Nat. Chem. 2026 | 氨基酸组成在肽合成过程中驱动聚集现象
今天介绍的是发表在 Nat. Chem. 2026 上的一项关于固相肽合成中聚集机制的研究。该工作聚焦一个长期困扰肽化学的核心难题:生长中的肽链在树脂上容易发生聚集,进而导致偶联效率下降、粗品纯度变差,甚至使目标序列难以顺利获得。不同于以往主要从序列顺序解释聚集的思路,这项研究结合机器学习与合成实验提出,在固相肽合成条件下,真正更关键的因素可能是氨基酸组成而非排列顺序。通过对大量合成数据的建模与验证,研究不仅识别出哪些氨基酸和保护基更容易促进聚集,还进一步发展出可用于预测聚集并指导抗聚集修饰位点选择的策略。该成果为理解肽合成过程中的聚集本质提供了新的视角,也为提高困难肽段的合成成功率提供了更具可操作性的方案。

获取详情及资源:
- 📄 论文: https://doi.org/10.1038/s41557-026-02090-0
- 💻 代码: https://github.com/rxn4chemistry/AI4Aggregation
0 摘要
肽在化学合成过程中发生的聚集问题由来已久,严重限制了合成效率与可靠性。尽管数据驱动方法已经加深了对多种序列相关现象的理解,但针对固相肽合成中所谓“非随机困难偶联”(通常与聚集密切相关)的问题,仍缺乏系统性的解决方案。相关研究基于已有的肽合成数据,并结合额外的实验数据,构建了一个预测模型,用于解析单个氨基酸在触发聚集过程中的作用。
分析首先发现并通过实验验证,相较于传统的序列模式,基于组成的聚集特征具有更强的预测能力。这一发现进一步推动了组成向量表征方法的发展,使得可以从整体组成角度解析不同氨基酸的聚集倾向。在此基础上,通过集成多个训练模型,实现了对肽聚集性质的预测,并可为降低聚集风险的策略提供优化建议。
通过系统刻画各类氨基酸对聚集行为的贡献,该方法为利用既有数据加速肽合成优化提供了可能,同时也为理解与调控肽聚集现象建立了一个稳健的分析框架。
1 引言
肽和蛋白质在生物体系中承担着多种功能,例如作为激素、酶以及信号分子,对维持生理过程至关重要。这种高度的多样性与特异性也使其成为重要的治疗分子,持续推动制药领域的发展。然而,仅依赖人类直觉难以从一级序列系统性理解蛋白质结构,这一难题长期以来被称为“蛋白质折叠问题”。随着大量数据的积累,计算方法逐渐成为预测蛋白质结构的关键工具,并最终推动了AlphaFold和RoseTTAFold等模型的发展,使得从序列精准预测结构成为可能。
尽管这些进展显著提升了对生理条件下蛋白质和肽折叠行为的理解,但在固相肽合成过程中的折叠与聚集问题仍研究不足。这主要源于两类体系条件的显著差异:其一,肽链的C端固定在固相载体上,改变了分子的柔性与极性;其二,大多数氨基酸侧链带有保护基,显著改变其空间与电子性质;其三,合成环境通常为如DMF等非质子溶剂,并伴随聚合物载体,整体极性远低于水相体系。在这种环境中,树脂或连接子结合的肽链容易发生聚集,并诱导形成类似折叠结构,从而降低合成效率,甚至使某些序列难以获得。
聚集通常源于固相上非期望的β折叠结构形成,这会导致肽链截断或缺失,使目标产物难以分离。值得注意的是,一旦发生聚集,即使增加偶联或脱保护步骤,或使用过量氨基酸,也难以实现完全转化。聚集行为受多种因素影响,包括反应温度、载体负载量以及氨基酸序列与保护基类型等。其中,靠近树脂连接点的C端区域尤为关键,已有研究表明聚集多发生在距锚定位点5–15个氨基酸范围内,且β支链氨基酸会加剧这一问题。
尽管已有研究尝试从序列角度或基于流动合成过程中的紫外数据分析聚集现象,但仍缺乏一种能够可靠预测聚集并指导替代合成策略的方法。相关研究基于自动化快速流动肽合成仪的在线紫外-可见光数据中的脱保护峰信息,这些信号与树脂上肽链的聚集状态直接相关。通过分析这些数据,可以解析不同因素,尤其是单个氨基酸对聚集的影响。
进一步通过对肽序列进行打乱分析发现,决定聚集特性的关键因素并非具体序列排列,而是氨基酸的组成比例。实验验证支持这一结论,并表明基于组成信息可以有效预测并规避聚集问题,从而为优化固相肽合成策略提供了新的思路。
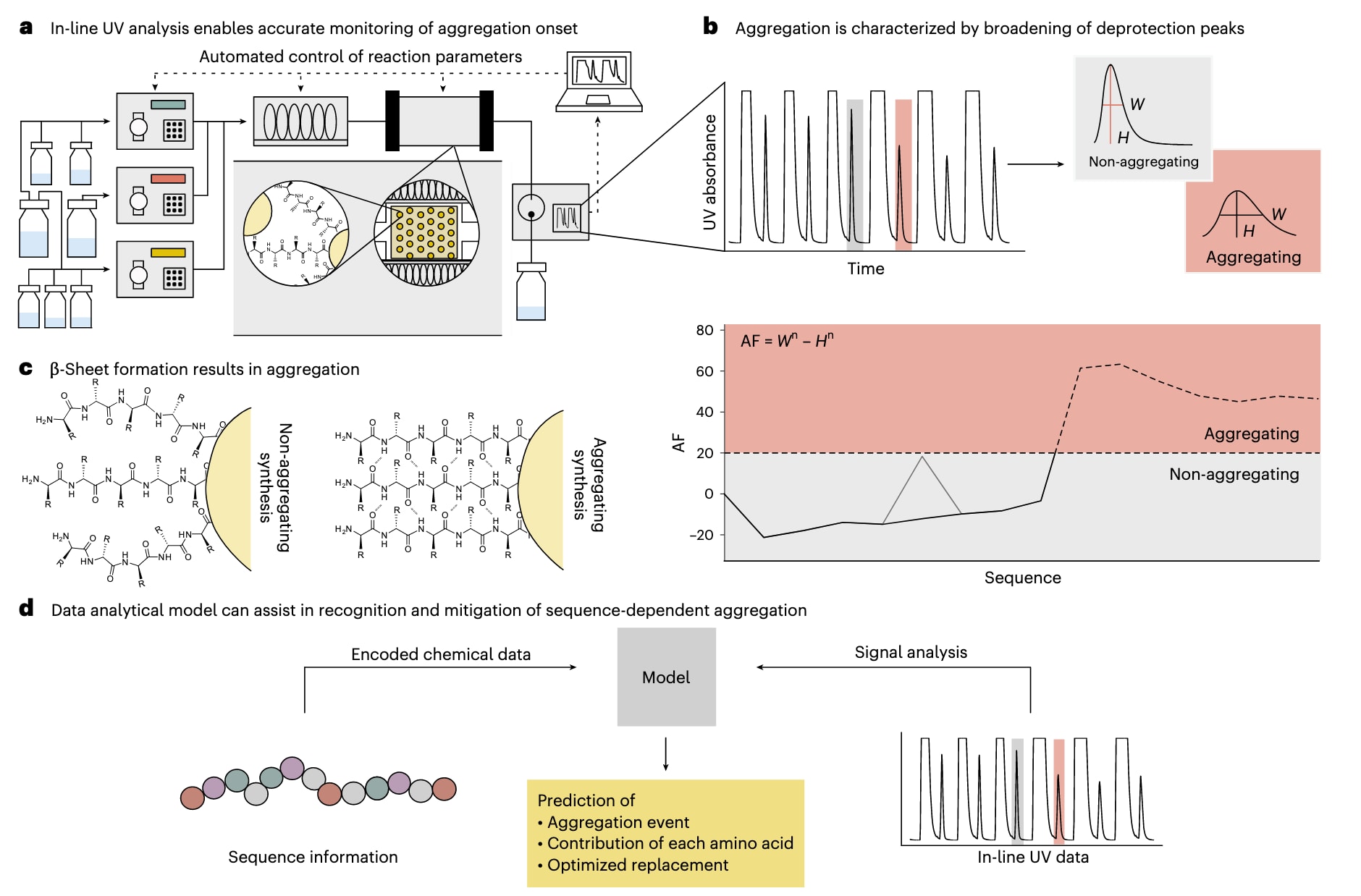
图1|基于在线紫外模块采集的分析数据可支持数据驱动的合成过程分析。 a,自动化快速流动肽合成系统能够对反应动力学进行精确监测,而这些动力学特征与肽序列的聚集行为密切相关。b,在在线紫外信号中,聚集表现为脱保护峰的展宽。聚集程度通过聚集因子AF进行量化,其计算公式为
2 结果与讨论
2.1 固相肽合成中的聚集预测
在预测肽聚集行为之前,需要首先建立区分“发生聚集”与“未发生聚集”的判定标准。相关数据均来源于配备在线紫外-可见检测器的自动化快速流动肽合成平台,该系统在合成过程中实时监测偶联与脱保护峰。脱保护峰源于Fmoc基团的去除,其峰面积反映偶联或脱保护效率,而峰形则直接指示聚集状态。为保证数据可比性,采用聚集因子对信号进行表征,即通过归一化后的峰高与峰宽差值来量化聚集程度,并将脱保护峰相较首次偶联展宽超过20%的情况定义为发生聚集。一旦合成过程中任意一步超过该阈值,则将整个序列标记为聚集型,这一现象在实际中对应粗产物纯度的下降。
在此基础上,引入机器学习方法对肽的聚集特性进行预测,所使用的数据包括公开数据集以及内部实验数据,两者均基于相似的合成平台与条件,因而具有较高一致性。经过筛选与整合后,共获得539条肽序列,其中约一半表现出聚集行为。考虑到聚集通常发生在距树脂锚定点5–15个氨基酸范围内,对长度超过20的序列进行了截断,同时剔除了过短序列及包含非标准氨基酸的数据,以提高建模稳定性。
围绕这一问题构建了多种模型与数据表征方式,并采用五折交叉验证评估性能,将任务定义为二分类问题,即判断序列是否发生聚集。由于数据来源于逐步合成过程,设计了两种预测策略:一种基于完整序列进行整体预测,另一种利用逐步延伸的合成信息进行阶段性预测。在逐步策略中,初始若干步标记为非聚集,一旦检测到聚集事件,其后步骤均视为聚集状态。
在模型选择上,既包括将氨基酸序列视为文本并采用语言模型的方法,如微调蛋白质语言模型ESM以及通用模型BERT,也包括从零训练的语言模型。此外,还将逐步合成过程视为时间序列,采用时间序列分类模型进行建模。同时,传统机器学习方法如随机森林与XGBoost也被用于不同形式的数据表示,包括数值编码、one-hot编码以及基于分子指纹的表示。已有方法同样被纳入比较。
结果显示,无论采用何种模型结构、数据表示或超参数配置,各方法的预测性能均较为接近。这一现象部分源于不同建模目标之间的差异,例如已有方法主要预测紫外信号曲线,而非直接进行聚集分类。由于紫外信号中存在大量稳定区域,这些区域虽然有助于降低回归误差,却对区分聚集与否贡献有限,从而导致分类性能受限。
进一步分析表明,在序列中最关键的信息集中于聚集发生之前的区域,而聚集发生之后的序列对预测贡献较小。通过系统评估聚集点前后不同长度的序列片段,发现只要序列长度足以形成二级结构(通常超过6个氨基酸),模型性能基本保持稳定。这一结果暗示,聚集行为可能不仅由序列本身决定,或者现有模型尚未能够充分捕捉数据中的关键信号。
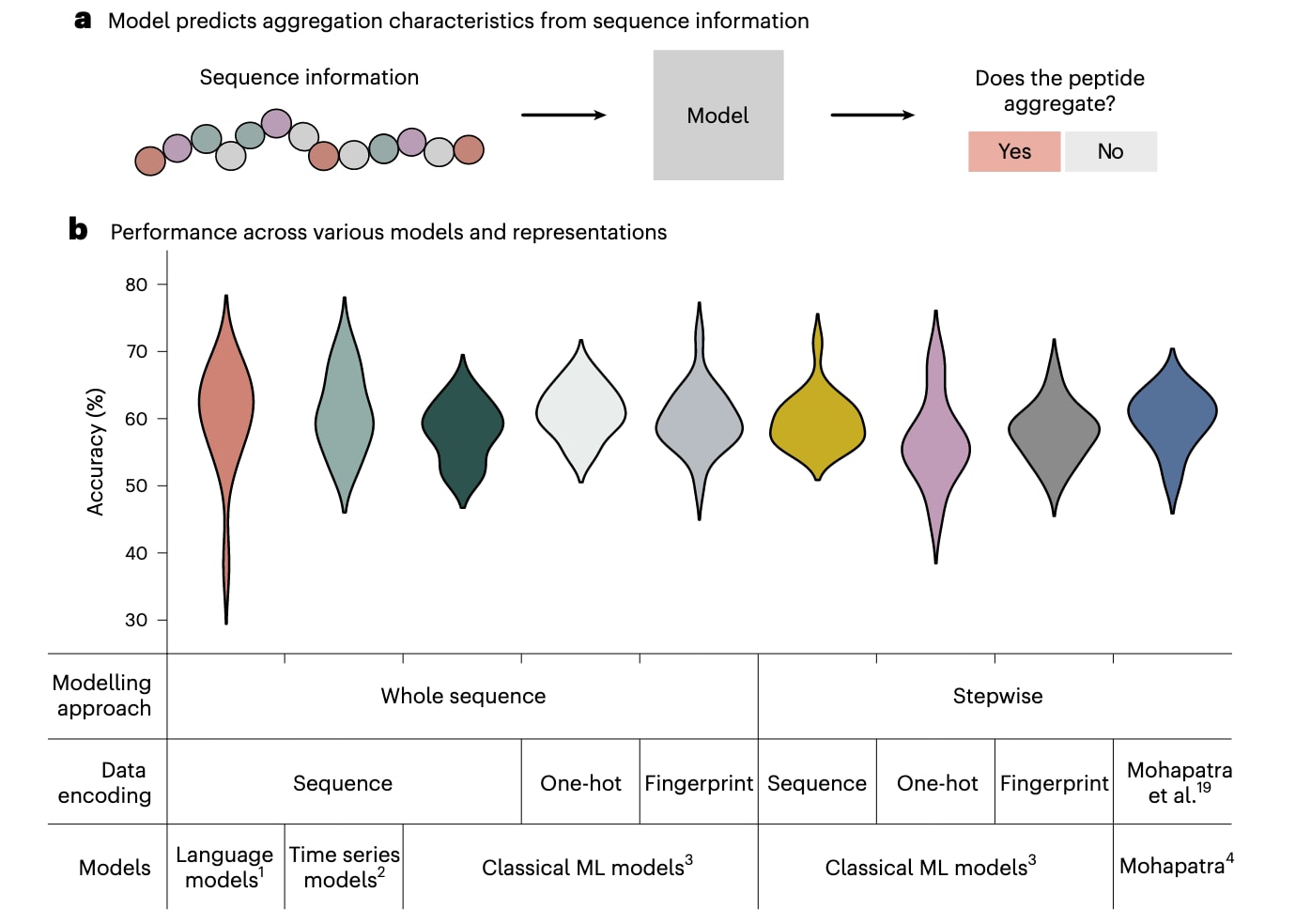
图2|预测准确率与模型类型或数据表征方式基本无关。 a,研究中采用了多种不同类型的模型,从语言模型到传统机器学习模型,用于预测给定肽序列是否发生聚集。b,无论模型架构、化学表示方式,还是输入方式(逐步输入或整体序列输入),各类方法均表现出相似的预测准确率。小提琴图展示了不同建模策略与数据表征下的准确率分布情况,每一种颜色对应某一类模型在五次测试划分中的表现。上标标注了模型类别:(1)语言模型(ESM2.0、BERT);(2)时间序列模型(HIVE-COTE2.0、WEASEL、time forest);(3)传统机器学习模型(XGBoost、随机森林、KNN、高斯过程);(4)Mohapatra等提出的模型变体(完整版本与简化版本)。
2.2 氨基酸组成对聚集的影响
不同模型和不同表示方法得到的结果高度一致,这促使研究进一步反思数据集本身的质量与一致性。作为验证实验,研究将所有模型都训练在经过随机打乱的肽序列数据上。按照传统观点,如果聚集高度依赖序列顺序,那么在打乱数据上模型表现应当明显变差;若没有出现这种差异,则说明模型并未捕捉到真正依赖序列顺序的聚集信号。为此,研究采用全序列表示方式,分别在原始序列数据和随机打乱后的数据上训练XGBoost模型。结果发现,两者在准确率上几乎没有实质性差别,原始序列的准确率为58.0%±3.5%,打乱序列的准确率为57.7%±3.3%。这一现象在所有测试过的表示方式和模型中都保持一致。这样的结果对聚集是高度依赖肽序列的普遍观点提出了挑战。
为了进一步探究这一问题,研究构建了一种更为简化的编码方法,即用一个20维向量表示每条序列中氨基酸的归一化组成。即便采用这种极简表示,模型准确率仍然保持在相近水平,达到59.5%±1.9%,进一步支持了这样一种判断:在影响聚集的因素中,氨基酸组成的重要性可能超过了序列顺序(图3a)。
为了从实验上检验究竟是组成而不是顺序决定聚集行为,研究选取了8条文献中已知性质明确的测试肽,并为每条肽合成了5种随机打乱顺序的变体(图3b)。其中,Barstar[75-90]、hGH[176-191]Y176F(简称hGH)、GLP-1和MYC[123-243]被选为易聚集序列;NBDY[53-68]、GHRH、MYC[421-439]和PCP-4[43-62]则被选为不易聚集序列。这些打乱后的序列通过可重复的随机化流程生成,以避免选择偏差。随后,这些长度在16到28个氨基酸之间的肽在自动快速肽合成过程中接受了聚集行为评估。
实验结果与计算分析结果一致。在20条打乱后的易聚集肽中,有19条依然保留了原有的聚集特征;而在20条打乱后的非聚集肽中,也有14条继续保持非聚集性质(图3)。这表明,只要整体氨基酸组成不变,大多数肽即使改变了氨基酸排列顺序,仍会保留原本的聚集行为。此外,这些打乱序列发生聚集的位置也与原始序列相近(图3c)。这些结果说明,决定肽聚集的关键因素并不只是序列信息本身,氨基酸组成很可能发挥了更加突出的作用。
表1|准确率结果:该表展示了不同数据表示方式与模型组合下获得的预测准确率。 每一种组合均采用五折交叉验证进行训练,并报告了五次划分结果的平均准确率及其标准差。

2.3 单个氨基酸对聚集的贡献
为了理解单个氨基酸如何影响聚集,研究使用Shapley Additive Explanations(SHAP)对各氨基酸的贡献进行了分析。为确保SHAP值提取具有稳健性,研究在50种不同的随机数据划分上训练了XGBoost分类器。肽序列采用组成向量进行表示,并利用SHAP值来解释和量化每种氨基酸对聚集的贡献。SHAP值为正表示该氨基酸会提高模型预测聚集的可能性,SHAP值为负则说明其倾向于降低聚集风险。通过这一方法,氨基酸组成与模型预测结果之间建立了直接联系,从而为理解肽聚集的分子决定因素提供了依据。
分析结果表明,不同氨基酸对聚集的影响呈现出明显差异(图4)。当Ser(t-Bu)、Ile、Val和Thr(t-Bu)在序列中的比例较高时,最容易提高聚集发生的可能性,其中Gln(Trt)和Leu也是重要的促进因素。相反,Phe、Asp(t-Bu)、Tyr(t-Bu)和Arg(Pbf)的存在最倾向于降低聚集,其次是Cys(Trt)、His(Trt)和Pro。其余氨基酸整体上表现得较为中性,既没有明显促进作用,也没有明显抑制作用。虽然这一分析显示,肽的组成是驱动聚集的主要因素,但聚集过程仍然会受到其他因素影响。基于这一点,研究进一步考察了二肽基序对聚集的作用,结果发现,Gly–Ser和Leu–Leu对聚集的促进作用最为明显。
总体来看,促进聚集的氨基酸通常带有脂肪族、非极性侧链,这类结构似乎更容易促进肽链之间的分子间相互作用与紧密堆积。相较之下,抑制聚集的氨基酸往往具有芳香族或极性侧基,这些结构可能会增加肽链之间的间距,并破坏容易导致聚集的构象。
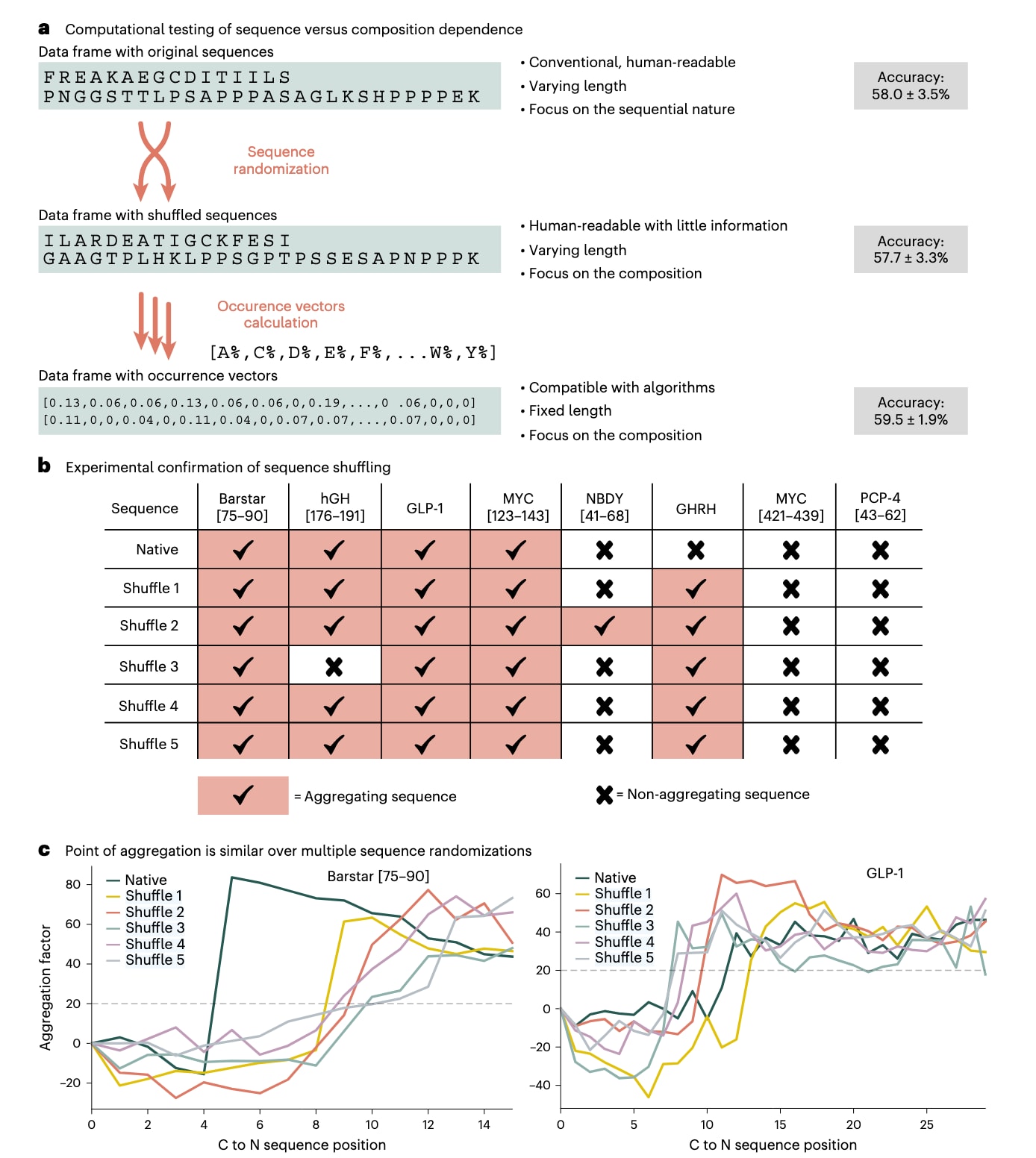
图3|通过计算与实验方法系统研究序列打乱对聚集行为的影响。 a,在随机打乱序列或仅使用氨基酸组成向量进行训练时,模型的预测准确率并未低于基于原始序列训练的结果。b,为验证计算分析结果,选取4条易聚集和4条不聚集的肽序列,并为每条序列构建5个可重复的随机打乱版本进行合成实验。c,对于易聚集的测试肽,其聚集发生的位置在不同打乱序列中保持一致。Barstar的原始UV–vis数据来源于文献20,GLP-1的数据来源于文献13。
2.4 基于模型的合成优化策略
训练后的模型为改进固相肽合成提供了可操作的条件建议。肽合成优化往往是一个相当繁琐的过程,因为聚集难以预测,常规流程通常依赖反复合成,并通过试错方式使用已知的抗聚集工具。训练得到的XGBoost分类器不仅能够预测给定肽序列的聚集倾向,还能够进一步提示,通过有策略地进行结构修饰,应当如何缓解聚集。通过分析特定氨基酸对聚集的贡献,模型可以预测如何更有效地使用不同的抗聚集手段,例如主链和侧链上的不同保护基。
所开发的算法流程如下(图5a)。首先,在不同的数据划分上训练100个XGBoost分类器,构成一个集成模型,以降低数据集规模相对较小所带来的偏差。使用者输入肽序列,以及可进行抗聚集替换的氨基酸位点信息,随后模型会预测该序列是否可能发生聚集。如果序列被预测为易聚集,则会进一步分析靠近树脂一侧的第2至第12位氨基酸,识别其中哪些氨基酸可以被其抗聚集对应形式替代。之后,这些潜在替换位点会根据它们对聚集的相对贡献进行排序,从而帮助优先选择最有影响的改动。
为了检验这一能力,研究选择了两条已知会发生聚集的序列hGH和GB1,并选用假脯氨酸保护的氨基酸构件作为一种广泛应用的抗聚集工具。值得注意的是,假脯氨酸正是对聚集贡献最高的两种受保护氨基酸Ser(t-Bu)和Thr(t-Bu)的抗聚集等价形式(图5b)。对于hGH,由100个XGBoost分类器构成的集成模型中有74%预测其会发生聚集;对于GB1,这一比例进一步上升到90%。接下来,模型评估了从树脂端即C端开始数第2至第12位氨基酸中Ser(t-Bu)和Thr(t-Bu)的贡献,最终为GB1给出了3个潜在替换位点,为hGH给出了2个潜在替换位点,并按照其缓解聚集的潜力进行了排序。
随后,两条肽的天然版本以及所有引入假脯氨酸替换的变体都被实际合成,所得粗品纯度与模型预测的排序一致。引入最理想的假脯氨酸后,hGH的粗品纯度由23%提高到69%,GB1则由17%提高到75%(图5c)。总体而言,这一算法能够利用训练后的模型预测肽的聚集性质,并指出最适合引入抗聚集工具的位置,从而提高合成效率。
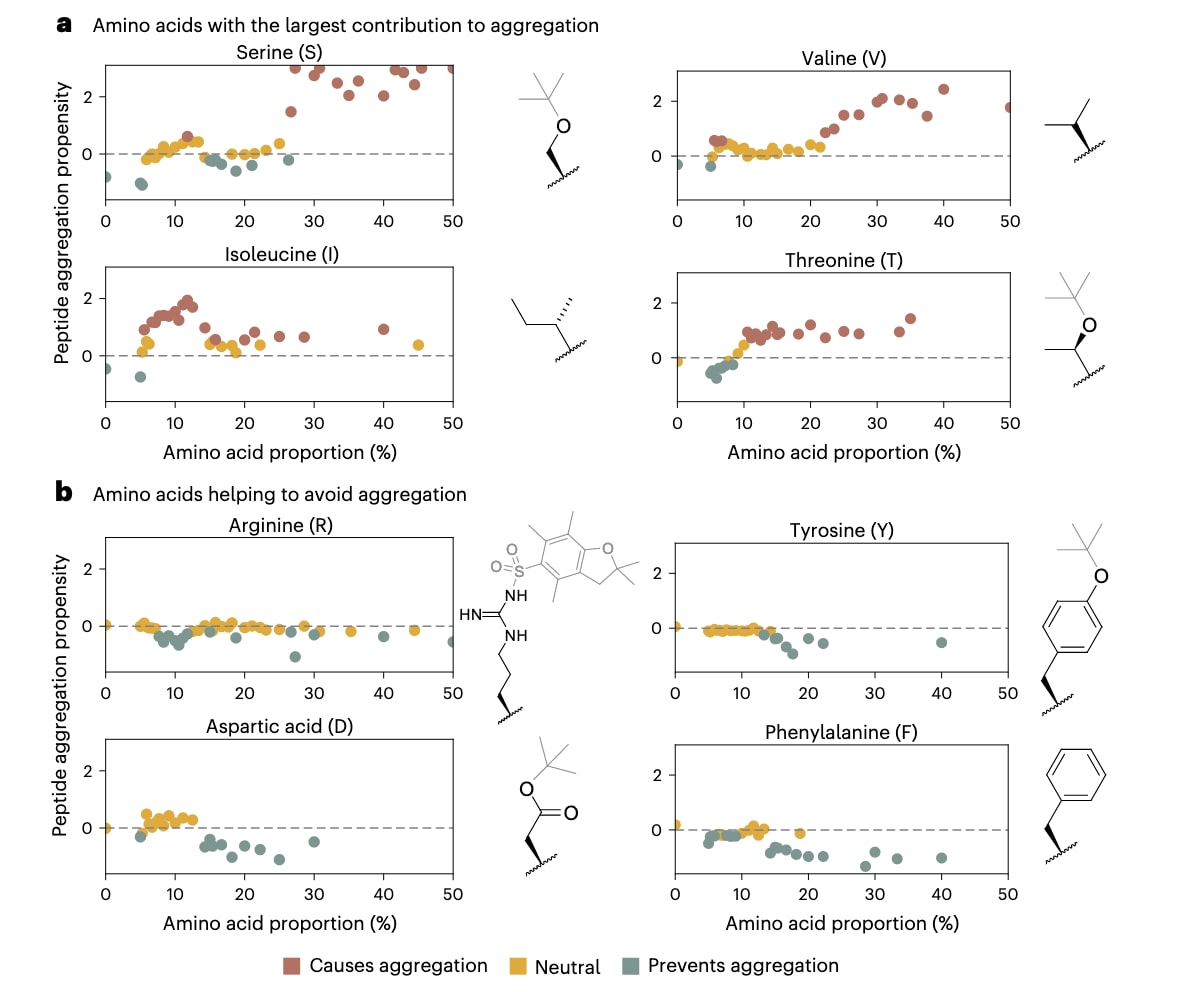
图4|对XGBoost分类器决策影响最大的氨基酸分析。 横坐标表示各氨基酸在序列中的比例,纵坐标为对应的平均SHAP值。SHAP值为正表示该氨基酸会提高模型预测为聚集的概率,负值则表示降低聚集发生的可能性。a,对聚集贡献最大的氨基酸包括丝氨酸、缬氨酸、异亮氨酸和苏氨酸。b,对聚集贡献最小的氨基酸包括精氨酸、酪氨酸、天冬氨酸和苯丙氨酸。
3 结论
在这项研究中,机器学习被用作一种发现工具,揭示了肽聚集对氨基酸组成存在出人意料且非常显著的依赖关系。为验证这一发现,研究中实验合成了40条序列,具体包括8条已知序列及其各自5种随机打乱后的变体。与此同时,还构建了一种简单的组成向量,作为表征肽序列的方法,用于研究固相肽合成过程中的聚集特征。借助这种表示方式较强的可解释性,研究发现,体积更大且极性更强的侧链或保护基通常有助于降低聚集倾向,而具有典型脂肪族特征的侧链则会提高聚集发生的可能性。
进一步地,这一发现展现出明确的实际应用价值。研究通过识别目标肽中对聚集贡献最大的关键氨基酸位点,并在这些位置有策略地引入假脯氨酸,成功降低了聚集,同时使两条测试序列的纯度分别提高了58%和46%。这些结果对当前关于聚集机制的传统理解提出了挑战。过去通常认为,聚集主要是由序列决定的事件,其根源在于分子间主链通过氢键相互作用形成
然而,这项研究显示,在固相肽合成过程中,真正主导聚集的主要是脂肪族侧链氨基酸,例如缬氨酸和异亮氨酸。与这些结构相似的保护基,例如叔丁基保护的丝氨酸或苏氨酸,在固相肽合成中也表现出类似行为。与天然肽的情况不同,具有芳香族侧链或芳香族保护基的氨基酸,例如苯丙氨酸或酪氨酸,反而倾向于降低聚集发生的概率。更重要的是,尽管聚集长期以来被普遍视为由序列决定,但研究结果表明,在固相肽合成条件下,氨基酸组成的影响实际上更大。正是基于这一认识,研究提出了组成向量这一简化的肽表示方法,不仅能够预测聚集开始发生的时点,还能够进一步给出缓解聚集的策略建议。
总体来看,这种由机器学习驱动的方法揭示了此前未被识别的肽聚集规律。肽组成与聚集之间这种强相关性,只有借助计算分析才得以显现,这凸显出机器学习在发现化学体系复杂关系方面的独特能力。这项工作说明,机器学习在化学中的价值并不局限于常见的性质预测或分子生成,它同样能够作为强有力的发现工具,挑战已有模型,并从分子数据中挖掘隐藏的规律。
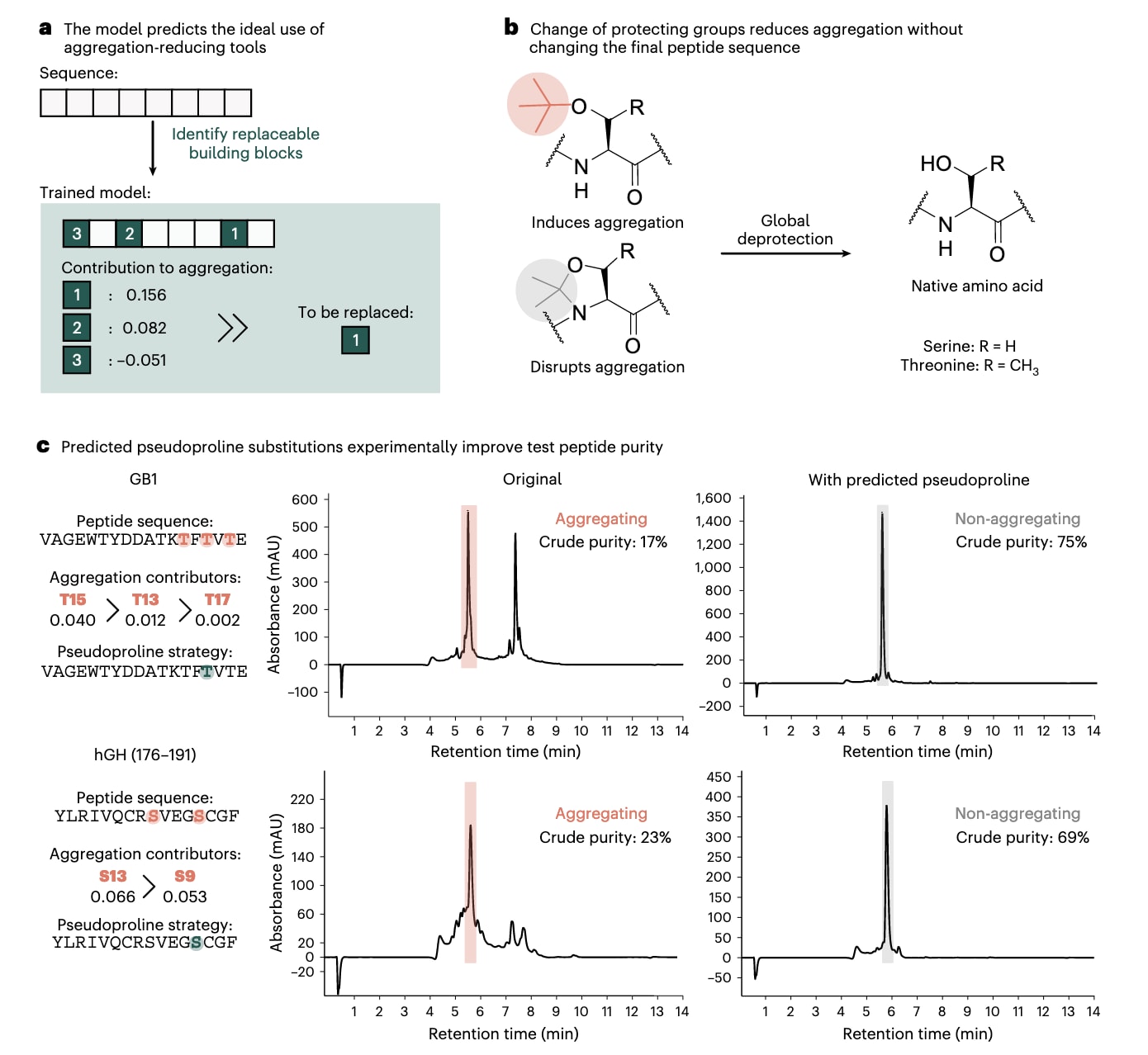
图 5 | 利用 XGBoost 集成模型指导合理使用聚集抑制工具,以优化合成条件。 a,基于用户输入的序列以及可替换的氨基酸,训练好的模型集成能够预测并评分该序列的聚集特性,同时预测肽链早期片段(第 2–12 位)中现有氨基酸对聚集的贡献。这使得能够更有效地引入抑制聚集的基团。b,丝氨酸和苏氨酸这两种带有叔丁基(t-Bu)保护基的氨基酸,在模型中被预测对聚集具有较大贡献,也可以以假脯氨酸(pseudoproline)的形式引入。后者是一类成熟的抗聚集工具,在全局脱保护后可恢复为天然氨基酸。c,该模型在两个已知易发生聚集的序列中进行了验证:GB1 和 hGH。选择对聚集贡献最大的丝氨酸和苏氨酸进行替换后,GB1 片段的粗产物纯度显著提高了 58%,hGH 片段提高了 46%。