白上,这些蛋白在AlphaFold2 DB中的模型置信度均为pLDDT < 0.7。图4a给出了DMFold与AlphaFold2 DB在这些困难蛋白上的整体折叠层级pLDDT分布直方图,可以清晰观察到DMFold模型整体向更高pLDDT值方向偏移。平均而言,DMFold模型的pLDDT为0.663,相比AlphaFold2 DB模型提高了约11%,且单侧Student t检验的P值小于2.2 × 10−16。共有94%(4,738/5,042)的DMFold模型pLDDT高于对应的AlphaFold2 DB模型。总体上,DMFold为1,934条AlphaFold2未能成功建模的人类蛋白生成了高质量的整体折叠结构,其pLDDT ≥ 0.7。
在补充图5中,进一步给出了这1,934条仅能由DMFold成功折叠的蛋白中,DMFold与AlphaFold2 DB模型之间TM-score的分布情况。其中80%(1,549/1,934)的DMFold模型与对应的AlphaFold2 DB模型整体结构差异显著,二者之间的TM-score < 0.6,表明DMFold带来的改进主要体现在拓扑层面。其余385条蛋白中,DMFold与AlphaFold2 DB模型的整体结构较为相似,因此DMFold的提升主要来源于局部结构修正。图4b进一步对这1,934条人类蛋白的残基级pLDDT进行了逐一比较,共涉及878,094个残基,结果显示DMFold在93%的残基上给出了高于AlphaFold2 DB的pLDDT评分。
图4c展示了一个来自未表征蛋白Q6ZQT0的示例。对于该蛋白,AlphaFold2仅在MSA中收集到9条同源序列,Neff为0.7;而DeepMSA2构建的MSA包含122条序列,Neff达到6.2。由于MSA信息极为稀疏,AlphaFold2生成的模型置信度较低,pLDDT仅为0.51,且呈现出不规则的二级结构。相比之下,借助DeepMSA2提供的更丰富MSA信息,DMFold生成了一个置信度显著更高的模型,pLDDT达到0.92,其整体折叠更加稳定,并形成了清晰的氢键网络与合理的二级结构。图4d给出了该蛋白的残基级pLDDT分布,其中DMFold模型几乎所有残基的pLDDT均高于0.7,而AlphaFold2 DB模型中对应残基全部低于0.7。两种模型的整体结构相似性很低,TM-score仅为0.44,表明DMFold显著提升了全局折叠质量。
图4e给出了另一个互补示例,来自推测的二酰甘油O-酰基转移酶2样蛋白Q6IED9,其结构属于α/β三层夹心折叠。尽管DMFold与AlphaFold2模型在整体折叠上较为接近,TM-score为0.88,但DMFold模型的pLDDT为0.83,明显高于AlphaFold2 DB模型的0.68。图4f所示的残基级pLDDT分布表明,DMFold在多个区域构建了更可靠的局部结构,这些区域在图4e中以红色标注,对应于两个更加完善的β折叠片层。上述示例表明,通过提供更具信息量的MSA,DMFold能够在全局折叠与局部结构两个层面同时改进AlphaFold2的建模结果。
在这5,042条缺乏高置信度AlphaFold2结构的人类蛋白中,有48条在AlphaFold2训练截止日期(2018年5月1日)之后于PDB中解析出了实验结构,且覆盖了天然蛋白序列的80%以上。对于这48条蛋白,AlphaFold2 DB模型的平均TM-score为0.630,而DMFold模型达到0.679,单侧Student t检验的P值为1.46 × 10−4。补充图6进一步分析了这48条蛋白中DMFold模型TM-score与pLDDT之间的关系。在所有pLDDT ≥ 0.7的DMFold模型中,85%的预测可视为真正例,即模型被预测为可折叠且其实际TM-score > 0.5。然而,基于0.7的pLDDT阈值,仍存在较高的漏报率(76%),说明许多pLDDT较低的模型同样可能具有正确折叠。总体而言,尽管在这一小规模、近期解析的人类蛋白集合上取得了积极结果,DMFold预测的人类蛋白质组模型的绝对质量仍有待未来更多实验结构的验证。
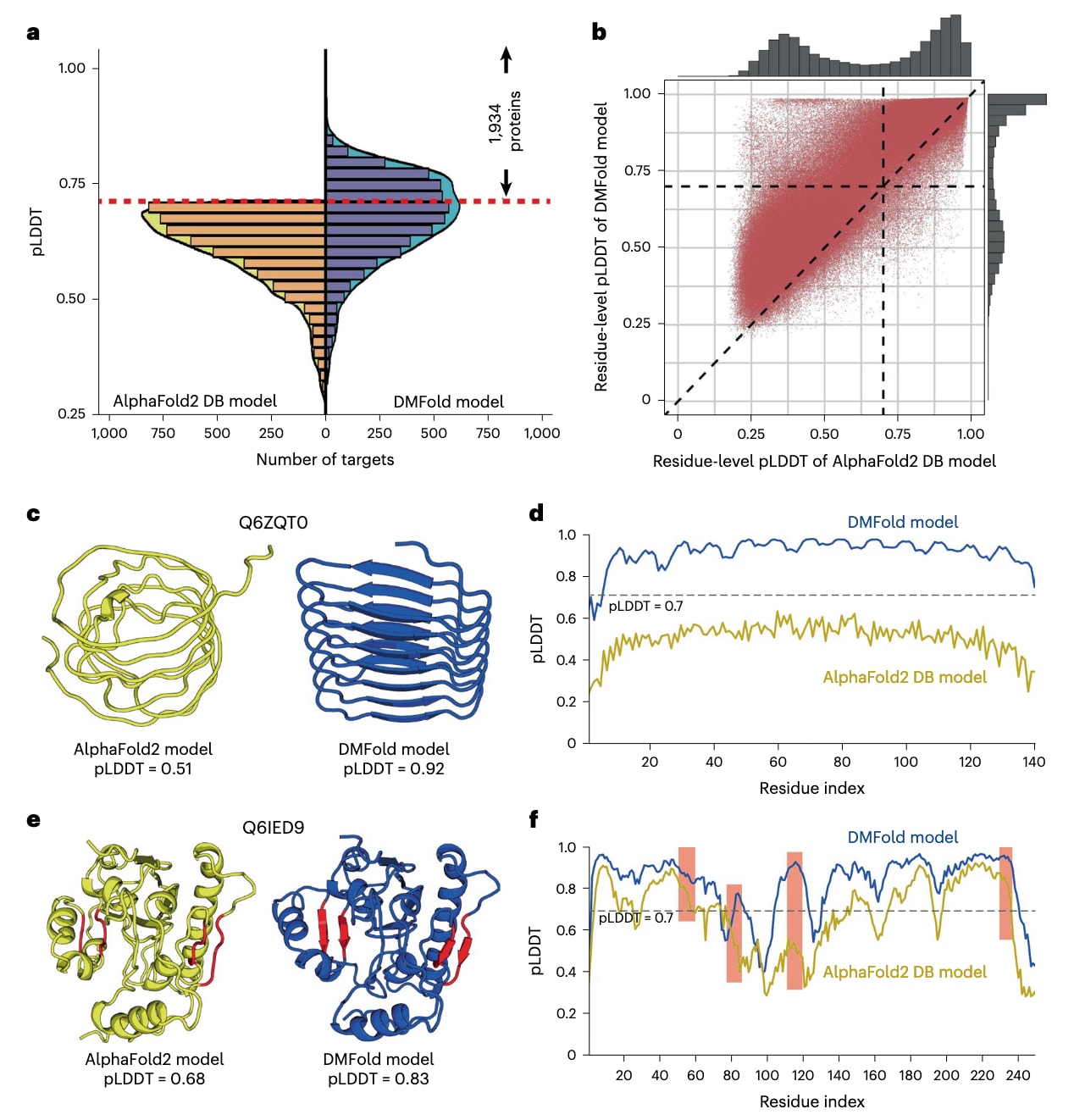
图4 | DMFold在人类蛋白质组5,042个困难目标上的结构建模结果。 a,针对AlphaFold2结构数据库中pLDDT < 0.70的5,042个目标,比较DMFold模型与AlphaFold2 DB模型的pLDDT分布。红色虚线表示置信预测阈值pLDDT = 0.70,其中DMFold在1,934个目标上获得了pLDDT ≥ 0.7。b,对这1,934个由DMFold高置信建模的人类蛋白进行残基级pLDDT逐一比较,共涉及878,094个残基。c,推测的未表征蛋白FLJ45035(Q6ZQT0)的结构模型对比,AlphaFold2 DB模型以黄色表示,DMFold模型以蓝色表示。d,Q6ZQT0在AlphaFold2 DB(黄色)与DMFold(蓝色)下的残基级pLDDT曲线。e,推测的二酰甘油O-酰基转移酶2样蛋白(Q6IED9)的结构模型对比,AlphaFold2 DB模型以黄色表示,DMFold模型以蓝色表示,其中由DMFold构建的两个更加完善的β折叠二级结构以红色标出。f,Q6IED9在AlphaFold2 DB(黄色)与DMFold(蓝色)下的残基级pLDDT曲线,其中与四条β链对应的pLDDT区域以红色背景标注。
2.3 蛋白复合物结构预测的改进
为评估DeepMSA2-Multimer对蛋白复合物结构建模的影响,该研究收集了来自CASP13与CASP14的54个复合物目标,每个目标包含2至8条蛋白链,其中40个为同源多聚体,14个为异源复合物。补充表7给出了这些复合物的详细信息。在补充表8中,对AlphaFold2-Multimer与DMFold-Multimer生成的复合物模型TM-score进行了系统比较,其中DMFold-Multimer通过使用DeepMSA2-Multimer生成的多聚体MSA替换了AlphaFold2-Multimer的默认MSA。结果显示,DMFold-Multimer在所有复合物、异源复合物及同源复合物上的平均TM-score分别为0.834、0.930和0.801,相较于AlphaFold2-Multimer的0.743、0.895和0.690,分别提高了12.2%、3.9%和16.1%。所有比较在单侧Student t检验中P值均小于0.05,表明差异具有统计显著性。
图5a给出了模型TM-score的逐一对比,DMFold-Multimer在70%的案例中优于AlphaFold2-Multimer。类似于单体建模的结果,这一提升主要体现在较困难的复合物上。对于26个两种方法均取得TM-score > 0.9的简单目标,两者的平均TM-score几乎一致,分别为0.961和0.960。然而在其余28个更具挑战性的目标上,DMFold-Multimer的平均TM-score为0.716,显著高于AlphaFold2-Multimer的0.542,P值为1.05 × 10−4。
相较于AlphaFold2-Multimer的默认MSA,DeepMSA2-Multimer带来的性能提升主要来自两个方面。一是其集成化的MSA生成、配对与筛选机制,二是引入了额外的大规模内部宏基因组数据库。为区分这两类因素的贡献,图5b比较了在不同序列数据库条件下AlphaFold2-Multimer与DMFold-Multimer的建模性能。即便在使用相同序列数据库(基因组序列、BFD与Mgnify)的情况下,DMFold-Multimer的TM-score仍由0.743提升至0.784,表明DeepMSA2-Multimer自身的MSA构建、配对与筛选策略具有独立价值。在引入扩展的宏基因组数据库后,模型质量进一步从0.784提升至0.834,说明大规模宏基因组数据同样对复合物建模具有重要作用。图5b还显示,相较于同源复合物,DMFold-Multimer在异源复合物上的TM-score提升幅度较小。这主要源于序列连接机制的限制,DeepMSA2-Multimer依据UniProt中的物种注释,仅在序列来自同一物种时进行连接,以确保直系同源的相互作用配对。由于宏基因组序列通常缺乏物种注释,这些序列无法用于异源复合物的连接,从而削弱了宏基因组数据带来的优势。相反,在同源复合物中,由于组分蛋白完全相同,单体MSA中的所有序列均可自连接,从而充分利用基因组与宏基因组数据库,并带来更显著的结构改进。未来,随着更完善的分类注释数据库或新的序列连接算法的发展,DMFold-Multimer在异源复合物上的性能有望进一步提升。
DeepMSA2提供的多聚体MSA对结构预测的积极影响主要体现在链间取向与距离图的预测上。作为案例分析,图5c展示了目标T0988o,这是一个由三条蛋白链组成、总计612个残基的同源三聚体。AlphaFold2-Multimer生成的模型TM-score仅为0.18,主要原因在于其默认MSA流程仅为每条链检测到8条同源序列,导致链内距离图预测误差较大(MAE = 5.93 Å,图5d),从而产生质量较差的单体模型(TM-score = 0.20)。相比之下,DeepMSA2检测到71条同源序列,生成了更准确的距离图(MAE = 0.81 Å,图5e),并为每条链构建了高质量单体结构(TM-score = 0.95)。在复合物层面,DMFold-Multimer为T0988o生成了TM-score高达0.96的高质量复合物模型。
图5f给出了另一个示例T1038o,这是一个由796个残基组成的同源二聚体。尽管AlphaFold2-Multimer在单体层面生成了较高质量的模型(TM-score = 0.91),但其复合物的四级结构取向完全错误,导致整体TM-score仅为0.53。该问题主要源于AlphaFold2-Multimer构建的MSA质量较低(Neff = 0.2),从而造成链间距离预测精度极差(MAE = 4.11 Å,图5g)。对于该目标,DeepMSA2-Multimer生成了更深的MSA,Neff提升至1.4,从而在链内与链间距离预测上均取得较高精度(分别为MAE = 1.11 Å与1.80 Å,图5h),最终使DMFold-Multimer生成了TM-score为0.91的高质量复合物模型。这些结果表明,正确构建多聚体MSA对于四级结构约束的学习以及最终模型预测均至关重要。
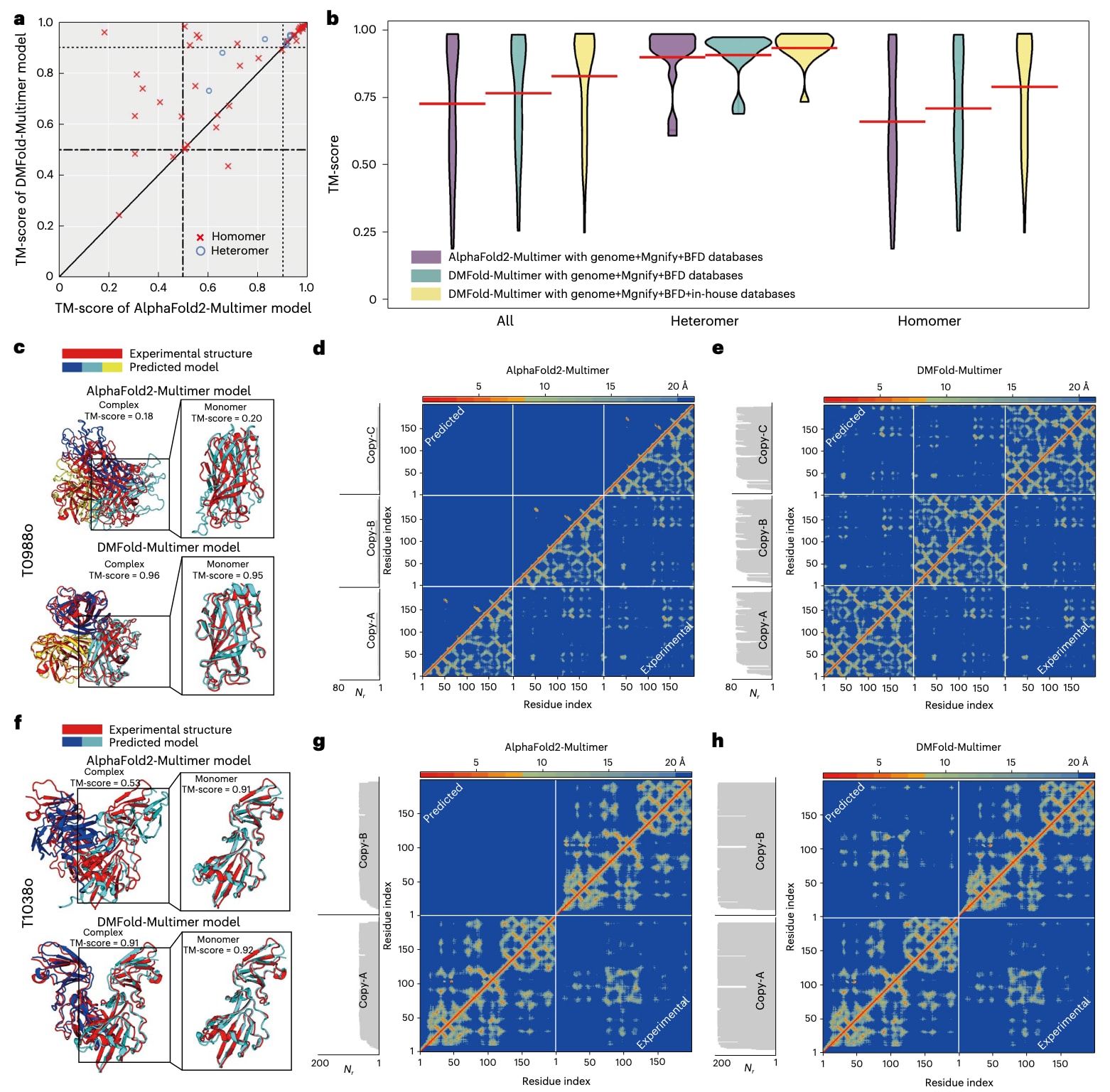
图5 | DMFold-Multimer在蛋白复合物结构预测中的基准评测结果。 a,在来自CASP13和CASP14的14个异源复合物与40个同源复合物上,DMFold-Multimer与AlphaFold2-Multimer预测模型的TM-score对比。b,针对a中目标,使用不同序列数据库的DMFold-Multimer与AlphaFold2-Multimer所得TM-score的小提琴图,红线表示各分布的均值。c,目标T0988o中,AlphaFold2-Multimer与DMFold-Multimer预测模型与实验结构的叠合结果,其中预测模型的单体以不同颜色表示。d,e,针对T0988o,AlphaFold2-Multimer(d)与DMFold-Multimer(e)预测得到的残基–残基距离图(热图),并在边缘标示每个位点的比对残基数Nr,红色对角线上方为预测结果,下方为实验结构计算得到的距离图。f,目标T1038o中,AlphaFold2-Multimer与DMFold-Multimer预测模型与实验结构的叠合结果。g,h,针对T1038o,AlphaFold2-Multimer(g)与DMFold-Multimer(h)预测的残基–残基距离图(热图)及对应的Nr分布,红色对角线上方为预测结果,下方为实验结构计算结果。
2.4 DeepMSA2/DMFold-Multimer在CASP15实验中的盲测表现
作为一次盲测,DeepMSA2/DMFold-Multimer流程参加了2022年举行的CASP15复杂结构预测评测。该实验共包含47个复合物目标,每个目标由2至27条组分链组成。图6a展示了其中38个已公布实验结构的复合物在DMFold-Multimer预测下的TM-score分布直方图。DMFold-Multimer在这些目标上的平均TM-score为0.83,其中36个复合物的TM-score高于0.5。尽管这一结果显示出较强性能,但复合物模型的TM-score仍明显低于对应单体模型的水平(TM-score = 0.89),表明链间取向仍然是蛋白四级结构预测中的关键难点。
图6b给出了27个预测TM-score > 0.8的复合物目标中,DMFold-Multimer模型与实验解析结构的叠合结果。其中包括7个大规模复合物,H1111、H1114、H1137、T1170o、H1171、H1172和T1181o,其序列长度分别为8,460、7,988、4,592、1,908、1,956、2,004和2,064个残基。DMFold-Multimer为这些目标构建了TM-score分别为0.98、0.91、0.94、0.93、0.93、0.91和0.85的高质量模型。值得注意的是,其中最大的三个目标均为异源复合物,其化学计量比分别为A9B9C9、A4B8C8和A1B1C1D1E1F1G2H1I1,DMFold-Multimer在这三种情况下均生成了TM-score > 0.9的高精度模型。这些结果表明,DMFold-Multimer在大尺度蛋白复合物建模方面具备显著能力,而这一直是传统四级结构预测方法面临的长期挑战。
在图6c及补充表9中,还给出了DMFold-Multimer(参赛名称Zheng)与另外86种CASP15多聚体建模方法的系统对比。根据评测方基于TM-score、LDDT、ICS和Interface Patch Score综合计算的Z-score总和,DMFold-Multimer在所有参赛方法中排名第一。TM-score和LDDT用于衡量整体折叠质量,而ICS和Interface Patch Score用于评估复合物界面建模质量。DMFold-Multimer的累计Z-score为35.30,几乎是NBIS-AF2-multimer组的三倍。NBIS-AF2-multimer对应于Elofsson实验室在CASP15目标上运行的AlphaFold2-Multimer公共服务器2022年3月版v.2.2.0,其累计Z-score为12.27。DMFold-Multimer的得分也比排名第二的方法高出21.1%(29.15)。DMFold-Multimer与AlphaFold2-Multimer在各子评分项上的详细对比列于补充表10中。
补充图7展示了三个示例目标H1140、H1141和H1144,它们均为纳米抗体–抗原复合物。纳米抗体是单结构域抗体,通过与抗原相互作用触发关键免疫反应,这三个目标代表了纳米抗体与同一小鼠2’,3’-环核苷酸3’-磷酸二酯酶相互作用的三种典型模式。结果显示,AlphaFold2-Multimer生成的模型TM-score均低于0.7,而DMFold-Multimer分别为三种复合物生成了TM-score为0.92、0.95和0.99的高质量模型。相应地,DMFold-Multimer模型的ICS F1分数分别为0.51、0.79和0.74,显著高于AlphaFold2-Multimer的0.02、0.06和0.09,说明DeepMSA2-Multimer构建的多聚体MSA在免疫蛋白–抗原复合物的链间相互作用建模中起到了关键作用。
在补充图8中,以目标H1144为例进一步分析了两种方法的差异。DMFold-Multimer采用多MSA配对策略生成了25个配对MSA,其中最佳模型(TM-score = 0.99)来自Neff最高的MSA(16.3)。相比之下,AlphaFold2-Multimer仅使用一个Neff为8.1的MSA,未能生成任何TM-score > 0.8的模型。尽管如此,DMFold-Multimer并未成功折叠CASP15中的所有纳米抗体–抗原复合物。补充图9给出了一个失败案例H1142,即使使用多个MSA,DMFold-Multimer仍未生成正确模型。关于DMFold-Multimer在纳米抗体–抗原复合物建模中成功与失败案例的深入分析,总结于补充讨论文本2中。
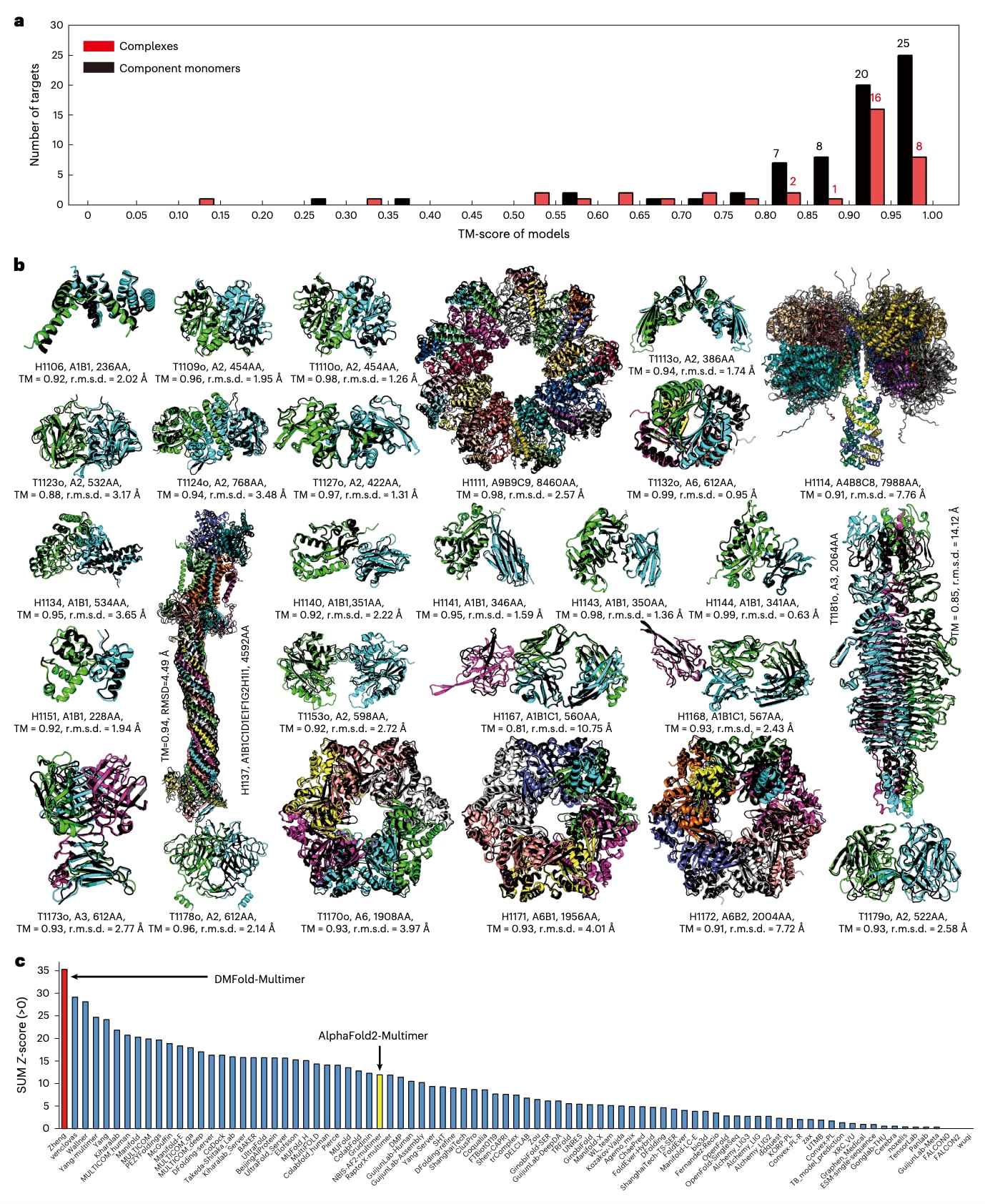
图6 | DMFold/DMFold-Multimer流程在CASP15实验中用于蛋白复合物结构预测的整体表现。 a,在38个已公布实验结构的复合物目标上,DMFold-Multimer预测模型的TM-score分布直方图。b,对27个TM-score(TM) >0.8的复合物目标,展示DMFold-Multimer生成的首个模型与实验解析结构的叠合结果,其中预测模型的各组成单体以不同颜色表示,实验结构以黑色标注。c,基于CASP15官网数据,比较87个参赛装配组在41个多聚体目标上的Z-score总和,其中DMFold-Multimer(注册名Zheng)以红色标出,AlphaFold2-Multimer公共服务器2022年3月版(注册名NBIS-AF2-multimer)以黄色标出。r.m.s.d.表示均方根偏差。
3 讨论
随着深度机器学习技术的快速发展,MSA在现代蛋白结构预测中的重要性日益凸显。基于在多种基因组与宏基因组序列数据库中进行的迭代式比对搜索,该研究提出了用于蛋白单体与多体MSA构建的分层流程DeepMSA2。大规模测试表明,DeepMSA2能够显著提升蛋白结构预测的整体精度。与现有MSA构建方法相比,DeepMSA2的一个核心优势在于其迭代搜索与基于模型的预筛选策略,能够生成在比对覆盖度与同源多样性之间更加平衡的MSA。迭代搜索机制还使得多个内部宏基因组数据库得以充分利用,进一步扩展了MSA所覆盖的序列空间。
系统基准测试显示,由此获得的进化与共进化信息能够明显提升结构模板识别以及深度学习距离和取向约束预测的准确性。将DeepMSA2与当前最先进的AlphaFold2建模方法结合后形成的DMFold,在缺乏PDB同源模板的自由建模结构域上,使AlphaFold2模型的TM-score平均提高了5%。在对人类蛋白质组的应用中,DMFold使5,042条AlphaFold2未能给出高置信度模型的困难蛋白的pLDDT平均提高了11%,显著扩展了可提供可靠结构预测的人类蛋白范围。
DeepMSA2-Multimer生成的多聚体MSA同样显著提升了多链蛋白复合物的结构预测性能。在CASP13和CASP14的54个复合物目标上,DMFold-Multimer相对于默认的AlphaFold2-Multimer实现了12.2%的TM-score提升。在最新的CASP15社区盲测中,根据评测方标准,DMFold-Multimer在复杂结构预测中取得了最高精度,其平均TM-score比AlphaFold2-Multimer公共服务器高15.4%,平均ICS分数高27.5%。尤其值得注意的是,DMFold-Multimer能够为规模高达8,460个氨基酸的巨大寡聚复合物构建TM-score为0.98的高质量模型,显示出其在大规模蛋白复合物建模方面的能力,这一问题长期以来一直困扰着传统四级结构预测方法。
尽管性能取得了显著提升,DeepMSA2仍面临一些挑战。其中一个明显的改进空间在于异源复合物建模,其相对于AlphaFold2的提升幅度小于同源复合物。由于当前多聚体MSA是由单体MSA构建而来,如何有效地将不同组分的MSA序列连接起来,形成最优的相互作用同源序列配对MSA,仍是一个基础性难题。目前基于物种注释的序列连接策略仅适用于基因组序列,使得宏基因组数据库中信息丰富的同源序列难以充分用于多链装配。对于同源复合物,当前方法简单地将单体MSA中的所有序列与自身连接,但并非所有同源序列都会发生自相互作用,若能进一步区分相互作用与非相互作用的同源体,例如结合蛋白–蛋白相互作用预测信息,有望进一步提升同源复合物MSA的质量。
鲁棒的配对MSA构建方法不仅对异源接触预测至关重要,也将对蛋白相互作用判别等相关任务具有重要价值。除MSA配对外,另一个潜在方向是重新训练类似AlphaFold2的模型,显式利用信息量更高的单体MSA,从而在网络学习过程中直接缓解序列配对问题。此外,DMFold-Multimer在运行前需要已知复合物的化学计量比,这在实际应用中可能构成限制。将基于深度学习的化学计量比预测方法整合进流程,有望缓解这一问题。进一步而言,DeepMSA2/DMFold是否能够扩展至RNA以及RNA–蛋白复合物结构预测,也是正在探索的方向,其中需要克服RNA序列与结构数据库相对稀缺的限制。
总体来看,DeepMSA2/DMFold流程所展现出的强大性能表明,蛋白结构预测问题远未被彻底解决。对于同源序列稀少的蛋白以及多蛋白复合物,当前方法仍存在显著改进空间。DeepMSA2/DMFold在若干此类困难目标上的成功进一步证明,在基于深度学习的蛋白结构预测中,输入信息内容的优化应当与预测模型本身的设计同等受到重视。