Cell Chem. Biol. 2025 | MSanalyst: 通过高度定制的分子网络平台实现具有双重解毒作用的芳香族糖苷的靶向发现
活性小分子的挖掘是药物发现中的关键环节,但由于这些分子通常存在于复杂的生物提取物中,其发现、优先排序及功能表征仍然十分困难。基于质谱的非靶向代谢组学以及MS2谱图相似性分析,是建立基于谱图推断的结构关系并发现未知代谢物的重要策略。然而,现有分析平台大多依赖单一算法对MS2数据进行比较,这往往会导致具有价值的代谢物被遗漏注释。
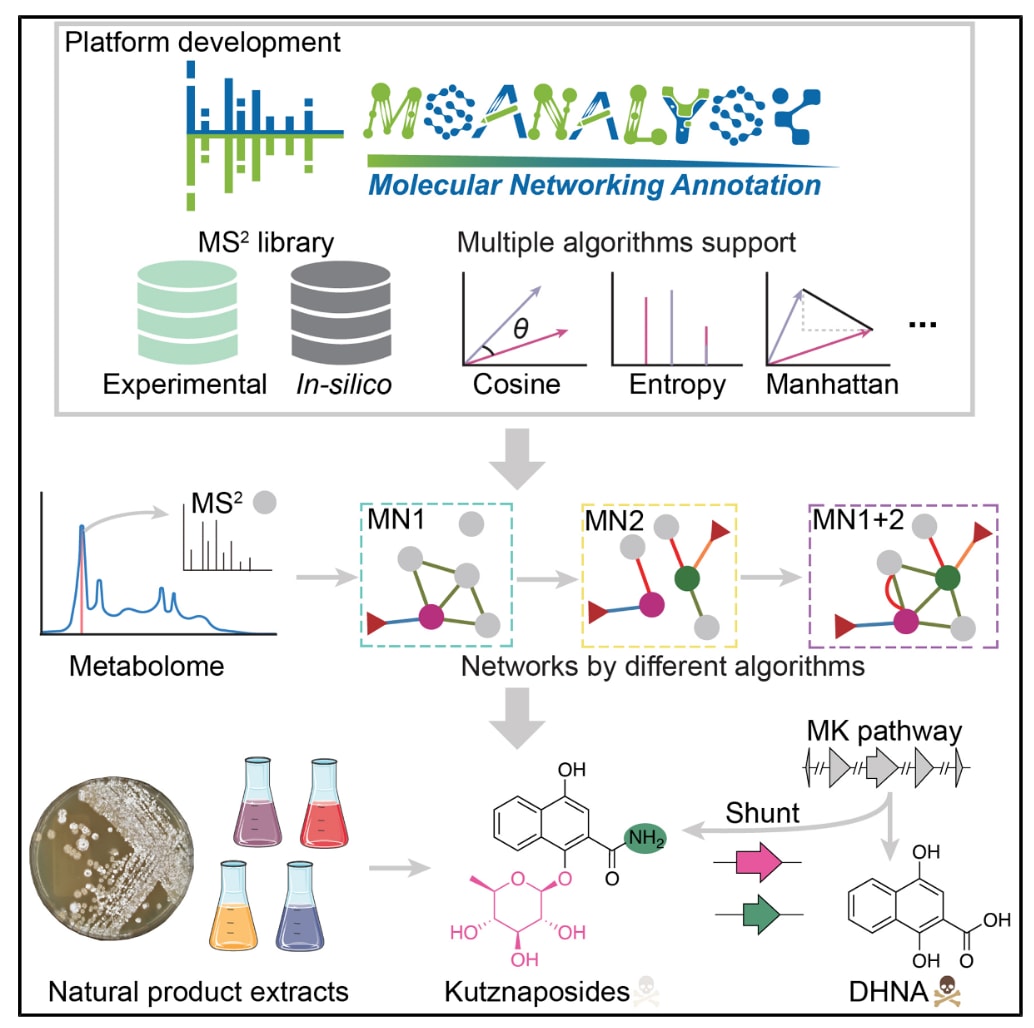
MSanalyst通过整合多种质谱相似性算法,有效弥补了这一局限,从而实现更全面的代谢物注释。将该平台应用于已被深入研究的菌株Kutzneria viridogrisea后,成功发现了一类此前未被描述的芳香族糖苷,并将其命名为kutznaposides。进一步结合多组学分析与体外实验,揭示这些分子来源于一条此前尚未被表征的甲萘醌旁路代谢途径。该途径作为一种关键的防御机制,使宿主能够清除活性氧并避免自身毒性。
通过展示算法整合的优势,MSanalyst推动了对隐藏代谢物、具有重要生物学功能的代谢通路以及潜在生物医学应用的系统性识别。

获取详情及资源:
0 摘要
复杂代谢组中蕴含的天然产物是重要的药物先导来源。基于余弦相似度的MS2比较的非靶向代谢组学方法,已被广泛用于活性分子的发现。为提升注释的准确性和分辨率,研究者开发了多种算法以补充传统的余弦相似度比较方法。
在此背景下,提出了MSanalyst这一易于使用的平台,其整合了46种不同的质谱相似性算法。通过在微生物代谢物数据集以及超过三百万对MS2谱图上的基准测试,结果表明,这些互补算法能够显著增强代谢物之间谱图关联的检测能力。将MSanalyst应用于Kutzneria viridogrisea DSM 43850后,发现了一类芳香族糖苷,即kutznaposides。
进一步的生物学实验与多组学分析表明,kutznaposides C–F来源于一条此前未被识别的甲萘醌旁路代谢途径,该途径能够帮助宿主缓解氧化应激并避免自身毒性。整体来看,这些结果凸显了MSanalyst在挖掘隐藏代谢物、解析代谢通路及其生物学功能方面的潜力。
1 引言
天然产物在多种生物学情境中发挥着重要作用,不仅可作为潜在药物候选物和疾病生物标志物,还参与微生物之间及其与宿主之间的信号传递,并作为初级代谢物的储存形式或非活性副产物。然而,在复杂基质中获取天然产物往往十分困难,其发现过程通常依赖耗时费力的分离纯化步骤。因此,发展先进的去重复鉴定或注释策略,对于靶向发现未被描述的次级代谢物及其相关代谢通路尤为关键,这不仅有助于避免已知化合物的重复发现,也能减少不必要的实验投入。
液相色谱-高分辨率质谱联用技术在非靶向代谢组学中被广泛应用,能够从少量样品中检测微量代谢物,并产生数以万计具有信息量的串联质谱MS2谱图。全球天然产物社交分子网络平台GNPS已成为质谱数据分析与知识共享的重要生态系统,提供了包括MassQL、MASST以及PanREDU在内的多种工具,支持复杂分子数据的探索与解析。其中,分子网络方法通过比较MS2谱图来构建结构相关分子的网络,已成为极具影响力的分析策略。基于特征的分子网络进一步结合液相色谱-串联质谱数据中的特征检测与提取过程,从而提升分析精度并拓展应用范围。随着GNPS 2.0的发布,其数据库已收录超过20亿条实验MS2谱图,为高精度谱图比对提供了丰富资源。
与此同时,多种基于计算的方法被开发用于代谢物注释,例如CSI-FingerID和CFM-ID等工具,以及扩展MS2搜索空间的ISDB数据库,这些方法为天然产物的去重复鉴定及已知化合物结构类似物的注释提供了有效途径。在GNPS体系中,分子网络通过MS2谱图比对,将具有相似结构基元的分子归类于同一簇,通常依赖改进的余弦相似度评分来衡量谱图相似性。近年来,越来越多新的MS2比较与谱库搜索算法被提出,包括SIMILE、t-SNE、spec2vec、entropy以及neutral loss等。这些方法在计算效率、受试者工作特征表现以及谱图关联能力等方面,均展现出优于传统余弦评分的性能。因此,整合多种质谱相似性算法,对于充分发挥分子网络在非靶向代谢组学中的潜力具有重要意义。
在这一背景下,提出了MSanalyst这一计算平台,用于实现多种评分指标在分子网络分析中的无缝切换与整合。该平台提供独立版本与网页版,分别可通过GitHub仓库和在线网站访问。在微生物天然产物数据集以及由2571个小分子构成的超过三百万对谱图上进行基准测试后发现,共有46种算法中有29种能够为传统改进余弦评分提供互补信息。MSanalyst整合了这些经过验证的相似性指标,并构建了涵盖实验谱图与预测谱图的综合谱库。
通过对模式放线菌Kutzneria viridogrisea DSM 43850的代谢组样本进行分析,该平台成功实现了目标天然产物的识别。进一步的生物活性与多组学研究表明,该菌株在甲萘醌代谢通路中演化出一条旁路通量,用于应对氧化应激,从而实现自我解毒。
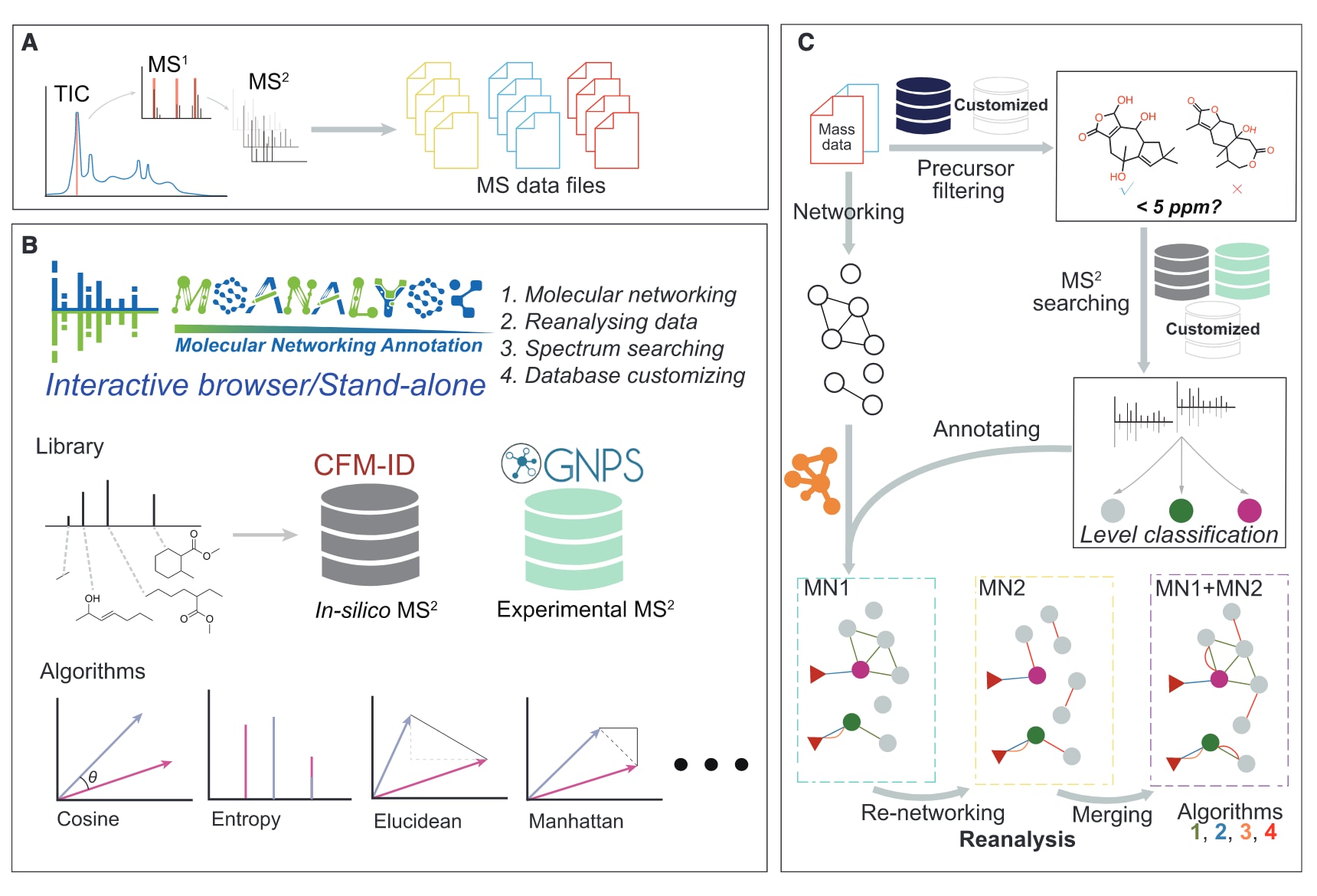
图1|MSanalyst工作流程概述 (A) 获取未知代谢组特征并进行预处理,作为MSanalyst的输入数据。(B) 平台整合了全面的质谱谱库、多种谱图相似性算法以及附加功能模块。(C) 在MSanalyst流程中对特征进行整合,从而提升注释能力并扩展谱图之间的关联性。
2 结果
2.1 MSanalyst整合了全面的质谱谱库与多种谱图相似性算法
在MSanalyst的工作流程中,原始数据首先被预处理为分子特征列表,这些特征构成后续分析的基础。由于质谱容差、噪声水平等关键参数在不同仪器与方法之间存在差异,建议直接使用预处理后的数据作为输入。同时,MSanalyst也集成了UmetaFlow与matchms,可对常见的质谱数据格式(如mzML、mzXML和mgf)进行预处理(图1A)。
分子网络是MSanalyst的核心模块,用于实现分子特征的注释、聚类与可视化,从而支持目标分子的优先筛选。在此基础上,通过扩展谱库并引入多种谱图相似性算法,对该功能进行了增强(图1B)。针对实验MS2谱图增长相对缓慢的问题,利用CFM-ID为467126个非冗余结构生成了计算模拟谱图,这些结构主要来源于CMNPD、NPAtlas以及COCONUT数据库。将这些模拟谱图与GNPS中的实验谱图整合后,MSanalyst能够更全面地覆盖天然产物的化学空间。
平台共整合了46种谱图相似性算法以优化MS2比较,其中许多算法的性能优于传统的余弦相似度方法。在主分子网络流程中(图1C),待分析特征在设定的质量容差范围内与谱库进行MS2比对。未达到设定相似性阈值的特征被标记为“未匹配”,而匹配成功的特征则根据参考谱来源被分类为实验谱或模拟谱。由于模拟谱图与实验谱图之间往往存在较大差异,其引入虽然提高了注释效率,但也可能带来假阳性。因此,仅当模拟谱注释与同一簇中共现的实验注释之间满足化学Dice相似度≥0.75时,才予以保留。最终,所有注释结果被整合进分子网络中。
在谱库搜索及后续自聚类过程中支持多种相似性算法,因为不同算法在谱图关联上既存在重叠也具有互补性,有助于增强网络构建的可靠性(图2)。附加注释会被链接至对应的特征,从而清晰展示不同算法在网络中的贡献。为提升可用性,MSanalyst同时提供网页版与独立版本,并配套教学视频与文档。
除核心流程外,平台还包含多个功能模块以改善用户体验。每项任务均分配唯一标识符以便数据管理与整合。“reanalysis”模块支持通过调整阈值快速优化结果并补充单一算法遗漏的谱图连接;“spectrum searching”模块可用于快速检索目标代谢物;“database customizing”模块则允许构建私有谱库以满足特定研究需求。这些集成功能使MSanalyst成为一个高效且灵活的非靶向代谢组学分析平台。
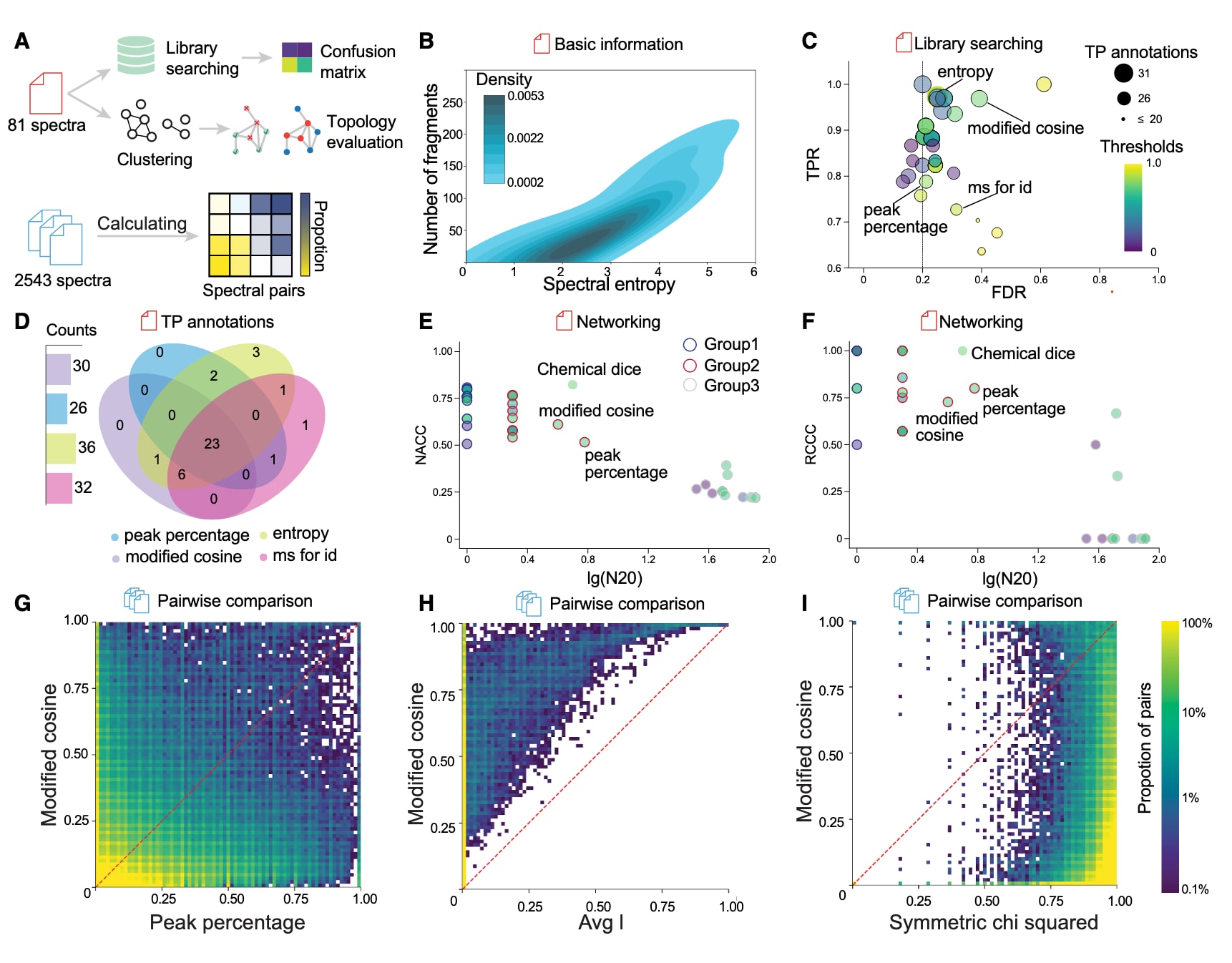
图2|MSanalyst中谱图相似性算法的比较分析 (A) 基于微生物天然产物实验数据集(n=81)对谱库搜索与聚类进行评估,同时利用整理后的数据集(n=2543)计算两两谱图相似性分布。(B) 在微生物天然产物数据集中,谱图熵与峰数量呈现显著差异。(C) 不同算法在采用最优阈值并筛选最高评分匹配后,其谱库搜索结果具有互补性,(D) 从而提高真阳性注释数量。(E) 不同谱图算法在最优阈值下构建网络的准确性,以及(F) 基于N20指标的正确分类簇比例;以化学Dice相似度构建的网络作为理想参考。(G) 在2543个谱图数据集中,peak percentage在37%的谱图对中得分高于改进余弦方法。(H) avg_l可视为改进余弦方法的子集,(I) symmetric_chi_squared在反映结构相似性方面表现较差。
2.2 多种谱图算法揭示额外的基于谱图推断的结构关联
改进余弦相似度仍是推断结构相似性中最常用的谱图比较算法。然而,已有研究表明,部分新算法在准确性与计算效率方面表现更优,但不同算法之间产生的谱图关联差异尚未得到系统评估。为此,采用81个微生物天然产物谱图对46种算法进行基准测试,并结合由2543个谱图构成的三百万对谱图进行比较分析(图2A)。
在初始谱库搜索中,采用常见阈值≥0.7时,各算法获得的真阳性注释数量差异显著(1–34不等),说明统一阈值并不适用。不同谱图的碎片峰数量差异也较大(4至275不等),固定匹配峰数阈值可能导致部分真阳性被忽略(图2B)。为优化性能,引入Youden指数以确定能够最大化真阳性率与假阳性率差值的最优阈值。结果表明,不同算法的最佳阈值确实存在差异,但均可将假发现率控制在约20%(图2C)。
在所有算法中,entropy表现最佳,获得36个真阳性注释,同时有39种算法能够补充改进余弦方法未检测到的真阳性结果。entropy、peak percentage以及ms_for_id等算法优于传统方法,这主要得益于其对强度归一化处理或仅基于匹配碎片进行比较的策略(图S5和S6)。混淆矩阵分析显示,多数算法在较高阈值下真阳性才显著下降,而假发现率则随阈值提高逐渐降低;但也有部分算法在各阈值下始终保持较高的假发现率。
值得注意的是,在各自最优阈值条件下,entropy与ms_for_id的组合能够覆盖所有单一算法检测到的真阳性结果(图2D)。因此,Youden指数为不同算法的阈值优化提供了有效依据,并可在此基础上进一步微调,以平衡真阳性与假阳性。
进一步通过网络分析评估各算法在识别结构类似物方面的表现。基于N20、分类簇网络准确性以及正确分类簇比例三个指标,将算法划分为三类(图2E和2F)。第一类算法生成少量但高准确度的小规模簇;第二类算法(包括传统余弦方法)生成数量更多且规模更大的簇,其中改进余弦与peak percentage最接近理想情况;第三类算法则产生包含无关结构的大型簇,表现出较低的结构关联能力。
在扩展至三百万对谱图的分析中,许多算法在相当比例的比较中优于改进余弦方法,其中peak percentage可覆盖高达37%的谱图对(图2G等)。部分算法仅为改进余弦的子集,而另一些则几乎无法有效反映结构相似性。综合来看,这些分析筛选出29种具有互补性的有效算法,尤其是peak percentage与entropy,在增强谱图关联构建方面表现突出。
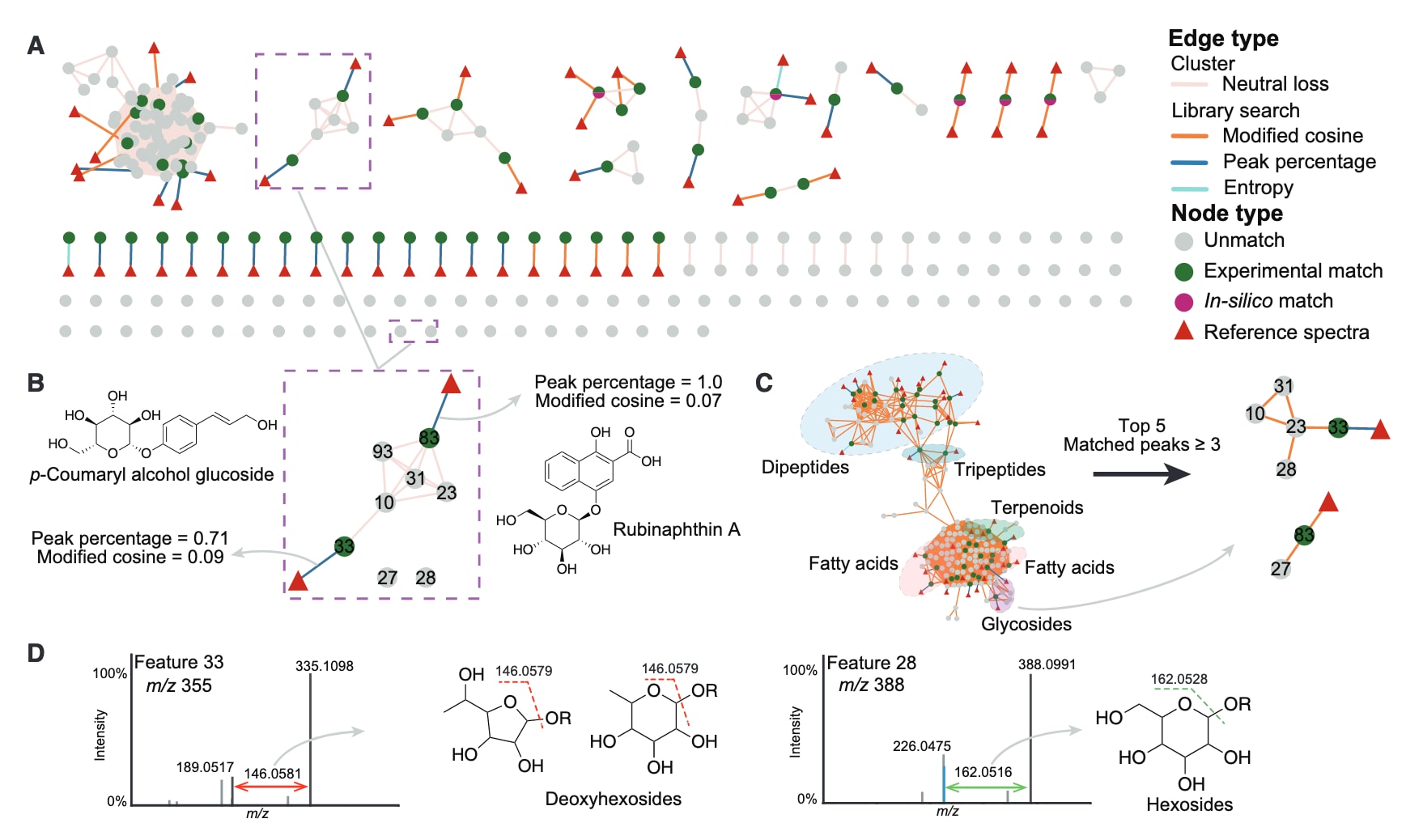
图3|多算法整合提升谱图关联能力 (A) 在MSanalyst中构建分子网络时,谱库搜索采用改进余弦、peak percentage和entropy算法,聚类步骤采用neutral loss算法,均使用各自最优阈值。(B) peak percentage识别出额外的糖苷类注释,这些结果在单一算法中会被遗漏;图中节点标注为特征ID。(C) 改进余弦方法生成较为致密的分子网络,并通过匹配峰和topK参数限制簇规模,有助于区分不同分子家族的边界。(D) 人工分析表明,146 Da的中性损失可能来源于脱氧己糖苷,而162 Da的中性损失则可能来源于己糖苷。
2.3 MSanalyst促进具有抗菌与抗氧化活性的kutznaposides A–F的发现
MSanalyst通过整合多种算法,实现对未知代谢组的全面解析,从而捕获单一方法可能遗漏的分子特征,同时为深入理解生物合成逻辑与生物学功能提供了新的可能。为验证该平台的能力,研究选择了富含生物合成基因簇且代谢组尚未被充分解析的微生物作为研究对象。在此前针对具有潜在强抗菌活性微生物的筛选中,Kutzneria属被发现拥有丰富的生物合成基因簇,能够编码合成复杂天然产物的酶体系。然而,尽管其基因簇丰富,已报道的天然产物却相对较少,提示大量基因簇在实验室条件下处于沉默状态,或因分析手段有限而未被发现。因此,选取商业可获得菌株中基因簇最丰富的Kutzneria viridogrisea DSM 43850进行深入研究。
为最大程度激活其生物合成潜力,该菌株在七种不同培养基条件下培养,并结合LC-MS/MS分析以全面挖掘其化学多样性。对粗提物质谱数据进行MSconvert和MZmine处理,并去除背景后,共鉴定出214个分子特征,随后利用MSanalyst进行系统分析。在谱库搜索中,选用与改进余弦方法具有互补性的算法。结果显示,改进余弦、peak percentage与entropy三种算法联合共识别出57个实验匹配和1个模拟匹配,覆盖了其他算法检测到的所有注释结果(图3A)。经人工筛查,排除了氟轻松缩酮、9-DHAB III等合成药物,以及线性或环状二肽、咪唑嘧啶和脂类等常见细菌初级代谢物(图S15)。
在剩余候选中,两个来源于植物的糖苷尤为突出:特征83被注释为rubinaphthin A,特征33被注释为对香豆醇4-O-葡萄糖苷(图3B)。这两类化合物在peak percentage评分中表现较高(>0.7),但在改进余弦评分中却极低(<0.1),显示仅依赖余弦算法可能会遗漏此类分子。酚类糖苷在细菌中较为罕见,可能具有重要的生物学与药理意义。因此,基于谱图相似性网络对这些特征进行扩展,以寻找更多类似物。
在各算法的最优阈值下,这两个糖苷特征在多数网络中表现为孤立节点,仅在neutral loss与改进余弦构建的网络中形成关联(图S17)。改进余弦网络能够连接这些类似物,但由于允许前体离子质量偏移匹配,网络结构较为拥挤,可通过topK与匹配峰数等参数优化分子家族边界(图3C)。相比之下,基于中性损失的网络能够形成结构清晰的糖苷簇(图3A)。综合分析共识别出7个潜在糖苷特征,其中28号特征含有己糖基(中性损失162.0528 Da),其余特征则含有脱氧己糖或脱氧硫代己糖基(中性损失146.0579 Da,图3D)。此外,由糖基丢失产生的5种碎片离子提示可能存在5种不同的苷元结构(图S18)。
将培养基信息整合至网络后发现,这些酚类糖苷仅在Gym Streptomyces Medium培养条件下检测到(图S19)。为验证MS2预测结构并评估其生物活性,在该培养基中进行放大发酵,最终分离得到6个此前未被报道的芳香族糖苷,命名为kutznaposides A–F(图4A)。通过系统的一维和二维核磁共振及质谱分析,明确解析了其平面结构与绝对构型(图4B及图S20–S26)。COSY、HSQC与HMBC谱进一步证明了各化合物中糖基的存在(图4C)。水解实验及异头质子J值分析表明,kutznaposides A、B、D和E含有α-L-鼠李糖,而C与F分别含有β-脱氧-D-葡萄糖与β-D-葡萄糖(图4D)。结构分析显示,1和2的苷元为取代苯环,而3、4、5和6则共享三取代萘骨架。鉴于二维核磁在某些共轭体系中可能存在误导,通过化学合成与晶体学方法进一步确认3、4和6的骨架为1,4-二羟基萘-2-甲酰胺(DHNC)。此外,在化合物5中检测到甲氧基信号,并通过二维核磁确认其位于C-4位置。这些结果不仅验证了kutznaposides的结构,也为后续活性研究提供了足量DHNC。
考虑到对位二羟基苯结构的氢醌类衍生物具有显著抗菌活性并与氧化还原反应密切相关,进一步评估了kutznaposides C–F及其前体的生物活性。结果显示,DHNA、DHNC以及kutznaposides C、D和F对鲍曼不动杆菌、枯草芽孢杆菌、粪肠球菌及耐甲氧西林金黄色葡萄球菌均表现出不同程度的抑制作用,其最低抑菌浓度为8–64 μg/mL(图4E)。此外,ABTS自由基清除实验表明,DHNA、DHNC及kutznaposide D的抗氧化能力与维生素C相当,IC50值为6.5–9.2 μM(图4E及图S27)。
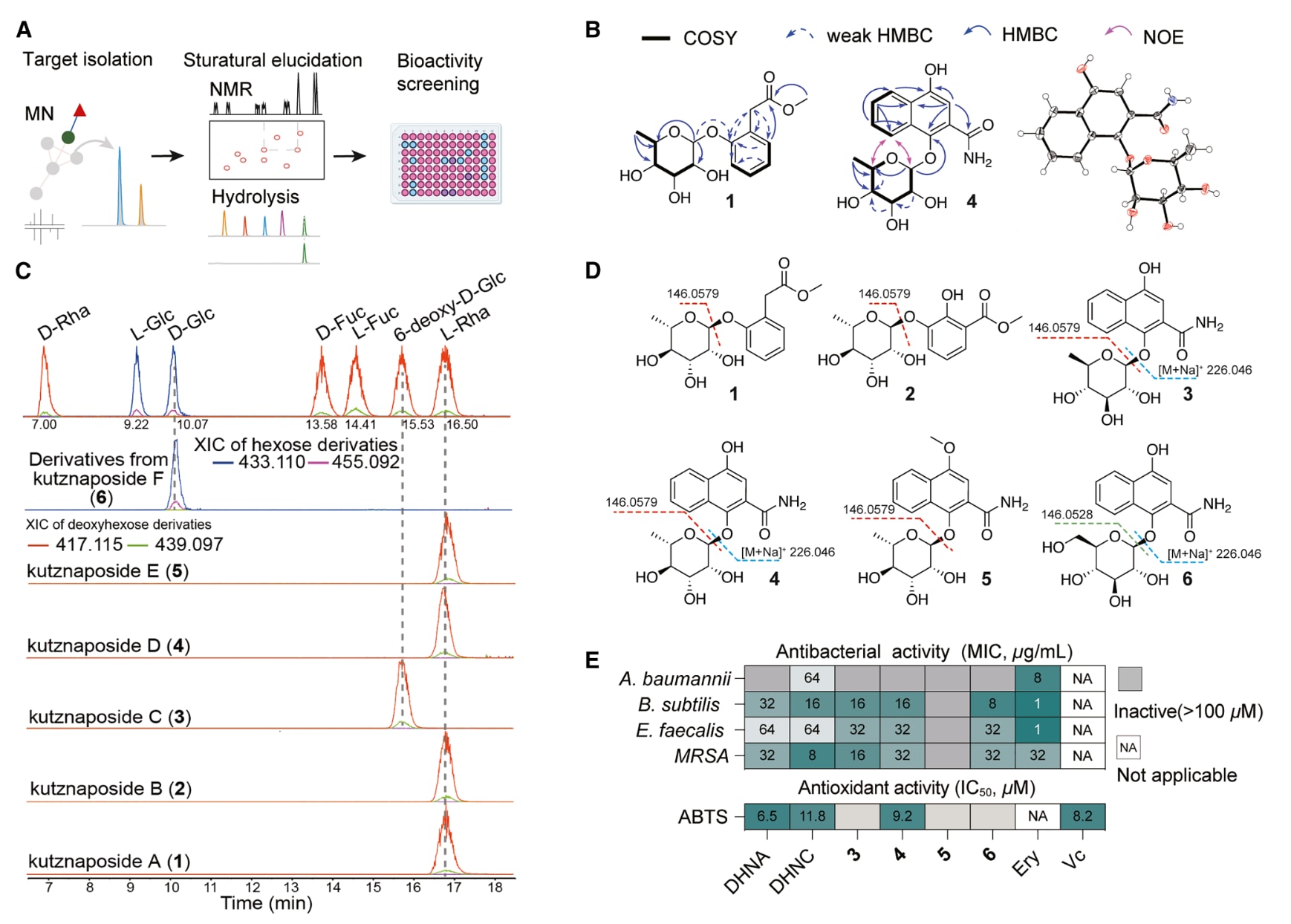
图4|kutznaposides A–F(1–6)的结构解析及其抗菌和抗氧化活性 (A) 代表一类酚类糖苷的分子簇由MSanalyst完成注释,随后进行了靶向分离、结构解析和生物活性筛选。(B) 采用核磁共振与晶体学方法确定代表性苯环和萘环糖苷的骨架结构。(C) 单糖及水解样品衍生物的提取离子色谱图。己糖对应
2.4 kutznaposides C–F来源于K. viridogrisea中的甲萘醌旁路代谢
kutznaposides C–F代表了一类来源于微生物的萘酚糖苷,其生物合成依赖于一条非常规的旁路代谢途径,该途径包含多种不同于经典细菌甲萘醌合成路径的酶促反应。其生物合成以甲萘醌通路中的中间体DHNA为起点,该分子由莽草酸途径经menB至menF、menH及menI等酶催化生成。随后,推测一类II型谷氨酰胺酰胺转移酶参与转氨反应,将氮基引入DHNA的羧基位置。最终,由尿苷二磷酸糖基转移酶或胸苷二磷酸糖基转移酶催化完成酚类分子的糖基化修饰。这些对DHNA的修饰不仅改变了分子的生物活性,也暗示其在翻译调控或解毒等方面可能具有功能。
为解析这一隐匿代谢途径,结合生物信息学与蛋白质组学进行分析。结果显示,在菌株DSM 43850中存在men基因编码蛋白的同源物,统称为knps。以已报道的链霉菌GAT和糖基转移酶为参考序列,在该菌株中筛选潜在候选蛋白,并结合蛋白质组数据筛选出2个GAT候选和16个潜在糖基转移酶。代谢组分析表明,kutznaposides C–F仅在GSM培养基中检测到,而DHNC在R2A培养基中的含量显著低于GSM,提示相关酶在不同培养条件下存在差异表达。
进一步的蛋白质组分析发现,仅有一个GAT(GAT1)在GSM条件下显著上调,同时有9个糖基转移酶表达上升,其中5个仅在GSM条件下表达,另外4个表达量提高2至6倍,表明其可能参与O-糖基化反应。随后对GT1–GT9进行异源表达实验,结果显示GT5和GT6分别可将DHNC转化为kutznaposides D和F。值得注意的是,这些生物合成相关基因分散于基因组不同位置,并未被antiSMASH预测为典型生物合成基因簇。此外,GT5和GT6均无法对DHNA进行糖基化,说明糖基化步骤发生在酰胺转移之后。
综合蛋白质组学与酶学实验结果,提出kutznaposides的生物合成路径:首先由knp编码的酶系生成DHNA相关中间体,随后经GAT与糖基转移酶进一步修饰形成最终产物。这一结果表明,kutznaposides来源于非典型代谢途径,传统基于基因组的挖掘策略难以识别,突显了MSanalyst在直接发现此类代谢物方面的优势。
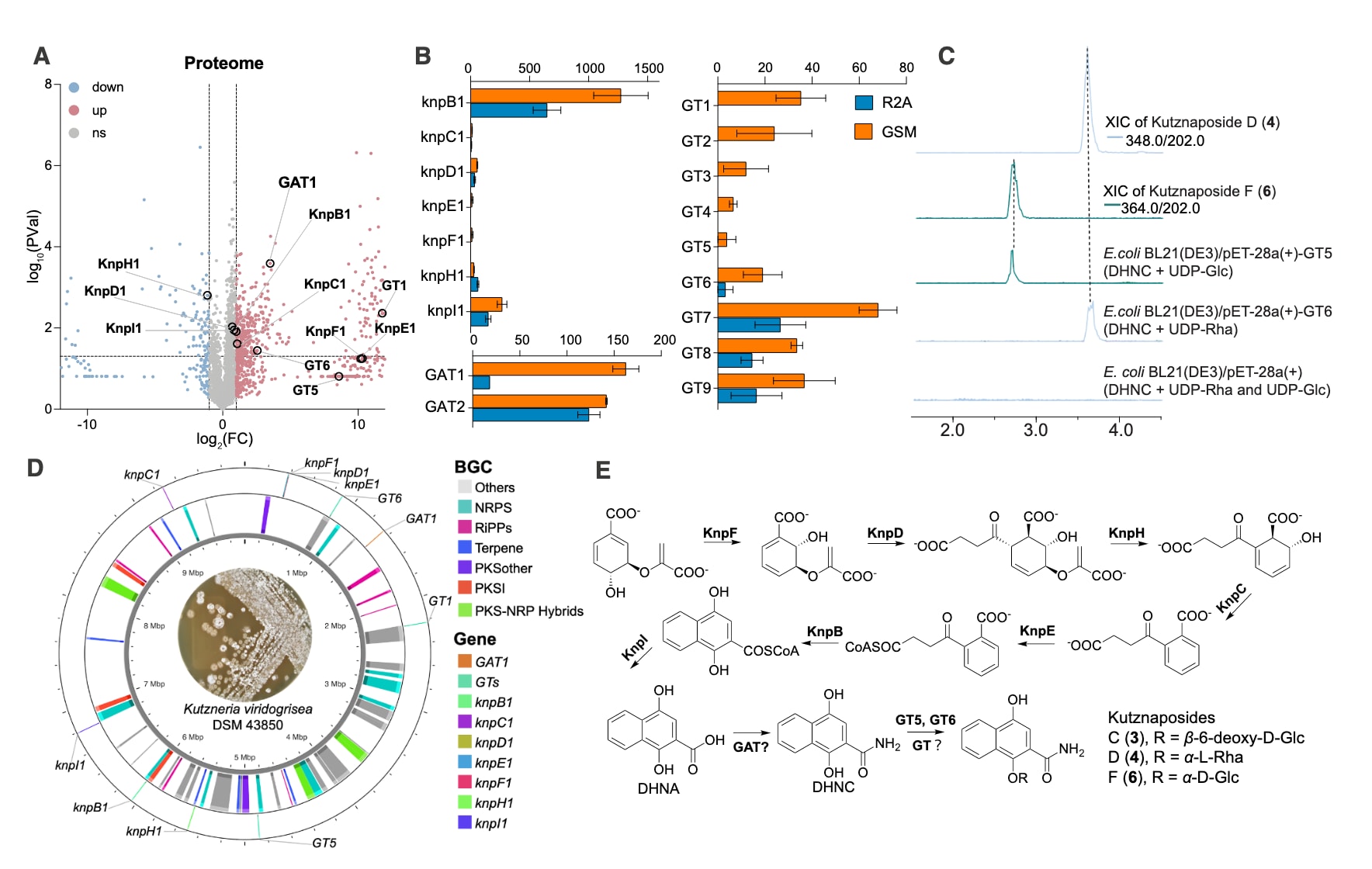
图5|kutznaposides C、D和F生物合成关键酶的蛋白质组学分析 (A) 展示了在GSM与R2A培养基条件下比较得到的火山图,(B) 以及蛋白质组中相关蛋白的表达情况,右侧柱状图表示其绝对表达量(n = 3)。(C) 在大肠杆菌BL21(DE3)中分别表达GT5和GT6,并以DHNC及UDP-Glc或UDP-Rha为底物进行全细胞催化反应后,检测到糖基化产物(4为推测的4-O-葡萄糖苷,6为推测的4-O-鼠李糖苷)。(D) 全基因组组装结果中,内环以不同颜色标示检测到的生物合成基因簇,外环则显示参与(E) kutznaposides C、D和F(3、4和6)生物合成的分散分布相关基因。
2.5 甲萘醌旁路代谢通量作为自我解毒机制
kutznaposides C–F是甲萘醌通路的旁路产物,而甲萘醌本身对微生物生长至关重要。因此,对关键中间体DHNA进行后修饰的GAT和糖基转移酶具有重要意义。已有研究表明DHNA具有潜在的自毒性,而糖基化与去糖基化过程常与微生物自我保护相关。因此可以推测,这些酶在K. viridogrisea中发挥保护作用,使其避免DHNA带来的毒性。
通过自抑制生长实验验证这一假设,结果显示DHNA对菌株的抑制作用最强,其次为DHNC及kutznaposides,说明该两步酶促反应确实构成一种解毒过程,相应基因也可视为自抗性基因。为进一步理解该旁路代谢的进化与激活机制,对蛋白质组数据进行了深入分析。富集分析显示,在GSM培养条件下,多种与解毒相关的GO功能显著富集,如对外源物刺激的细胞响应及外源物代谢过程。同时,KEGG分析表明丙氨酸、天冬氨酸和谷氨酸代谢以及丙酸代谢通路显著上调,且相关基因普遍表达增强,提示其与旁路代谢的激活密切相关。
进一步基于蛋白互作网络分析发现,参与NAD+合成初始步骤的喹啉酸合酶与L-天冬氨酸氧化酶与GAT1存在间接关联,其上调可能导致活性氧积累。在乙醛酸氧化过程中,过氧化氢酶缺失以及(S)-2-羟基酸氧化酶上调同样可能促进活性氧生成。这可能源于GSM条件下更强的有氧代谢状态,例如三羧酸循环相关蛋白普遍上调,且菌体生物量明显高于R2A培养条件。
据此提出一种机制:K. viridogrisea在应对氧化压力时合成DHNA以清除过量活性氧,但DHNA本身对细胞具有毒性。为避免自损伤,该菌进化出旁路代谢,将代谢通量转向生成kutznaposides,从而既维持抗氧化能力,又降低自身毒性。因此,该来源于细菌的甲萘醌旁路代谢通过一系列非常规反应,实现了在氧化环境中的生存与自我解毒的双重策略。
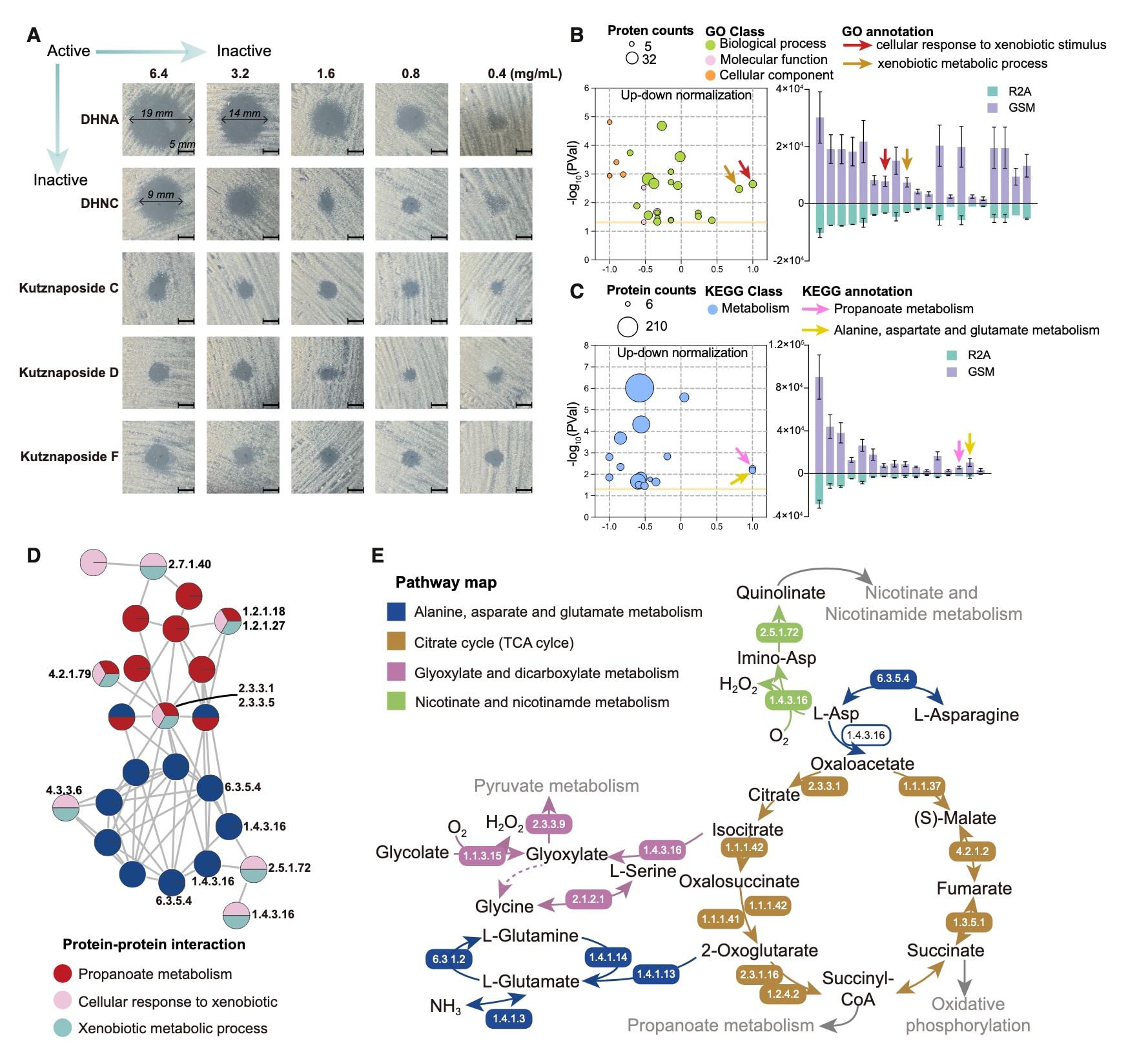
图6|多组学分析揭示甲萘醌旁路代谢作为一种非常规自我解毒策略 (A) 展示了K. viridogrisea在DHNA及其下游代谢物作用下的影响(黑色柱,5 mm),并与图S29中的DMSO对照进行比较。(B) 基于蛋白质组中差异表达蛋白得到的前20个GO功能注释,以及(C) 前17条KEGG代谢通路。气泡图中,纵轴表示
3 讨论
分子网络是基于LC-MS的非靶向代谢组分析中的重要工具,能够实现清晰的分子注释并传播结构信息。然而,其应用受到谱库规模有限以及依赖单一谱图算法的限制。为克服这些问题,研究通过引入计算模拟谱图扩展参考谱库,并整合多种替代算法。尽管模拟谱图的引入提高了注释覆盖率,但假阳性问题依然突出。通过利用同一分子簇中经实验验证的注释对模拟结果进行筛选,可以显著降低假发现率。
在微生物天然产物数据集及标准谱图对上的评估表明,entropy和peak percentage等算法能够有效补充改进余弦方法,从而提升注释深度。多算法组合使得K. viridogrisea中未知代谢组得到了更全面的解析。基于多算法构建的分子网络揭示了更多潜在的谱图关联与分子注释,而这些信息在单一算法下往往难以发现。为提高平台的可用性,MSanalyst提供了网页版和独立版本,并附带详细文档与教学视频,所有代码与数据均公开以确保可重复性。
由于未知代谢物的谱图特征具有高度多样性,固定的相似性阈值与匹配峰数标准并不适用于所有情况。因此,MSanalyst支持通过调整阈值或切换算法进行快速再分析。谱库搜索评估表明,严格阈值虽可降低假发现率,但可能丢失真阳性,尤其是在碎片信息较少的谱图中,因此需要针对不同算法进行精细优化。此外,不同算法在计算方式上的差异,例如是否允许
整体而言,多种互补指标的整合能够显著提升未知代谢组空间的注释能力,并增强现有分析工具的性能。通过应用MSanalyst,从K. viridogrisea中成功发现了一类新的特化糖苷代谢物kutznaposides A–F。尽管中性损失算法在技术上属于改进余弦的子集,但在该研究中其更清晰地界定了糖苷分子家族边界,而单独使用改进余弦则会产生较为拥挤且难以解析的网络结构。kutznaposides是一类此前未被描述的细菌来源芳香族糖苷,其中C–F被鉴定为甲萘醌通路的旁路产物。生物信息学与多组学分析表明,其生物合成相关酶分散于基因组中,并未形成典型的生物合成基因簇,这意味着传统基于基因组挖掘的方法难以识别这类分子。相比之下,MSanalyst优化了分子网络分析流程,为传统自上而下的天然产物发现策略提供了重要补充,显著增强了对具有特殊生物功能的隐匿代谢物的发现能力。
尽管甲萘醌通路中的相关酶已被广泛研究,但其调控机制仍需进一步探索。通过蛋白质组学分析结合体外生物活性实验,揭示了该旁路代谢通量的潜在机制,推测K. viridogrisea通过一系列酶促反应适应氧化环境并避免自身毒性。糖基化作为常见的解毒方式,这些自抗性基因为挖掘其他类似基因簇提供了重要线索。鉴于甲萘醌在环境及人类病原体致病性中的关键作用,这些发现也为潜在药物靶点的识别提供了基础。
总体来看,MSanalyst在高效注释未知代谢物及推动天然产物发现方面展现出显著优势。通过整合并优化现有代谢组学方法,可以实现更深入的数据解析。结合多组学、生物活性研究及其他先进技术,该平台有望进一步推动化学生物学领域的发展。
3.1 研究局限性
当前方法与资源仍存在一定局限。首先,尽管MSanalyst整合了如CFM-ID等先进机器学习模型用于生成虚拟MS/MS谱库,但预测谱图与实验谱图之间仍存在系统性差异,这限制了其在天然产物注释中的准确性。其次,不同谱图相似性算法的注释性能高度依赖于数据集组成,而谱图噪声、低质量碎片以及缺乏标准化基准数据集,均限制了对算法的严格评估。最后,关于K. viridogrisea自我保护机制的提出仍停留在初步阶段,尚缺乏直接的遗传学或生化证据,有待通过基因敲除与体外重构实验进一步验证。