Science 2026 | FoldMason: 大规模蛋白质结构多重比对

获取详情及资源:
0 摘要
蛋白质结构在很大程度上超越序列而保持保守性,这使得多重结构比对在分析远缘同源蛋白时具有关键作用。随着计算预测方法的快速发展,可获得的蛋白质结构数量大幅增长,对快速且准确的多重结构比对方法提出了更高要求。该研究提出FoldMason,一种渐进式多重结构比对方法,结合了Foldseek和TM-align两种成对结构比对工具,能够对数十万规模的蛋白质结构进行多重比对,在比对质量上可达到或超过当前最先进的方法,同时计算速度快了两个数量级。以黄病毒科糖蛋白为例,研究表明FoldMason生成的多重结构比对能够支持超越“暮光区”的系统发育分析。FoldMason还能够计算比对置信度分数,提供交互式可视化,并在准确结构预测时代为大规模蛋白质结构分析提供了必要的速度与精度。FoldMason是一款免费且开源的软件。
1 引言
通过比对对多个蛋白质进行比较,有助于阐明它们的进化历史、多样性以及功能。已知最优多重序列比对问题在计算复杂性上属于NP完全问题,因此现有多重比对工具通常采用启发式策略,例如渐进式比对方法,将整体任务分解为一系列成对操作。在这种框架下,首先根据未比对的序列估计一棵引导树,然后按照由近及远的顺序依次进行成对比对,优先比对相似序列。基于这一思想,已经发展出多种构建多重序列比对的方法,其中较为典型的包括MAFFT、MUSCLE、Clustal Omega以及FAMSA。研究表明,引入结构信息能够获得比仅依赖蛋白质序列更高质量的比对结果。蛋白质结构在氨基酸序列层面之上仍具有较强的保守性,因此即使在序列一致性低于约25%的“暮光区”范围内,仍然可以进行有效比较。这一特点推动了多种多重结构比对方法的发展。
与序列比对类似,大多数结构比对工具也采用渐进式策略,通过逐步整合成对结构比对来构建多重结构比对。例如,US-align先利用TM-align对所有输入结构对进行成对比对,再基于UPGMA引导树进行渐进式整合;Caretta-shape通过统计重叠的Zernike骨架片段来生成引导树,并结合结构叠合与动态规划完成比对,这两种方法还额外融合了氨基酸序列相似性。3DCoffee在T-Coffee框架中同时引入了TM-align和非刚性叠合方法SAP;MUSTANG依据残基间接触关系和局部结构拓扑进行渐进式比对;Matt则通过对齐片段对的链式连接算法来刻画结构柔性。这些多重结构比对方法在精度方面表现突出,但通常需要付出较高的计算代价。
近年来,结构预测领域的突破性进展使得蛋白质结构数据库的规模迅速膨胀,例如包含数亿结构的AlphaFoldDB和ESMAtlas,这对比对方法在计算速度上的可扩展性提出了数量级上的新要求。此前提出的Foldseek能够在超大规模结构数据库中高效检索并识别成对结构相似性。该方法采用一种包含20种状态的结构字母表,将蛋白质三级结构中的相互作用编码为一维序列,从而可以利用快速的字符串比较算法进行处理。该编码方式按从N端到C端的顺序,为每个残基记录其与最近空间邻居之间的三级相互作用。随后,这种结构字母及其替换矩阵被用于将快速的序列多重比对方法改造为临时的多重结构比对方案。借助Foldseek,研究者已经将AlphaFoldDB聚类为数百万个结构簇,其中一些簇包含多达数十万个高度分化的蛋白质结构。要全面解析这些结构簇的内在关系,仍然需要同时具备高精度与高可扩展性的多重结构比对方法。
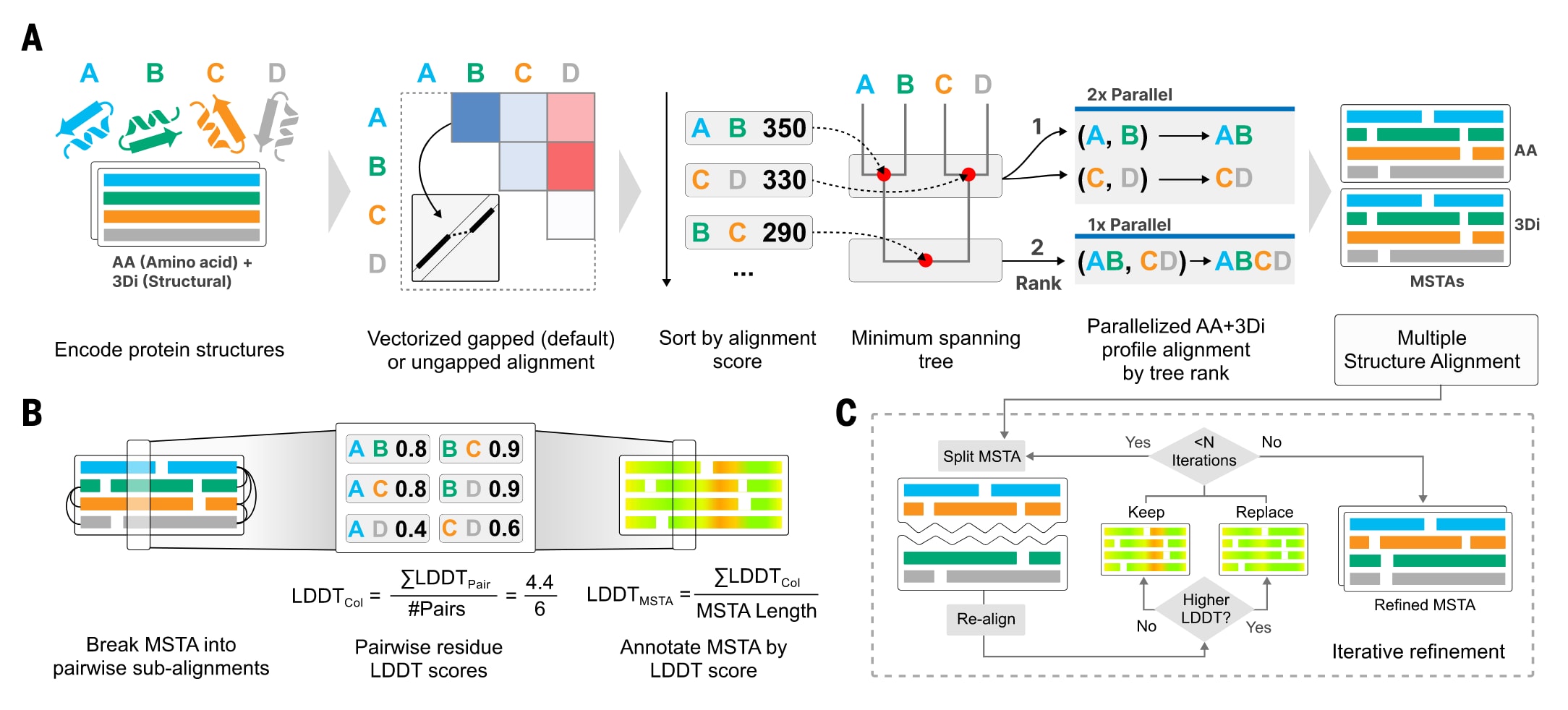
图1|FoldMason中的多重结构比对与评分流程。 (A) FoldMason使用3Di+AA字母表将输入的蛋白质结构表示为字符串,并对每一对结构计算成对比对得分。随后根据这些得分进行排序,构建最小生成引导树。沿着引导树从叶节点到根节点,逐步对3Di+AA结构轮廓进行渐进式比对,并依据引导树中的层级顺序并行计算相互独立的比对任务。(B) 基于LDDT的置信度评分计算流程。首先将输入的多重结构比对拆分为一系列成对的子比对,并在这些子比对上计算LDDT分数。随后将每个残基的LDDT分数映射回其在多重结构比对中的对应位置,并对同一列的残基分数取平均,得到列级别的LDDT分数。最终,对所有列的LDDT分数进行平均,获得整体多重结构比对的LDDT评分。(C) 通过迭代最大化LDDT对多重结构比对进行优化。输入的多重结构比对被随机划分为两个子多重结构比对,随后删除仅包含缺口的列,并对这两个子比对重新进行对齐。在每一次迭代中计算平均多重结构比对的LDDT分数,若分数得到提升,则用新的比对结果替换原始比对。该过程作为基础流程完成后的一个可选步骤提供。
2 FoldMason以渐进方式构建多重结构比对
为应对上述挑战,该研究提出了一种渐进式结构比对工具FoldMason。如图1A所示,FoldMason在基于参考和无参考的多种基准测试中,生成的多重结构比对质量可与公认的金标准方法相当。针对无参考评估,研究进一步设计了一种结构置信度评分,用于衡量比对结果的可靠性。在计算效率方面,FoldMason相较于其他基于结构的多重结构比对方法快了两到三个数量级,其速度也仅比最快的序列比对工具慢一个数量级。FoldMason既可作为命令行软件使用,也提供了网页服务器形式,并能够生成交互式可视化结果,便于对多重结构比对进行直观解读。
在具体流程上,FoldMason首先利用3Di+AA字母表对输入蛋白质结构进行编码,通过加速的全对全比对生成最小生成树,并以此为基础迭代式地、并行地引导多重结构比对的构建。在第一轮迭代中,成对结构比对的评分综合考虑了3Di+AA序列相似性、全局结构叠合以及局部残基邻域的相似性。在随后的迭代中,成对比对基于由先前合并的结构集合构建的结构轮廓进行,并不再包含全局叠合评分。此外,该研究还提出了一种强调速度的模式FoldMason Fast,该模式通过无缺口的全对全比对并在渐进式对齐过程中省略邻域相似性项,以进一步提升运行效率。
已有多种指标被提出用于评估无需结构叠合的成对局部相似性,其中局部距离差检验LDDT因在AlphaFold2中的应用而成为事实上的社区标准。FoldMason将LDDT推广到多重结构比对场景中,基于局部结构邻域保守性定义了一种无参考的比对质量置信度评分,如图1B所示。在此基础上,FoldMason还可以选择性地对多重结构比对结果进行迭代优化,以最大化平均LDDT得分,如图1C所示。
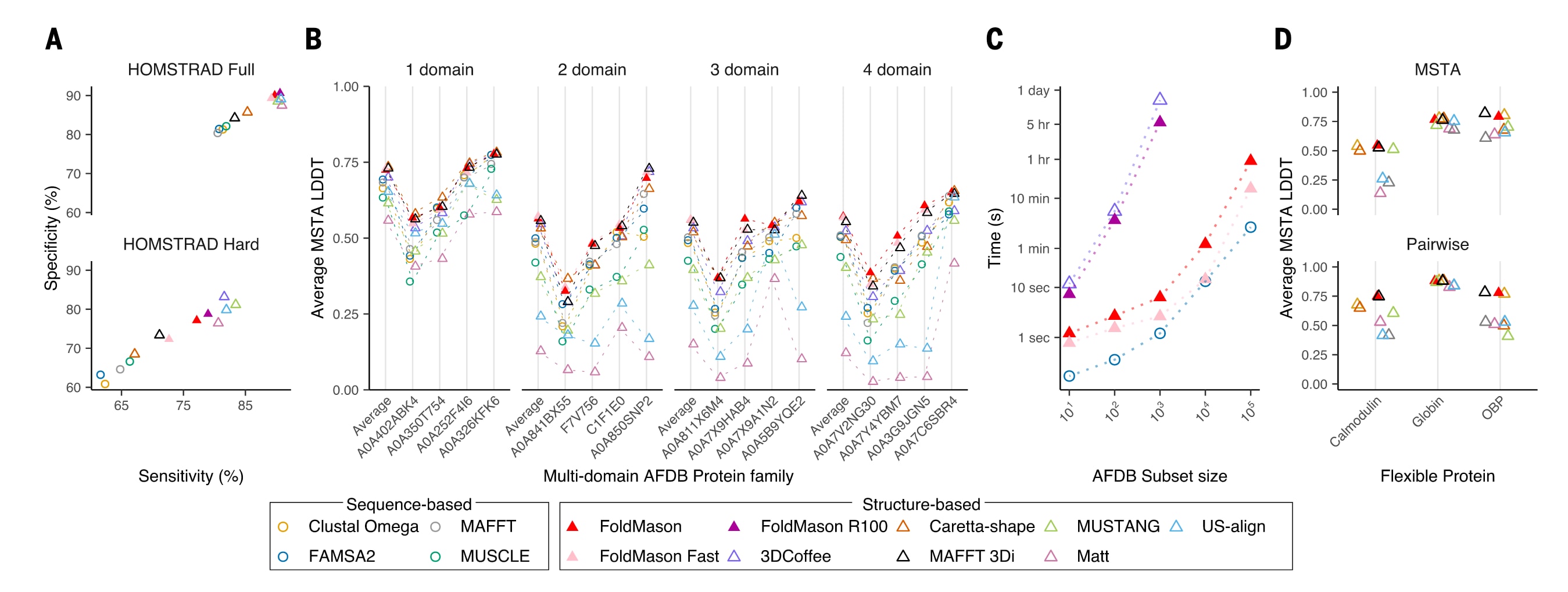
图2|FoldMason在基准测试中与其他方法在准确性和速度方面的比较。 (A) 测试比对相对于HOMSTRAD参考比对的sum-of-pairs得分,上图对应1032个蛋白质家族的完整数据集,下图对应一个包含55个蛋白质家族的子集,这些家族至少包含4条成员序列,且多重序列比对中的平均成对序列一致性低于25%。其中,灵敏度表示测试多重序列比对中成功恢复的HOMSTRAD参考残基对所占比例,特异性表示测试多重序列比对中的残基对在参考比对中出现的比例。(B) 在160个AFDB结构簇基准数据集上,不同方法生成比对结果的平均LDDT分数,展示了其中8种方法的结果,其余方法见图S5。图中同时给出了按CATH结构域数量划分后、每个子集内40个家族的平均得分,以及从全部40个家族中按所有方法的中位得分排序后每隔10个选取的4个代表性家族的得分。结果表明,FoldMason在保持高准确性的同时,其运行速度比其他结构比对方法快36倍到970倍。(C) 基于序列和基于结构的方法在LysR AFDB结构簇不同规模子集上的运行时间比较,该结构簇的代表性UniProt编号为A0A258JAY9。(D) 三类具有构象柔性的蛋白家族在多重结构比对层面和成对比对层面上的平均LDDT分数,其中上图对应多重结构比对,下图对应成对比对。
3 FoldMason在有参考与无参考基准测试中达到或超过最先进的结构比对方法
在基准评测中,研究将FoldMason与多种公认的基于结构和基于序列的对齐工具进行比较,测试数据包括有参考的HOMSTRAD蛋白质结构数据集以及一个由160个AFDB结构簇组成的无参考数据集。同时,还展示了FoldMason在大型AFDB结构簇上的可扩展性,并通过案例分析说明其在柔性蛋白结构比对以及基于结构的系统发育重建中的应用,以黄病毒科糖蛋白为例加以说明。
在HOMSTRAD数据集上,研究将FoldMason生成的比对结果与多种结构和序列比对工具的结果进行比较,包括3DCoffee、US-align、mTM-align、MUSTANG、Matt、Caretta-shape,以及使用氨基酸距离矩阵或3Di距离矩阵的MAFFT,还有MUSCLE、FAMSA2和Clustal Omega。HOMSTRAD包含1032个蛋白质结构家族,并提供了人工整理的高质量参考多重比对。其中还选取了55个“困难”家族子集,这些家族至少包含4条序列,且多重比对中的平均成对序列一致性低于25%,并被用于参数优化。基于sum-of-pairs指标的评估表明,结构比对方法整体上明显优于纯序列方法。该指标统计测试比对中与参考比对一致的残基对数量,并区分参考到测试方向的灵敏度以及测试到参考方向的特异性。在完整数据集和困难子集上,结构方法在灵敏度上平均分别提高了7.1%和13.1%,在特异性上分别提高了8.0%和14.1%。由于3DCoffee、US-align、mTM-align、Caretta-shape以及FoldMason同时利用了氨基酸序列信息,它们的比对结果反映了序列与结构信号的综合效果。FoldMason在完整数据集和困难子集上的准确性均达到了结构比对方法的水平,其F1分数分别为89.9和77.1。此外,在成对比对质量的评估中,FoldMason同样表现出与其他方法相当或更优的性能。
尽管HOMSTRAD提供了高质量的人工参考比对,但其主要由小规模、单结构域且长度偏短的蛋白质家族组成,难以全面代表真实蛋白质家族的复杂性。为此,研究构建了一个无参考的基准数据集,基于AFDB中高质量、全长且高度多样化的AlphaFold2预测结构。利用CATH结构域注释,筛选得到200个多结构域蛋白质结构簇,每个簇包含20个结构,具有1到4个结构域,在CATH拓扑层级上相同,但在同源超家族层级上不同,从而形成一组高度分化但仍可近似全局比对的结构集合。随后随机选取40个簇用于参数优化,其余160个簇用于正式评测。由于这些簇通过Foldseek在90%结构覆盖率条件下聚类且未设置序列一致性阈值,优化集与评测集之间的相似性较低,从而降低了过拟合风险。
在无参考评估中,研究采用平均LDDT分数衡量比对质量,该指标与HOMSTRAD上的有参考评估结果具有良好相关性,并可由FoldMason直接计算。结果显示,在全部AFDB结构簇上,FoldMason在准确性上达到或超过所有结构比对工具,并明显优于序列比对方法。依赖刚体叠合的结构比对工具在结构域数量增加时性能下降,而考虑局部残基邻域相似性的工具,如FoldMason和3DCoffee,则能够保持较高准确性。默认的平均LDDT指标通过对齐长度进行归一化,更强调灵敏度,即在覆盖更多残基的同时保持比对紧凑性。研究还提出了一种替代的平均LDDT指标,通过对非零得分列数归一化,更侧重于精确性,从而突出每一对齐列本身的结构支持度。在这两种指标下,FoldMason分别排名第一和第三,表明其在整体覆盖度和局部准确性之间实现了良好的平衡。
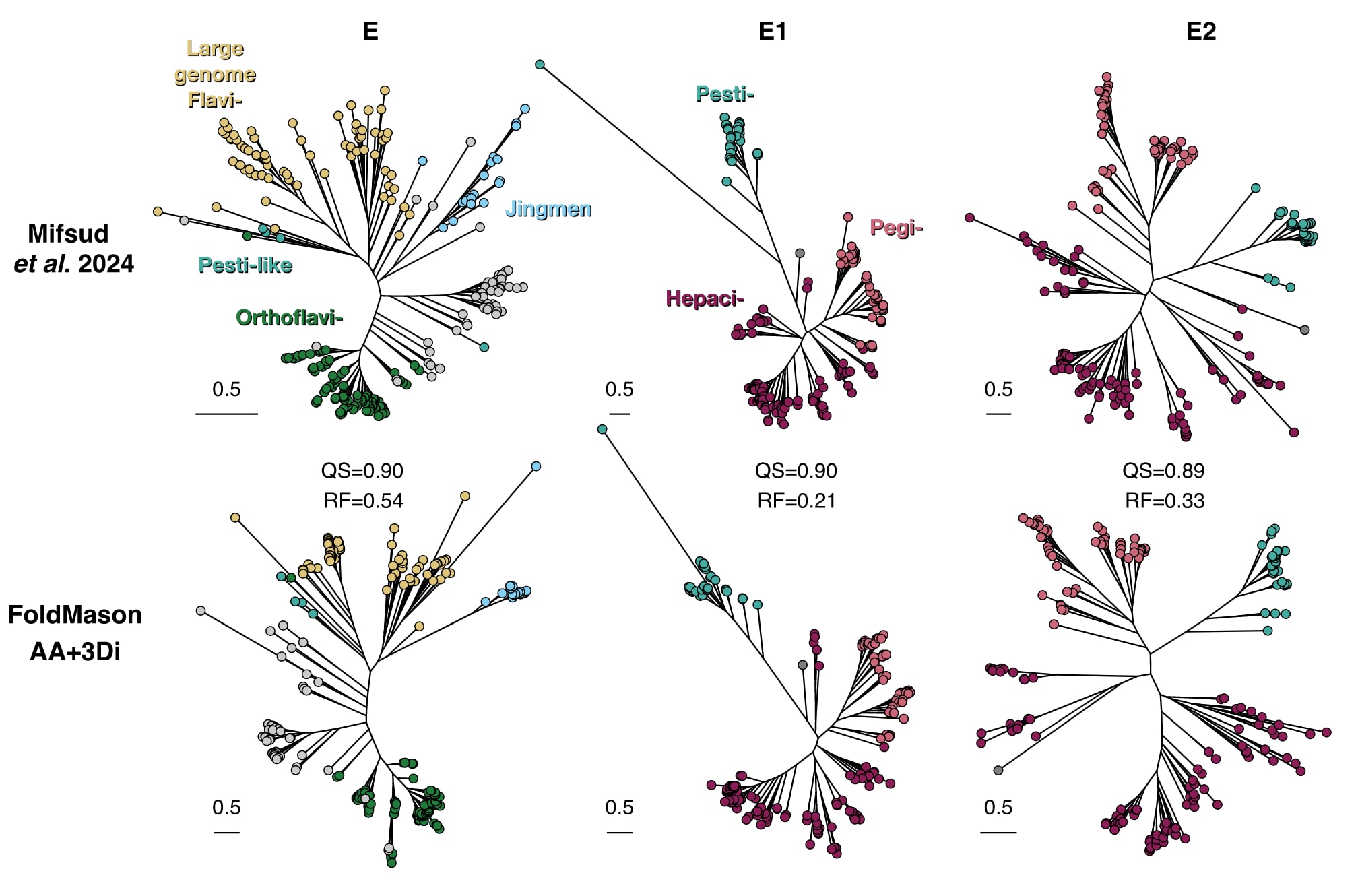
图3|FoldMason直接计算得到的多重结构比对支持此前通过人工多重结构比对推断的结构系统发育关系。 研究使用FoldMason对Mifsud等人报道的参考结构进行比对,并基于所得的多重结构比对重建系统发育树。由FoldMason生成的系统发育结果与既有结果之间具有较高一致性,这一点通过较高的四元组相似性以及较低的Robinson–Foulds距离得以体现。
4 FoldMason可扩展至大规模结构数据集并能够处理构象柔性
为展示FoldMason在大规模蛋白质结构数据集上的可扩展性,研究从一个大型AFDB结构簇中选取不同规模的子集进行比对,规模从10个到100000个结构不等。该结构簇主要由LysR转录激活因子蛋白组成,代表性UniProt编号为A0A258JAY9,平均长度为301.4个氨基酸,总计包含533657个结构。评测中分别选用3DCoffee和FAMSA2作为结构比对方法和序列比对方法的代表。结果表明,FAMSA2在扩展性方面表现最优,可在159秒内完成100000条序列的比对。在相同数据集上,FoldMason标准模式耗时3338秒,约慢21倍,而快速模式耗时941秒,约慢6倍,而3DCoffee在2天内甚至无法完成10000个结构的比对。总体而言,FoldMason在100000个结构上的运行速度,比3DCoffee在1000个结构上的速度快了多个数量级。
由于蛋白质具有柔性并可能发生构象变化,这种特性可能会影响多重结构比对方法的效果。因此,研究进一步测试了多种结构比对工具在处理构象柔性蛋白家族时的表现,这些家族此前已被用作柔性结构比对的经典案例,包括类钙调蛋白EF-hand蛋白、肌红蛋白以及气味结合蛋白,成员数量分别为4个、9个和5个。钙调蛋白属于EF-hand型Ca2+感知蛋白家族,由N端和C端两个结构域通过一段柔性连接区相连。肌红蛋白存在明显的内部柔性,使得结构比对较为困难。猪和牛中的气味结合蛋白在C端区域存在构象重排。尽管这三类蛋白在氨基酸序列上几乎完全一致,但基于刚体叠合的结构比对方法在这些情况下均表现不佳,而FoldMason则保持了稳健性能,始终获得较高的平均LDDT分数。研究还通过成对比对进一步展示了在柔性区域中不同方法的差异。然而,对于构象变化极为剧烈的情况,例如发生折叠切换的蛋白如RfaH,由于结构相似性显著降低,即便是FoldMason也更难实现可靠比对。
5 FoldMason多重结构比对支持超越暮光区的基于结构的系统发育重建
系统发育分析通常依赖多重序列比对,而多重结构比对能够将这类分析拓展到序列相似性低于暮光区的蛋白质。已有多项研究利用Foldseek及其3Di字母表开展远缘系统发育分析。例如,有研究表明,基于3Di序列的多重结构比对可为传统序列信息提供互补,从而改进系统发育重建。另有工作提出FoldTree,通过Foldseek计算由结构成对比对生成的序列一致性矩阵,用于比较远缘相关蛋白。还有研究针对病毒糖蛋白之间的远缘系统发育关系,构建了结合Foldseek、ColabFold以及基于3Di改造的FAMSA的分析流程。这些方法中,FoldMason通过直接从预测结构同时在3Di和氨基酸两个层面生成引导树和多重结构比对,显著简化了整体流程。
为验证FoldMason在该应用场景下的能力,研究基于FoldMason生成的多重结构比对,重新构建了此前报道的E、E1和E2糖蛋白的系统发育树。结果显示,FoldMason在E、E1和E2上的平均多重结构比对LDDT分数分别为0.434、0.497和0.324,均高于此前方法对应的0.324、0.491和0.237。基于FoldMason比对结果生成的系统发育树,其拓扑结构整体上与既有结果一致,所有已注释的进化分支均被成功恢复。通过四元组相似性评估,FoldMason生成的系统发育树与原有树之间的一致性分别达到90%、90%和89%,且自助法支持度水平相近。这表明,FoldMason生成的多重结构比对能够构建质量相当的系统发育树,证明了其在超越暮光区的系统发育分析中的实用价值。
6 展望与结论
在未来工作中,研究计划整合蛋白质语言模型ProstT5,直接从氨基酸序列预测3Di表示,从而避免耗时的结构预测步骤。与优化后的ColabFold预测相比,这一策略可将FoldMason输入生成速度提升3000倍以上,尤其适用于涉及长蛋白的研究场景。初步结果表明,FoldMason-ProstT5在HOMSTRAD困难子集上的表现优于Caretta-shape,但仍略逊于FoldMason快速模式。当前基于LDDT的比对优化过程依赖一种简单的随机双向划分策略,但已能够稳定提升比对质量,未来将探索更加系统化的优化方法。此外,FoldMason正在与Foldseek网页服务器进行深度整合,使用户能够直接从Foldseek搜索结果生成多重结构比对,并通过Foldseek-Multimer扩展到蛋白质复合物层面,从而构建一个用于探索生命之树中蛋白质结构及其进化关系的综合工具。总体而言,FoldMason在比对精度上达到或超过当前最先进的结构比对方法,同时在速度上实现了数量级提升,能够扩展至大规模数据集,并提供友好的可视化和网页服务,使其成为后AlphaFold时代中一项极具价值的蛋白质结构分析工具。
7 FoldMason 使用教程
FoldMason是一款用于从大规模蛋白质结构集合中构建高精度多重结构比对的软件工具,面向后AlphaFold时代的大规模结构分析需求,兼顾准确性、速度和可扩展性。
7.1 软件简介
FoldMason能够直接以蛋白质三维结构为输入,生成多重结构比对结果,并同时输出氨基酸序列和3Di结构字母表两种形式的比对。其核心优势在于可以高效处理成百上千甚至数万规模的蛋白质结构,同时提供基于LDDT的结构一致性评分以及交互式可视化结果,便于结果解读和后续分析。
7.2 Webserver使用方式
如果不希望本地部署,可以直接使用FoldMason官方网页服务:
https://search.foldseek.com/foldmason
在网页端上传PDB或mmCIF结构文件即可快速获得多重结构比对结果及可视化报告,适合快速测试和小规模分析。
7.3 安装方式
FoldMason提供多种平台的预编译版本,可根据硬件环境选择合适的安装方式。
Linux AVX2版本
wget https://mmseqs.com/foldmason/foldmason-linux-avx2.tar.gz
tar xvzf foldmason-linux-avx2.tar.gz
export PATH=$(pwd)/foldmason/bin/:$PATH
Linux SSE2版本
wget https://mmseqs.com/foldmason/foldmason-linux-sse2.tar.gz
tar xvzf foldmason-linux-sse2.tar.gz
export PATH=$(pwd)/foldmason/bin/:$PATH
Linux ARM64版本
wget https://mmseqs.com/foldmason/foldmason-linux-arm64.tar.gz
tar xvzf foldmason-linux-arm64.tar.gz
export PATH=$(pwd)/foldmason/bin/:$PATH
macOS通用版本
wget https://mmseqs.com/foldmason/foldmason-osx-universal.tar.gz
tar xvzf foldmason-osx-universal.tar.gz
export PATH=$(pwd)/foldmason/bin/:$PATH
Conda安装
conda install -c conda-forge -c bioconda foldmason
更多预编译版本可在 https://mmseqs.com/foldmason 获取。
7.4 快速上手
1. 多重结构比对
最常用的方式是通过easy-msa模块,对多个PDB或mmCIF结构文件进行多重比对。
foldmason easy-msa <PDB/mmCIF文件> result.fasta tmpFolder --report-mode 1
该命令会生成FASTA格式的比对结果,并同时输出一个包含LDDT评分和结构映射的交互式HTML报告。
示例命令:
foldmason easy-msa ./lib/foldseek/example/d* example.fasta tmpFolder --report-mode 1
2. 输出结果说明
FASTA格式比对
FoldMason默认输出两种FASTA文件:
- _aa.fa:基于氨基酸字母表的多重比对
- _3di.fa:基于3Di结构字母表的多重比对
交互式HTML报告
当使用--report-mode 1时,会生成一个HTML文件,用于交互式查看多重结构比对、LDDT评分以及结构对应关系。
foldmason easy-msa <PDB/mmCIF文件> result tmpFolder --report-mode 1
内部对应流程为:
foldmason msa2lddtreport myDb result.fa result.html
如果需要生成可在网页端加载的JSON文件,可以使用:
foldmason msa2lddtjson myDb result.fa result.json
3. 常用参数说明
| 参数 | 类型 | 说明 |
|---|---|---|
| --gap-open | 比对 | 缺口开启罚分,默认10 |
| --gap-extend | 比对 | 缺口延伸罚分,默认1 |
| --refine-iters | 比对 | 初始比对后进行的迭代优化次数,默认0 |
| --output-mode | 比对 | 0表示氨基酸,1表示3Di字母表 |
| --pair-threshold | 评分 | LDDT计算时允许的最大缺口比例 |
7.5 自定义数据库与索引
当需要多次重复比对同一批结构时,建议先构建结构数据库,以提升效率。
foldmason createdb example/ structureDB
之后可直接基于该数据库进行多重结构比对。
7.6 核心模块简介
FoldMason主要模块包括:
- easy-msa:从结构文件直接完成多重结构比对的完整流程
- structuremsa:基于结构数据库进行多重结构比对
- msa2lddt:计算多重结构比对的平均LDDT评分
- refinemsa:基于LDDT的迭代式比对优化
7.7 使用示例
基础多重结构比对流程
最简用法如下:
foldmason easy-msa <PDB/mmCIF文件> result tmpFolder
默认会生成:
- 氨基酸和3Di格式的FASTA比对文件
- Newick格式的引导树文件
等价的底层流程为:
foldmason createdb <PDB/mmCIF文件> myDb
foldmason structuremsa myDb result
大规模数据集比对
对于结构数量较大的数据集,可启用预聚类功能以提升效率:
foldmason easy-msa <PDB/mmCIF文件> result tmpFolder --precluster
对已有MSA计算LDDT评分
如果已经有其他工具生成的多重比对,只要结构存在于数据库中,也可以直接计算其LDDT评分:
foldmason msa2lddt myDb result.fa
foldmason msa2lddtreport myDb result.fa result.html
多重结构比对优化
FoldMason支持基于LDDT的迭代优化,用于进一步提升比对质量:
foldmason refinemsa myDb result.fasta refined.fasta --refine-iters 1000
也可以在easy-msa中直接通过--refine-iters参数启用该功能。
7.8 小结
FoldMason提供了一套完整而高效的多重蛋白质结构比对解决方案,既适用于小规模探索性分析,也能够扩展到大规模结构数据库。结合LDDT评分、3Di结构表示以及交互式可视化功能,使其成为当前结构生物信息学研究中的一项实用工具。