BIB 2025 | cyclicpeptide: 用于环肽药物设计的Python软件包
今天介绍的这项工作来自 Briefings in Bioinformatics 。环肽是一类具有闭环骨架结构的多肽分子。由于其结构中不存在自由末端,并且环状拓扑能够限制构象自由度,这类分子通常表现出更高的结构稳定性、较强的抗蛋白酶降解能力以及更优异的靶标结合能力。因此,环肽在抗感染、抗肿瘤、免疫调节以及蛋白-蛋白相互作用调控等多个领域逐渐受到关注,并被认为是连接传统小分子药物与生物大分子药物的重要桥梁。然而,环肽在计算建模和数据处理方面仍然面临一些独特挑战。与线性肽相比,环肽具有更加复杂的拓扑结构,其环化方式可能包括头尾环化、侧链-侧链连接以及其他类型的化学交联结构。这些结构特征使得环肽在序列表示、结构建模以及数据库存储方面缺乏统一标准。同时,现有的大多数生物信息学工具主要针对线性肽或小分子化合物开发,难以直接适用于环肽研究,从而在一定程度上限制了环肽在人工智能驱动药物设计流程中的应用。针对这一问题,该研究提出了一个名为cyclicpeptide的Python软件包,旨在为环肽药物设计提供系统化的数据处理与分析工具。该软件包整合了多种功能模块,包括环肽序列与结构之间的转换、不同数据格式的标准化处理、基于图结构的环肽比对与搜索以及分子性质分析等。通过提供统一的数据表示方式和便捷的计算接口,该工具能够帮助研究人员更加高效地处理环肽相关数据,并将其整合到现代计算药物设计流程之中。总体而言,cyclicpeptide软件包为环肽研究提供了一个专门化的计算平台。通过标准化环肽数据表示并整合多种分析工具,该软件不仅降低了环肽数据处理的复杂度,也为将人工智能方法应用于环肽药物设计奠定了基础,从而有助于加速新型环肽治疗药物的发现与开发。

获取详情及资源:
- 📄 论文: https://academic.oup.com/bib/article/26/1/bbae714/7948245
- 💻 代码: https://github.com/dfwlab/cyclicpeptide
0 摘要
环肽独特的环状结构赋予其出色的稳定性与生物活性,使其在多种疾病治疗中展现出重要潜力。然而,目前缺乏针对环肽数据的标准化工具,这在一定程度上限制了其在当今以人工智能驱动的高效药物设计中的应用。为弥补这一空缺,该研究开发了一个名为cyclicpeptide的Python软件包,专门用于环肽药物设计。该软件包提供了一系列标准化工具,例如Structure2Sequence、Sequence2Structure以及多种格式转换功能,用于处理、转换并规范化环肽的结构与序列数据。此外,该软件包还包含GraphAlignment模块,用于实现针对环肽特性的序列比对与相似性搜索,以及PropertyAnalysis模块,用于分析环肽的类药性质并评估其潜在应用价值。通过整合上述功能,该工具为环肽数据的处理与分析提供了一套完整的计算框架,从而有助于将环肽更高效地整合进现代药物发现流程,加速基于环肽的治疗药物研发。
1 引言
环肽是一类具有环状结构的肽分子,由于其独特的结构特征以及良好的类药性质,在药物设计领域受到越来越多的关注。环肽通常具有较高的生物稳定性以及对特定靶标的高选择性,并在抗菌、抗肿瘤以及神经系统相关疾病等多个方向展现出广泛的生物活性。目前,美国食品药品监督管理局(FDA)已经批准了40余种环肽类药物,其中包括2023年10月获批用于治疗全身型重症肌无力的药物zilucoplan。此外,较早获批的环肽药物环孢素被广泛用于预防器官移植后的排斥反应以及治疗自身免疫性疾病,并已成为全球范围内预防移植后移植物排斥反应的一线治疗药物。
尽管环肽在药物开发中具有重要潜力,但其设计过程仍然面临诸多挑战。这些挑战主要来源于环肽复杂的氨基酸组成以及多样化的环化方式,例如头尾环化、侧链连接以及内部环化等结构形式。近年来,计算能力的提升以及人工智能技术的发展,使研究者能够开发基于数据驱动的预测算法,例如人工智能驱动的药物设计(AIDD)以及计算机辅助药物设计(CADD)。然而,这类方法通常依赖于大量标准化的环肽知识信息,例如氨基酸组成及其修饰方式、环化位点以及理化性质等。这些要求在一定程度上增加了环肽药物设计中的数据处理与标准化难度。在计算机辅助药物设计流程中,研究者通常会使用传统的化学信息学工具包进行初步分析,例如RDKit和Biopython。然而,这些工具主要针对小分子或线性肽开发,对于结构更加复杂的环肽分子并不完全适用。近年来也出现了一些新的工具,例如pyPept,该工具在自动生成复杂肽分子的二维和三维结构表示方面展现出一定潜力,但其在处理非天然氨基酸以及预测肽分子生物活性构象方面仍存在一定局限。同时,一些基于人工智能的环肽设计工具也逐渐出现,例如AfCycDesign,但这类工具通常集中于特定功能,例如环肽序列生成。虽然也存在专门用于识别环肽氨基酸单体的工具,例如2015年由Yoann Dufresne开发的Smiles2Monomers,但这些工具在功能范围、使用复杂度以及可扩展性方面仍存在限制,难以满足当前环肽药物开发的需求。
基于上述背景,该关研究提出了一个名为cyclicpeptide的Python软件包,用于支持环肽药物设计。该软件包提供了多种功能模块,包括Structure2Sequence、Sequence2Structure、GraphAlignment以及PropertyAnalysis等,能够覆盖环肽药物开发中的多种应用场景。同时,该软件包以开源形式发布,使用户能够根据实际需求对其功能进行扩展。通过整合这些工具,cyclicpeptide旨在将环肽更加顺畅地融入现代人工智能驱动的药物研发流程之中,从而加速基于环肽的治疗药物开发。
2 结果
2.1 cyclicpeptide工具包:核心功能
cyclicpeptide是一个基于RDKit和NetworkX开发的Python软件包,专门用于环肽分子的分析与处理。该软件包集成了多种功能模块,包括Structure2Sequence、Sequence2Structure、GraphAlignment、PropertyAnalysis,以及用于格式转换的StructureTransformer与SequenceTransformer,还包含用于生成环肽序列的SequenceGeneration模块。该软件包以开源形式发布,代码托管于GitHub,同时提供了完整且详细的在线文档,方便研究人员理解和使用相关功能,从而在环肽数据处理与药物设计研究中更加高效地应用这些工具。(图1A)
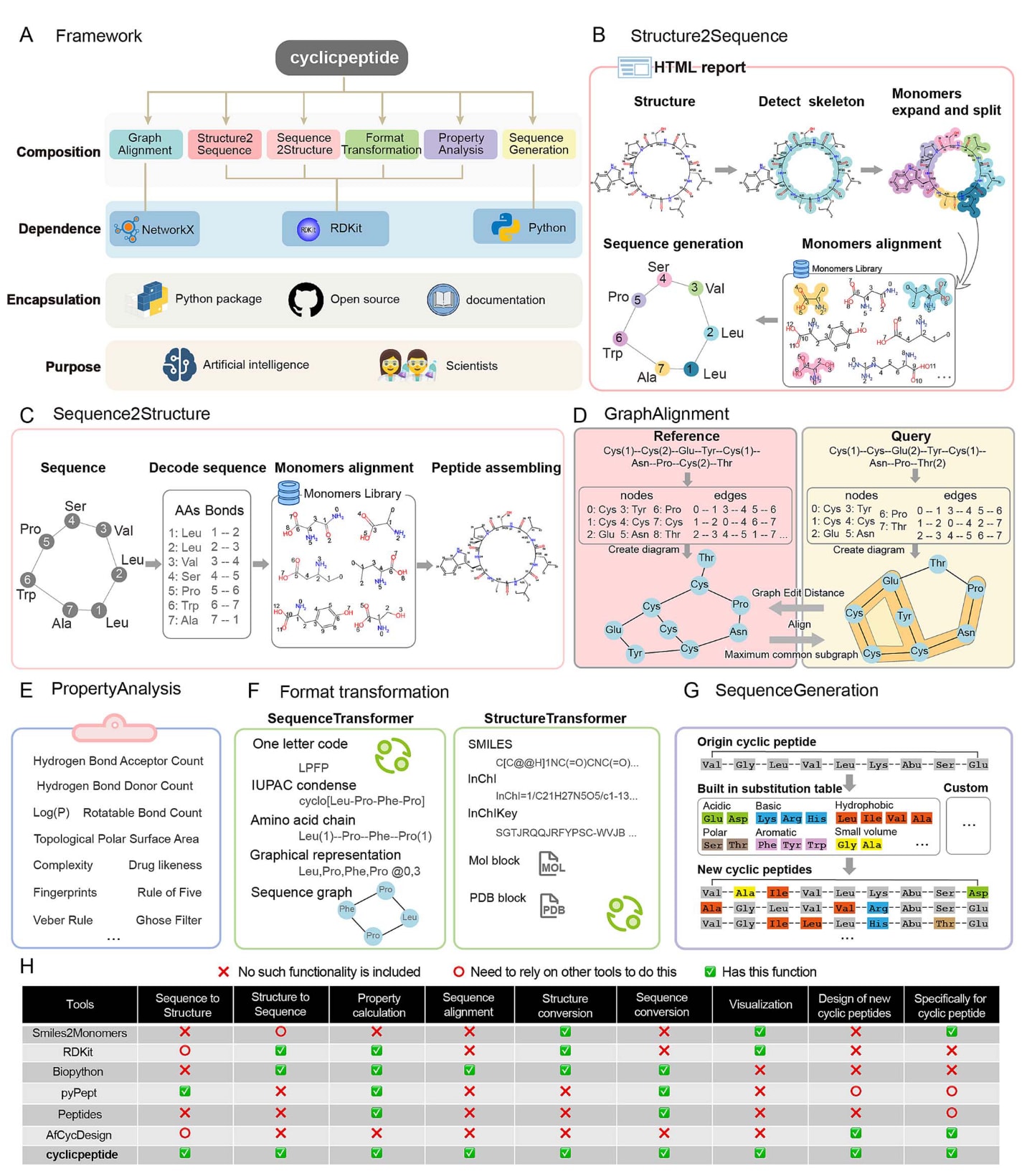
图1|cyclicpeptide Python软件包的工作流程与方法概览。 (A) cyclicpeptide框架概览。cyclicpeptide基于RDKit和NetworkX开发,包含多种功能模块,包括Structure2Sequence、Sequence2Structure、GraphAlignment、格式转换工具(StructureTransformer和SequenceTransformer)、PropertyAnalysis以及SequenceGeneration。(B) Structure2Sequence的工作流程示意图。该过程首先从环肽结构中识别主链(backbone),随后对主链进行扩展以定位各个氨基酸单体。之后通过单体比对与组装生成序列,并允许在比对过程中自定义单体库。Structure2Sequence还能够生成HTML格式的输出报告。(C) Sequence2Structure的工作流程示意图。通过对序列进行解析,确定各个氨基酸的顺序及其连接关系。随后通过单体匹配,将肽链组装为头尾环化(head-to-tail)的环肽结构。(D) GraphAlignment工具示意图。氨基酸序列首先被转换为由节点和边构成的集合,并利用NetworkX进行可视化表示。该工具提供两种相似性计算算法:Graph similarity()用于计算图编辑距离(graph edit distance),mcs similarity()用于确定最大公共子图(maximum common subgraph)。(E) PropertyAnalysis模块可以计算多种理化性质指标。(F) 结构格式与序列格式转换。StructureTransformer用于结构格式转换,并提供三维结构预测功能。(G) SequenceGeneration工具示意图。该工具基于氨基酸替换表自动生成新的环肽序列。(H) 不同工具功能对比。cyclicpeptide提供较为全面的功能体系,适用于环肽开发过程中多种分析与处理任务。
2.2 Structure2Sequence
Structure2Sequence工具用于对环肽的SMILES(Simplified Molecular Input Line Entry System)结构表示进行氨基酸单体识别,将其拆分为对应的氨基酸残基并转换为序列格式(图1B)。该功能适用于多种不同的环化方式,并支持超过500种天然与非天然氨基酸单体(表S1)。借助Structure2Sequence,研究人员可以根据已知结构快速推断环肽可能对应的氨基酸序列。为了更加直观地展示从结构到序列的转换过程,该工具还会生成HTML(HyperText Markup Language)报告,其中包含环肽结构图以及结构解析与序列转换过程的可视化结果(图1B)。该报告能够帮助用户检查转换结果的准确性,并在必要时进行进一步调整。
2.3 Sequence2Structure
Sequence2Structure工具能够将已知的环肽序列(例如单字母编码或氨基酸链表示)转换为对应的环状结构,并以SMILES格式输出。在这一过程中,环肽序列首先被转换为图结构,从而解析其中的单体组成以及相互之间的连接关系。随后,该模块从单体库中提取对应的氨基酸结构,并根据这些连接关系逐步组装出完整的环肽分子结构(图1C)。
2.4 GraphAlignment
GraphAlignment模块用于比较不同环肽分子之间的结构相似性(图1D)。该工具利用NetworkX将氨基酸序列表示转换为图结构,使得两个环肽分子之间的相似性可以通过节点与边的关系进行直观比较。这种方法能够有效克服传统线性序列比对算法在处理环状结构时存在的局限。GraphAlignment提供两种相似性度量方式,包括图编辑距离(graph edit distance)以及最大公共子结构(maximum common substructure)。
2.5 PropertyAnalysis
PropertyAnalysis模块用于计算环肽分子的多种理化性质指标,例如拓扑极性表面积(Topological Polar Surface Area)、分子复杂度(Complexity)、疏水性Log(P)、氢键供体数量以及氢键受体数量等(图1E)。这些指标能够帮助研究人员更好地理解环肽分子的类药性质及其潜在应用价值。
2.6 Format transformation
cyclicpeptide还提供了用于结构与序列格式转换的工具。其中,StructureTransformer用于结构格式转换,SequenceTransformer用于序列格式转换(图1F)。StructureTransformer支持多种结构表示形式,例如SMILES、InChI以及用于三维结构表示的Protein Data Bank(PDB)格式。SequenceTransformer则支持多种序列表示方式,包括单字母编码、一种由IUPAC(International Union of Pure and Applied Chemistry)定义的缩写格式、氨基酸链表示以及图形化表示形式。这两个工具能够对用户提供的环肽结构和序列数据进行标准化处理,从而确保这些数据能够与后续分析模块兼容,例如Structure2Sequence、Sequence2Structure以及GraphAlignment等工具。
2.7 SequenceGeneration
cyclicpeptide提供了一个基于序列的环肽生成工具SequenceGeneration(图1G)。该工具可以根据氨基酸替换表自动生成潜在的新型环肽序列。用户既可以使用内置的氨基酸替换表,也可以使用自定义替换表生成新的序列。这些生成的候选序列能够进一步输入到其他分析模块中进行后续研究与筛选。该研究还将cyclicpeptide与多种现有的肽分析工具进行了比较(图1H)。例如,smiles2monomers是一种基于网页的可视化工具,主要用于结构转换。RDKit提供可视化功能,同时支持序列与结构之间的相互转换、性质计算以及结构格式转换,但其设计主要面向小分子。Biopython提供多种基础生物信息学功能,但缺乏针对环肽设计的专门模块。pyPept与AfCycDesign等工具主要侧重于环肽结构生成,但其功能范围相对有限。相比之下,cyclicpeptide针对环肽研究提供了一套更加完整的工具体系,包括序列与结构之间的相互转换(Structure2Sequence与Sequence2Structure)、序列相似性比较(GraphAlignment)、结构与序列格式转换(StructureTransformer与SequenceTransformer)、环肽序列生成(SequenceGeneration)、分子性质计算(PropertyAnalysis)以及可视化功能。这些功能共同为环肽药物开发中的多种分析与处理任务提供了有力支持。
3 工具可靠性验证
为了验证cyclicpeptide软件包的可靠性,研究从CyclicPepedia知识库中提取了四组环肽数据:(i)包含结构与序列信息的830个环肽;(ii)包含结构与序列信息并具有头尾环化特征的484个单环肽;(iii)包含结构信息并具有头尾环化特征的670个单环肽;(iv)包含序列信息并具有头尾环化特征的4658个单环肽,其中4342条为单字母编码格式,316条为IUPAC缩写格式(图2A)。这些数据集被用于验证Structure2Sequence和Sequence2Structure两个模块的准确性与稳定性(图2B)。其中,准确性指通过序列到结构或结构到序列转换后得到的结果是否与已知结构或序列一致;稳定性则指序列(或结构)在转换为结构(或序列)后再转换回原始形式时是否保持一致。
数据集a根据氨基酸组成(天然与非天然氨基酸)以及环化类型(单环或多环)被划分为多个子集,用于验证Structure2Sequence模块的性能。结果显示,Structure2Sequence在现有数据集上的平均氨基酸检测率达到99.6%(图2C,表S2)。在剩余的0.4%误差中,0.1%源于原始序列与结构信息之间的不一致(图S1A),0.3%则由于酯键环化情况下氨基酸拆分不准确所导致(图S1B)。对于酯键环化氨基酸的错误分割,需要结合化学反应相关的先验知识进行进一步判断。目前,cyclicpeptide会为用户提供多种拆分策略的详细报告以供参考。
数据集b用于验证Sequence2Structure模块,其结构预测准确率达到97.7%(图2D)。错误预测主要来源于特殊的环化方式,例如二硫键、酯键以及辅基的存在(例如金属离子或乙酸)。这些差异主要源于该工具默认假设环肽通过头尾肽键形成环化结构(图S1C),因此在预测侧链环化、酯键环化或二硫键环化结构时效果相对有限。在这些情况下,通常需要借助外部编辑工具对结果进行进一步修正。
此外,该研究还利用数据集c和d对Structure2Sequence与Sequence2Structure的稳定性进行了验证(图2B)。结果显示,这两个转换工具均表现出较高的稳定性,其中数据集c的稳定性为98.2%,数据集d为97.5%(图2E与图2F)。这表明在转换过程中,除辅基信息外,环肽分子的其他信息基本不会丢失。
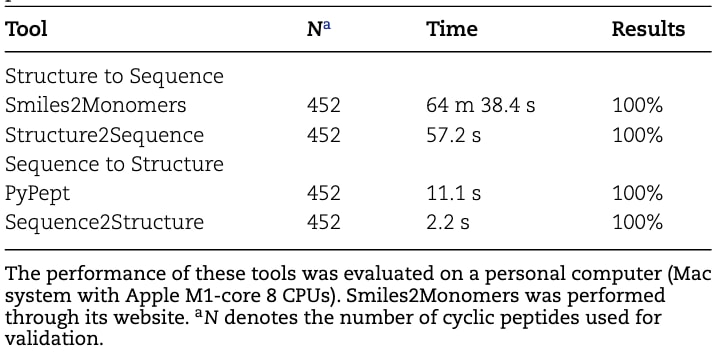
为了进一步评估cyclicpeptide在实际应用中的性能,研究还将其与多个现有分析工具进行了比较,包括Smiles2Monomers、Biopython、RDKit以及pyPept,并从处理时间与结果准确性两个方面进行了评估。如表1所示,在结构到序列转换任务中,Smiles2Monomers与cyclicpeptide中的Structure2Sequence均能够准确识别氨基酸。然而在相同数据规模下,Structure2Sequence的运行速度明显更快,在57.2秒内即可完成452个环肽的处理。另一方面,在序列到结构转换任务中,pyPept与cyclicpeptide中的Sequence2Structure均可以将环肽序列转换为SMILES格式,但Sequence2Structure在转换效率方面表现更优。pyPept需要将输入序列转换为Boehringer Ingelheim Line Notation(BILN)或Hierarchical Editing Language for Macromolecules(HELM)格式后才能进行处理,这增加了额外的步骤。
表1|结构与序列转换性能比较。
在数据库检索任务中,研究以头尾环化环肽“AAGFPVFF”为查询序列进行测试(表2)。数据库(N=452)中同时包含该序列本身“AAGFPVFF”以及其循环重排形式“PVFFAAGF”。研究评估不同工具是否能够同时识别这两个序列。Biopython由于主要针对线性肽设计,未能识别循环变体,其相似度评分仅为0.54。相比之下,RDKit与GraphAlignment均表现出较好的性能,但两者也各有局限。RDKit侧重于结构相似性识别,因此需要完整的结构信息;GraphAlignment虽然能够进行有效比较,但计算时间较长。为提高GraphAlignment的计算效率,研究进一步构建了一个基于图卷积网络的估计模型GraphAlignment GCN(GA GCN,图S2,表2)。GA GCN能够准确预测GraphAlignment得分(均方误差
表2|头尾环化环肽“AAGFPVFF”的数据库检索结果。
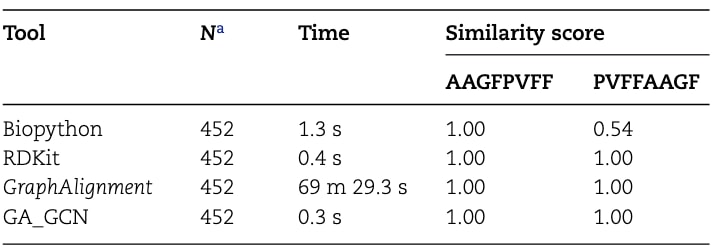
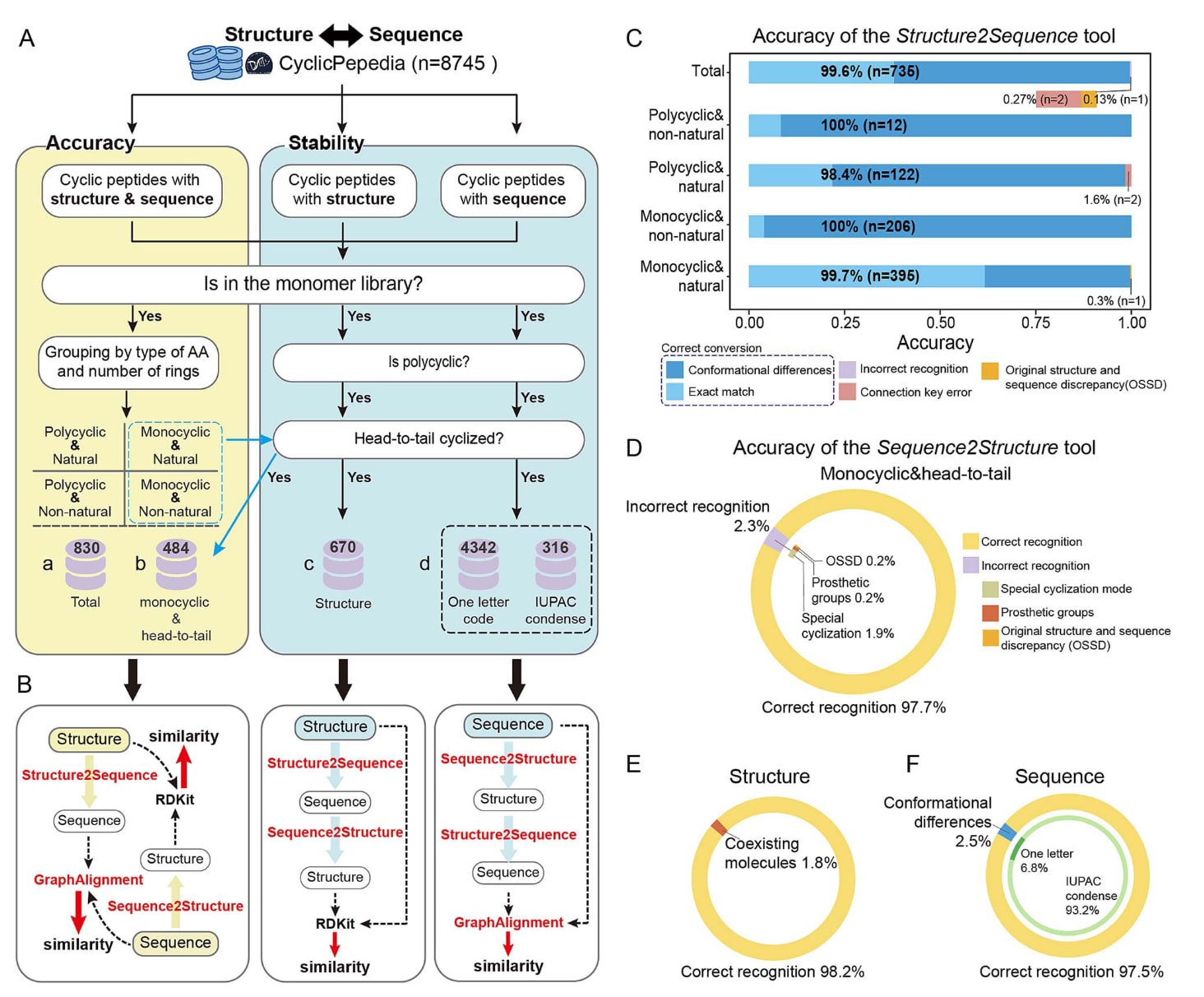
图 2|cyclicpeptide Python软件包的验证结果。 (A) 验证数据集的筛选流程。从CyclicPepedia知识库中收录的8745个环肽中筛选出若干数据子集,包括:830个同时具有结构与序列信息的环肽;484个具有结构与序列信息并具有头尾环化特征的单环肽;670个仅包含结构信息且具有头尾环化的单环肽;以及4658个仅包含序列信息且具有头尾环化的单环肽。(B) 可靠性验证分为准确性验证和稳定性验证。准确性验证是指利用转换工具Structure2Sequence或Sequence2Structure将结构(或序列)转换为序列(或结构),并将结果与已知对应的真实数据进行比较。稳定性验证则是在完成两轮转换后,将处理得到的结构或序列与原始数据进行比较,以评估在转换过程中是否存在信息丢失。(C) Structure2Sequence工具的准确性验证结果。构象差异指转换得到的序列与原始序列之间在氨基酸构象上的差异,这通常来源于原始序列中未明确指定构象或同时存在多种构象的情况。(D) Sequence2Structure工具的准确性验证结果。错误转换主要来源于特殊的环化方式以及辅基的存在,例如金属离子或乙酸等。(E) 结构转换的稳定性验证结果。(F) 序列转换的稳定性验证结果。在IUPAC缩写格式中可能出现构象差异,而单字母编码格式并不包含构象信息。
4 案例研究1:结构与序列之间的相互转换及相似性分析
在该案例研究中,通过三个示例展示了cyclicpeptide中基础功能的应用方式(图3)。首先利用Structure2Sequence工具将“Alpha-Amanitin”的结构转换为对应的序列(图3A)。在转换过程中,用户可以通过生成的HTML报告查看匹配到的氨基酸单体以及可能的序列结果。同时,Structure2Sequence还会使用实线与虚线区分肽键与非肽键,从而更加清晰地展示环肽结构中的连接关系。
Sequence2Structure工具支持多种序列输入格式,包括氨基酸链形式以及单字母编码格式。在示例中,将序列“AIPFNSL”输入到Sequence2Structure中进行序列到结构的转换(图3B)。该工具首先将序列拆分为单个氨基酸以及对应的连接关系,然后与单体库中的氨基酸结构进行匹配,并最终通过头尾环化方式将这些单体组装为完整的环肽结构。
在GraphAlignment算法的示例中,以“Cys(1)(2)—Cys—OH-DL-Val(2)—4OH-Leu—OH-Ile(1)”作为参考序列(Reference),并选择三个相似序列作为查询序列(Query)。结果显示,GraphAlignment能够正确识别Query1与Reference为相同的环肽结构。同时,该算法还能够准确指出Reference与Query2以及Reference与Query3之间在连接键以及氨基酸组成上的差异(图3C)。此外,在计算相似性时,用户可以根据需求选择图编辑距离(graph edit distance)算法或最大公共子结构(maximum common substructure)算法。
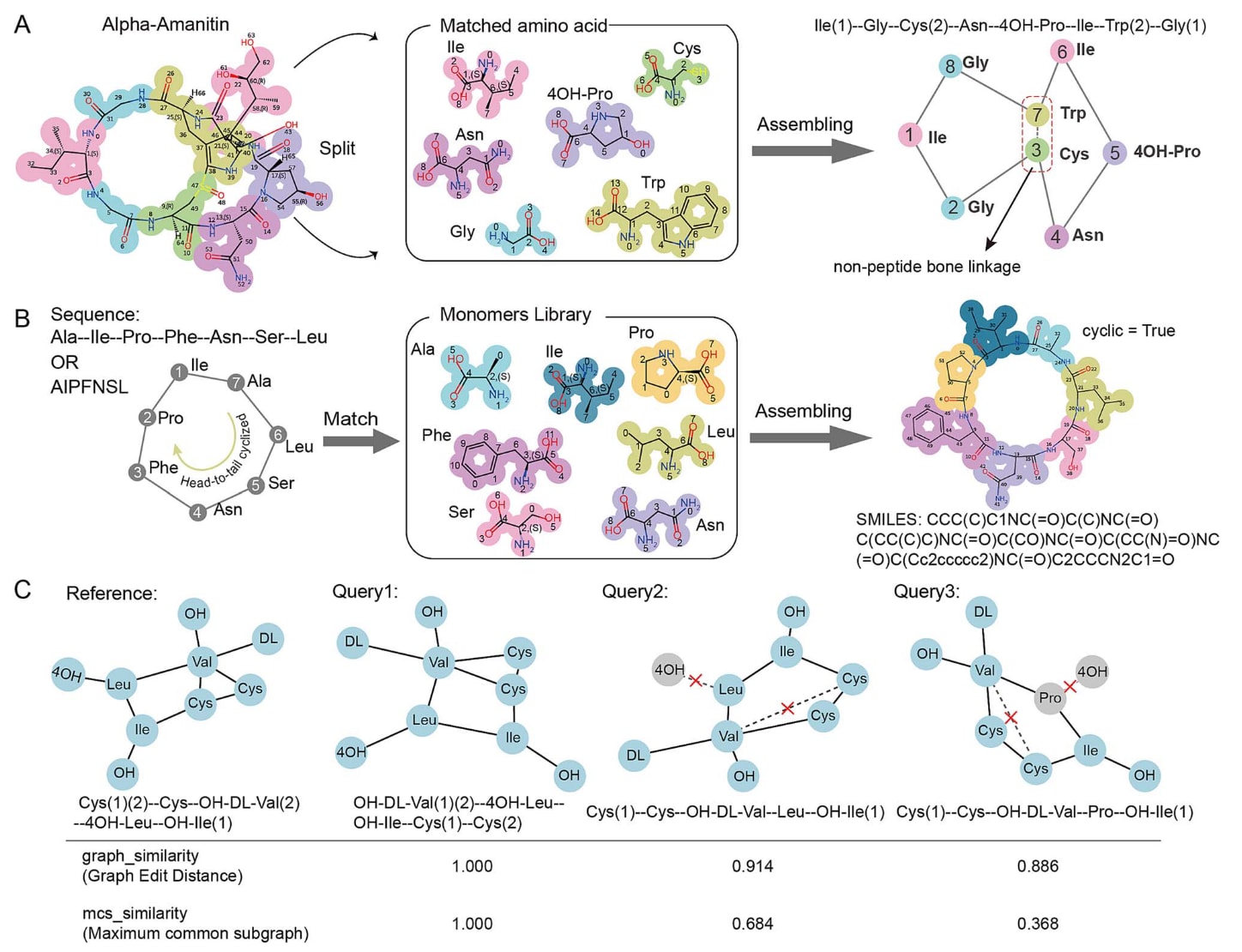
图 3|cyclicpeptide基础功能的示例结果。 (A) 使用Structure2Sequence将“alpha-Amanitin”的SMILES字符串转换为氨基酸序列。(B) 使用Sequence2Structure将序列“AIPFNSL”转换为SMILES字符串,其中参数“cyclic”设置为true。(C) 展示了GraphAlignment计算得到的“graph similarity”(图编辑距离)以及“mcs similarity”(最大公共子图)结果。
5 案例研究2:新型环孢素A类似物的设计
为了展示cyclicpeptide工具包在环肽药物设计中的应用,该研究开展了一个关于环孢素A类似物设计的案例研究。通过对环孢素A及其类似物进行序列比对,可以发现第2位和第3位氨基酸可能在决定免疫抑制活性方面起关键作用(图4A)。例如,一些衍生物如alisporivir和SCY-635在消除免疫抑制作用的同时,增强了抗病毒或其他药理活性(图4B)。
基于这些观察结果,在SequenceGeneration模块中建立了相应的序列修饰规则,从而生成了66条环孢素A类似物序列。这些序列在第2位和第3位氨基酸位置发生变化,同时在其他位置进行随机修饰,整体变异率小于30%(表S3)。随后利用Sequence2Structure和StructureTransformer预测这些序列对应的结构,并将所得结构与环孢素结合蛋白cyclophilin A进行蛋白-配体分子对接分析。在所有候选分子中,三个得分最高的类似物(CsA RP47、CsA RP29和CsA RP48)表现出与cyclophilin A最强的对接活性(图4B和图4D)。与环孢素A(图4C)相比,这三个类似物在空间构象上更接近alisporivir,表明它们可能同样具有非免疫抑制特性(图4D)。
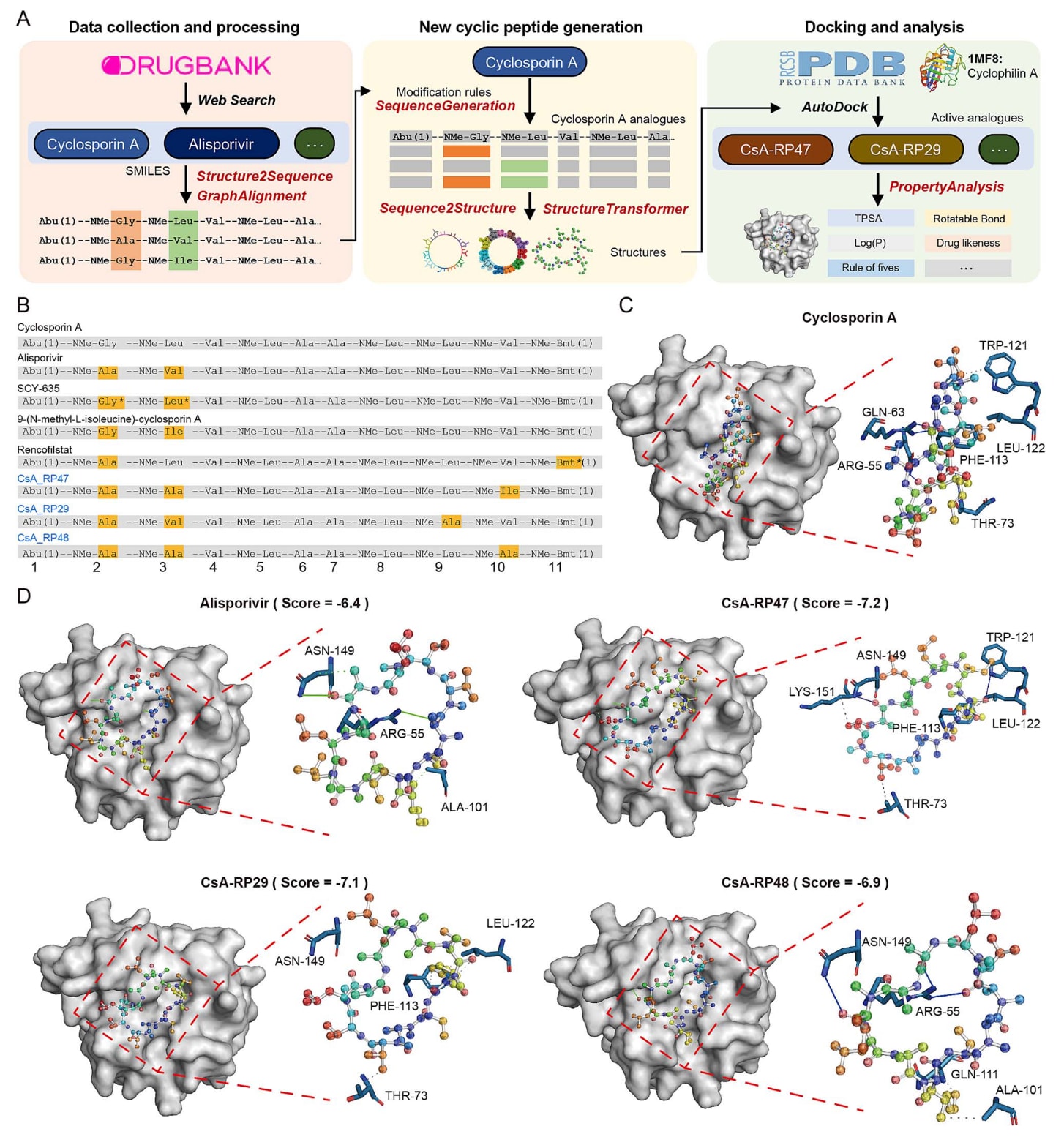
图 4|新型环孢素A(CsA)类似物的设计。 (A) 利用cyclicpeptide工具包进行药物设计的工作流程示意,以环孢素A(CsA)为例。首先从DrugBank等药物数据库获取CsA及其类似物(例如alisporivir)的结构信息。随后使用Structure2Sequence工具获得对应的氨基酸序列,并利用GraphAlignment比较序列之间的相似性,从而识别可能的关键修饰位点。在此基础上建立特定的修饰规则,并通过SequenceGeneration根据设定条件随机生成新的环肽序列。随后利用Sequence2Structure和StructureTransformer将这些序列转换为三维结构,并使用AutoDock与来自PDB数据库的已知靶标cyclophilin A(CypA)进行分子对接。筛选出具有更高对接效率的类似物,并使用PropertyAnalysis计算其理化性质,以供进一步研究。(B) CsA类似物的序列,其中高亮显示的氨基酸表示与CsA序列存在差异的位置。(C) CsA与CypA之间相互作用的示意图,其中标出了具体的结合位点。(D) CsA类似物(如alisporivir及其他类似物)与CypA之间相互作用的示意图,并标示出对应的结合位点。
5.1 Cyclicpeptide用户手册
在成功安装cyclicpeptide工具包之后,用户需要调用相应的功能模块(例如Structure2Sequence、Sequence2Structure、GraphAlignment等)来使用其各项功能。
在进行结构到序列的转换时,可以从cyclicpeptide软件包中导入Structure2Sequence模块,并调用transform()函数将结构(SMILES)转换为序列。其中,“monomers path”参数默认使用内置的单体库,但用户也可以通过该参数指定自定义的单体库。“report”参数用于控制是否输出转换结果报告(图5A)。
在进行序列到结构转换时,首先需要使用reference aa monomer()函数读取单体库,然后通过seq2stru non naturalAA()函数将序列(氨基酸链)转换为结构(SMILES)(图5B)。
在进行序列相似性比较时,首先使用GraphAlignment模块中的sequence to node edge()函数,从序列(氨基酸链)中提取节点与边的信息。随后通过create graph()函数构建NetworkX图结构,并选择相似性计算方法(graph similarity()或mcs similarity())来计算相似度(图5C)。
此外,cyclicpeptide还提供了IOManager模块用于绘图与图像导出,例如plot smiles()(图5B)和graph2svg()(图5C)。关于该工具包的详细使用说明,可以在官方文档中查阅。
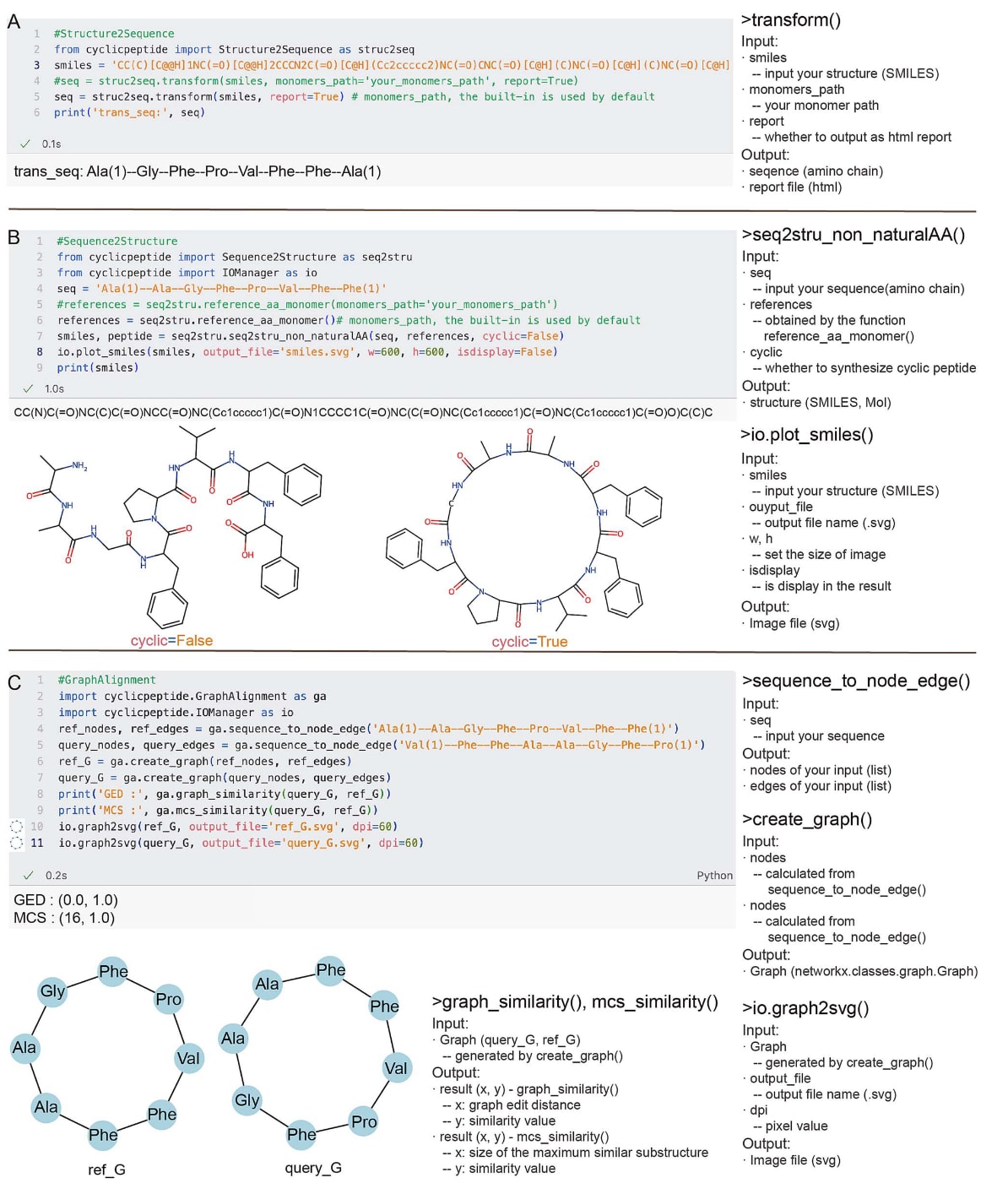
图 5|代码使用示例。 (A) Structure2Sequence工具的使用示例。transform()函数的输入结构必须为SMILES格式;如果为其他格式,可以先通过StructureTransformer工具进行转换。(B) Sequence2Structure工具的使用示例。seq2stru non naturalAA()函数的序列输入必须为氨基酸链格式,而seq2stru naturalAA()函数的序列输入既可以是单字母编码格式,也可以是氨基酸链格式。其他格式的序列可以通过SequenceTransformer工具进行转换。(C) GraphAlignment工具的使用示例。sequence to node edge()函数的序列输入必须为氨基酸链格式。
6 讨论
尽管环肽在药物研发领域逐渐成为重要研究方向,但目前仍缺乏专门针对环肽药物设计开发的分析工具。为了解决这一问题,该研究开发了一个名为cyclicpeptide的Python软件包,用于辅助环肽药物设计。该软件包提供了Structure2Sequence、Sequence2Structure、GraphAlignment等多种功能模块,为环肽药物早期设计阶段提供技术支持。
随着人工智能驱动药物设计(AIDD)和计算机辅助药物设计(CADD)的发展,这些方法正逐渐成为传统实验方法的重要补充。通过深度学习与机器学习技术,可以预测环肽的膜通透性,并针对特定靶标和疾病设计具有特异性的环肽药物,从而显著加速药物研发过程。这类数据驱动的药物设计方法高度依赖高质量且规模充足的数据集。cyclicpeptide通过Structure2Sequence、Sequence2Structure、SequenceGeneration以及格式转换等功能,为环肽药物开发早期阶段提供标准化数据处理能力,从而在一定程度上缓解了数据标准化与数据准备方面的困难。
cyclicpeptide中的GraphAlignment工具能够识别环肽分子之间在氨基酸组成和键类型等方面的结构相似性与差异,这对于发现新的药物作用机制以及潜在靶点具有重要意义。然而,由于图结构计算本身具有较高的计算复杂度,在处理大规模数据时可能带来较高的计算时间成本。因此,可以通过并行计算、使用GraphAlignment估计模型(GA GCN),或将GraphAlignment与其他工具(例如Biopython或RDKit)结合使用来优化计算效率。此外,SequenceGeneration模块能够生成新的环肽序列,而PropertyAnalysis模块则可以提供全面的理化性质分析,从而帮助研究人员发现新的环肽候选药物,并评估其类药性质及应用潜力。
尽管cyclicpeptide为环肽设计提供了多种功能,但仍存在一些需要进一步优化的方面。例如,当GraphAlignment算法应用于大规模数据集时,计算时间与内存消耗可能显著增加。为解决这一问题,研究提出利用图卷积网络模型对GraphAlignment得分进行近似预测。此外,环肽的合成过程通常涉及复杂且精细的化学反应以及多种分子相互作用。目前的工具主要能够生成环肽的二维结构,并基于能量最小化方法预测潜在的三维结构,这些预测结果可能与真实实验结构存在一定差异。在实际应用中,可以结合其他工具,例如AfCycDesign生成三维结构并对结果进行验证。未来版本计划进一步引入支持环肽合成分析的方法,从而扩展cyclicpeptide的应用范围。
总体而言,cyclicpeptide是一种功能强大的分析工具,能够显著加速环肽治疗药物在早期设计阶段的研究进程,并且随着持续优化,其计算效率与用户使用体验仍有进一步提升的空间。