NC 2020 | DIP/Dpr相互作用以及蛋白质家族中特异性的进化设计
今天介绍的是发表在Nature Communications上的一项研究,聚焦蛋白质家族中相互作用特异性的进化设计问题。该工作以果蝇DIP与Dpr神经识别蛋白为模型体系,探讨在序列和结构高度相似的背景下,蛋白质如何实现精确而选择性的结合。DIP/Dpr家族成员数量众多,但其相互作用并非任意组合,而是被严格限制在特定亚群之间,这一现象为解析特异性编码机制提供了理想案例。
研究结合序列分析、结构建模与能量计算,并辅以生物物理实验验证,提出“负约束”是驱动特异性的关键因素,即通过在界面上引入不利相互作用来阻止非目标结合。这种机制强调特异性并非仅来源于增强正确配对,而更多依赖于系统性削弱错误配对。相关结果不仅深化了对蛋白质相互作用网络的理解,也为多蛋白家族在进化过程中如何实现功能分化提供了重要视角。

获取详情及资源:
0 摘要
在许多生物学现象中,包括细胞间识别,紧密相关的蛋白质家族成员之间的差异性结合亲和力发挥着关键作用。果蝇中的DIP和Dpr蛋白通过高度特异性的蛋白质-蛋白质相互作用介导神经元的靶向连接。相关研究表明,DIP/Dpr可以根据其成员之间的结合偏好被划分为七个特异性亚群。
进一步结合基于序列、结构和能量的计算方法以及实验测定的结合亲和力数据,解析了这种特异性如何编码在经典的DIP/Dpr相互作用界面上。结果显示,DIP/Dpr亚群的结合特异性主要由“负约束”机制所控制,即通过干扰非目标结合来实现选择性。为了实现精确识别,每个亚群采用不同组合的负约束,这些约束广泛分布于蛋白质相互作用界面的各个区域,几乎覆盖了整个界面。
最后,还探讨了负约束在结构层面的来源,以及这一机制在多蛋白家族中结合特异性进化起源方面的潜在普适意义。
1 引言
在进化过程中,基因复制及随后的序列分化产生了大量蛋白质家族,这些同源成员逐渐演化出不同的结合特异性。尽管许多紧密相关的蛋白在序列和结构上仅存在细微差异,却往往带来显著的功能差别。细胞间黏附蛋白家族便是典型例子,微小的序列变化即可产生高度精确的蛋白质相互作用特异性。例如,无脊椎动物Dscam1/2以及脊椎动物簇状原钙黏蛋白在神经元“条形码”识别中表现出严格的家族内同源结合特异性。即使不同亚型之间序列相似性超过90%,少量差异仍足以确保仅发生同源识别,这对于神经元身份多样化及自我/非自我识别至关重要。相比之下,经典I型和II型钙黏蛋白的特异性较为宽松,除了同源结合外,还能够与部分家族成员发生异源结合;而nectin家族则主要表现为异源相互作用,参与形成不同细胞类型之间的棋盘状排列。这些不同的特异性模式决定了其生物学功能,并在进化中保持保守,因此解析多蛋白家族中结合特异性的编码机制具有重要意义。
围绕这一问题,研究对果蝇神经识别蛋白家族Dpr(21个成员)及其相互作用蛋白DIP(11个成员)开展了系统的计算与实验分析。这两类蛋白在结构和相互作用上已有较为充分的研究基础,并通过生物物理方法定量测定了结合亲和力,因而构成研究蛋白质界面特异性进化设计的理想体系。DIP与Dpr在发育中的神经系统中呈现细胞特异性表达,并优先形成特异性的异源结合网络,该网络与果蝇视网膜的突触特异性密切相关,提示其在神经模式建立中发挥关键作用。结构上,DIP和Dpr的胞外区域分别由三个和两个串联的Ig样结构域构成,其同源二聚体和异源二聚体均通过膜远端的Ig1结构域界面形成。
基于表面等离子共振实验结果,此前研究将DIP/Dpr划分为四个特异性亚群,而当前分析结合更强的异源结合偏好及序列相似性,将其扩展为七个亚群。值得注意的是,尽管不同成员之间序列同源性较高(DIP家族约>50%,Dpr家族约>40%,而两者之间约30%),其已解析的复合物结构界面几乎完全一致,空间叠合误差在1 Å以内。这引出了核心问题:在如此相似的序列与结构背景下,如何实现高度特异的相互作用?
**研究表明,“负约束”在这一过程中发挥核心作用,即界面中的某些氨基酸残基通过干扰非配对结合来实现选择性。**由于49种可能的DIP/Dpr组合中仅有7种表现出强结合,其余42种组合均受到负约束的限制。这些约束分布在约1900 Ų的Ig1–Ig1界面上,涉及DIP侧与Dpr侧各33个界面残基。虽然非界面残基也可能参与调控,但主要决定因素集中于界面区域。
在方法上,首先分析序列信息,发现其具有一定预测能力但仍不充分。随后构建所有可能DIP/Dpr复合物的同源模型,并利用已有结构计算界面突变的能量变化,评估不同氨基酸替换对结合自由能(ΔΔG)的影响。通过比较多种算法后,选择FoldX在准确性与计算效率之间取得最佳平衡。基于该方法,系统识别了各对组合中的负约束,并通过表面等离子共振实验对部分预测结果进行了验证。
结果显示,蛋白界面的大部分区域用于编码负约束,通过多种能量机制削弱非特异性结合,包括空间位阻、电荷排斥、埋藏电荷未满足以及极性基团不匹配等。不同亚群在界面上利用的约束位置和组合存在差异,通常需要多个负约束协同作用才能有效降低错误结合。此外,研究还讨论了特异性与亲和力之间的权衡关系,这一平衡对于形成多个特异性亚群至关重要。
总体来看,该研究揭示了在序列和结构高度相似的蛋白家族中,如何通过负约束实现精细调控的结合特异性,为理解多蛋白家族中相互作用特异性的进化设计提供了普遍性的理论框架。
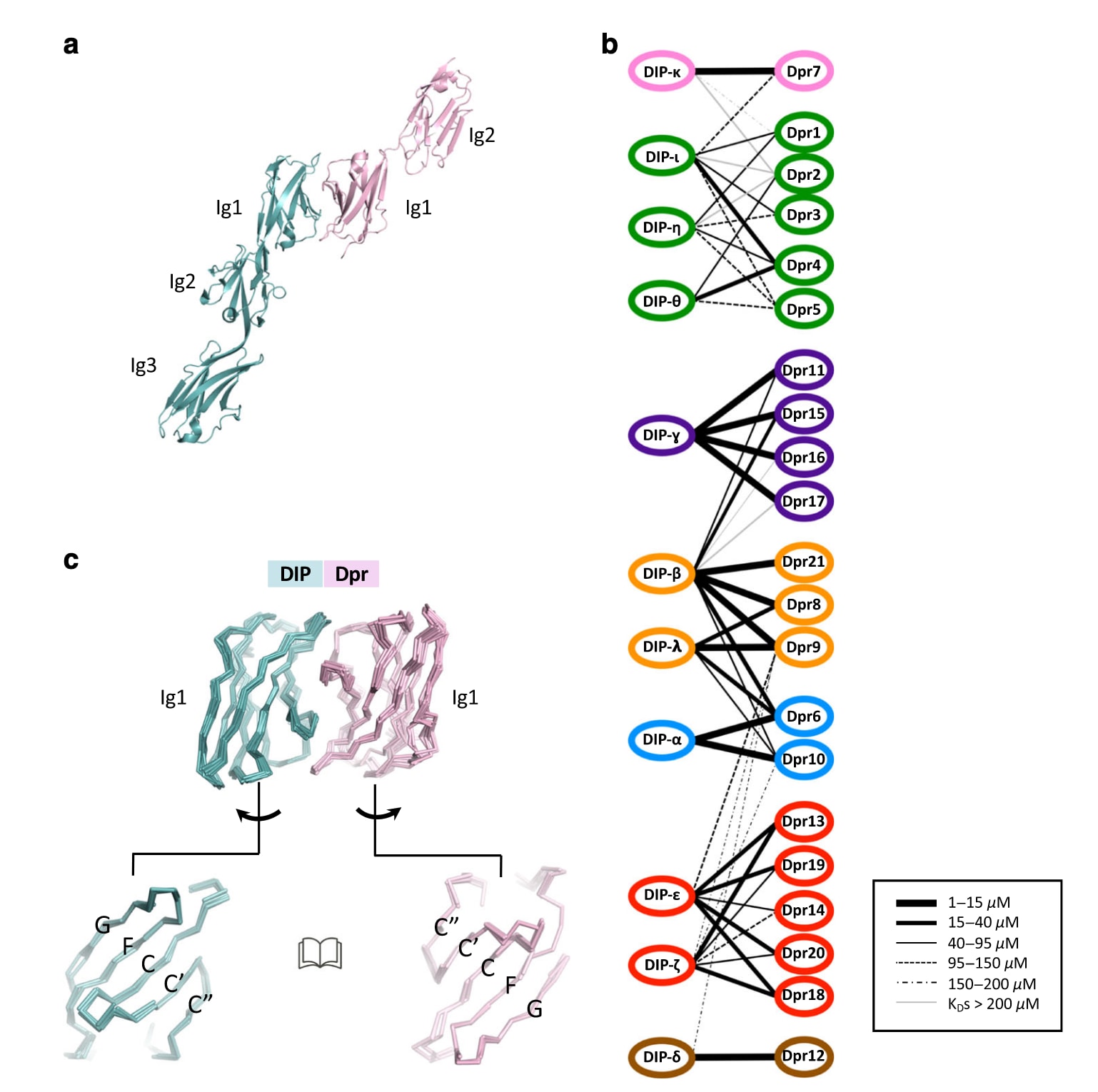
图1|DIP与Dpr的结构及其相互作用特性。 a 展示了DIP/Dpr异源二聚体的带状结构(PDBID: 6EG0),其中DIP以青色表示,Dpr以粉色表示。b 基于亲和力构建的果蝇DIP与Dpr相互作用网络,连线粗细表示对应蛋白对之间的解离常数
2 结果
2.1 DIP与Dpr的序列分析
图2展示了DIP和Dpr在昆虫中的同源蛋白界面残基的序列logo比对结果(具体方法见方法部分)。界面位点的定义基于已解析晶体结构中复合物形成前后每个残基可及表面积的变化,即当可及表面积发生非零变化时,该残基被视为界面位点。对于尚未解析结构的DIP/Dpr复合物,其界面位置通过多序列比对进行推断,依据与已知结构中界面残基的位置对应关系确定。文中统一采用家族范围的编号方式(i=1–33,标注于图2的序列logo上方),并在补充表1中给出了该编号与Uniprot编号之间的对应关系。
在这些界面位点中,有四个位置(图中以黄色标示)几乎完全由疏水性残基占据,共同构成DIP/Dpr相互作用界面的疏水核心。此外,还可以观察到两个高度保守的极性残基:在两个亚家族中均保守的谷氨酰胺(Gln)以及在DIP中保守的天冬酰胺(Asn)(图中以紫色标示)。这些残基通过形成埋藏的侧链-主链氢键,在Ig1–Ig1界面中起到“锁定”结构的作用。由于这些位点在不同亚群之间高度保守,它们本身并不能解释特异性差异,但其中个别疏水残基的具体类型变化仍可能对特异性产生影响。
相比之下,真正决定特异性的关键位点通常表现为:在至少一个亚家族中保持保守,但在不同亚群之间存在差异。基于这一原则,研究评估了多种基于序列的方法在识别七个特异性亚群决定性位点方面的能力,包括GroupSim、SDPpred、SPEER和Multi-Harmony。分析中使用的输入数据为仅包含界面残基的多序列比对,其中包括1732个DIP同源序列和2570个Dpr同源序列,并按七个特异性亚群进行划分。
综合四种方法的预测结果,当至少三种方法达成一致时,共识性识别出9个特异性决定位点;此外,还有14个位点至少被其中一种方法预测为潜在特异性位点(这些结果以灰色标注在图2的logo下方)。相比此前仅基于果蝇单一物种序列并通过人工观察获得的结果,该研究显著扩展了潜在特异性位点的范围。随后将这些基于序列的预测结果与基于能量的结构分析进行对比,以进一步解析其在特异性形成中的作用。
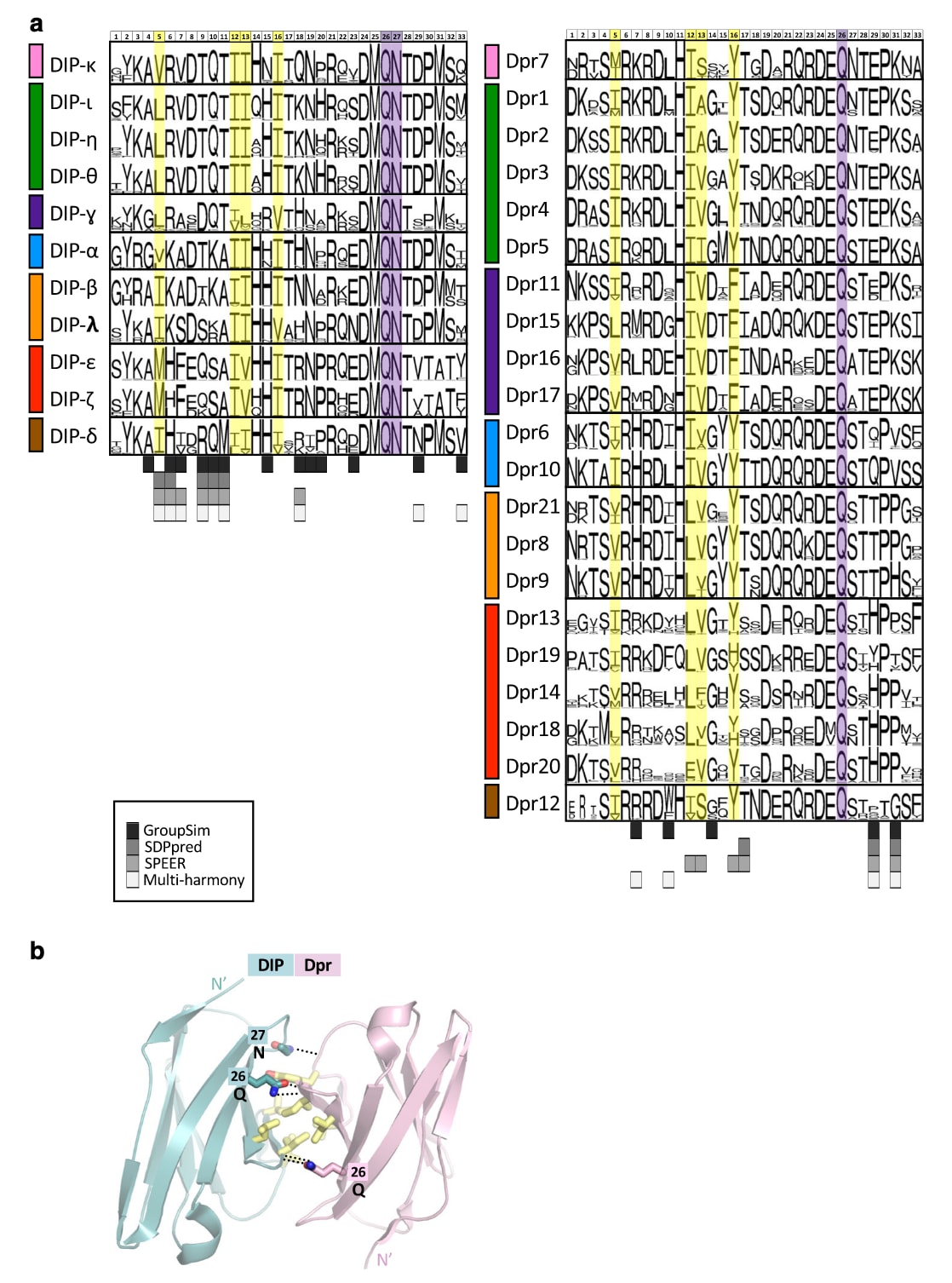
图2|基于序列的DIP/Dpr特异性位点预测及共同结合决定因素。 a 展示了DIP和Dpr在昆虫同源蛋白中界面残基的序列logo比对结果。图中logo下方以不同深浅灰色标注了多种序列分析方法(GroupSim、SDPpred、SPEER和Multi-Harmony)对特异性决定位置的预测结果,具体对应关系见图中标注框。每组logo左侧的色条表示对应的DIP/Dpr特异性亚群(参见图1b)。界面位点采用统一的家族编号(标注于logo上方),其与具体蛋白的Uniprot编号对应关系见补充表1。序列logo的构建方法详见方法部分。b 展示了DIP/Dpr二聚体Ig1–Ig1界面上的共同结合决定因素,以带状结构并辅以棒状表示。界面中的疏水性残基以黄色标示,构成结合的核心区域;保守的埋藏氢键以虚线表示。图中用方框标出的界面位置对应家族统一编号,并标注了这些位置上保守氨基酸的具体类型。
2.2 负约束的识别
基于前述分析,研究提出负约束是决定DIP/Dpr特异性分组的核心因素,并据此构建了一套计算策略用于系统识别与表征这些潜在约束。由于每个特异性亚群(如i和j)同时包含DIP和Dpr成员,可以将相关蛋白划分为四类:DIP-i、DIP-j、Dpr-i和Dpr-j。关键问题在于解释为何DIP-i不会与Dpr-j结合,以及反之DIP-j不会与Dpr-i结合。
分析从Dpr侧入手:由于Dpr-i能够与DIP-i结合,而Dpr-j不能,因此通过FoldX计算将Dpr-i的界面残基突变为Dpr-j对应残基,评估这些突变对Dpr-i与DIP-i结合能力的影响。如果某些突变显著削弱结合,则说明这些位点在Dpr-j中起到负约束作用。实验验证亦遵循相同逻辑,通过测定突变体Dpr-i与DIP-i的解离常数
具体计算流程如下:首先选取每个亚群的一个或多个DIP/Dpr复合物结构作为模板;(1)将复合物中DIP一侧的每一个界面残基突变为其他亚群DIP在相同位置出现的所有氨基酸;(2)计算每种突变对其与对应Dpr结合自由能变化
在定义负约束时,首先采用基于能量的筛选标准:若某一界面位点在突变后对所有模板复合物均表现出一致的去稳定化作用(即显著提高
需要强调的是,能量筛选基于果蝇蛋白结构的FoldX计算结果,而进化筛选则基于多物种同源序列比对,二者相互补充。部分预测结果随后通过实验进行了验证。最终,在42种不发生相互作用的DIP/Dpr亚群组合中均识别出了对应的负约束,系统揭示了其在特异性形成中的关键作用。
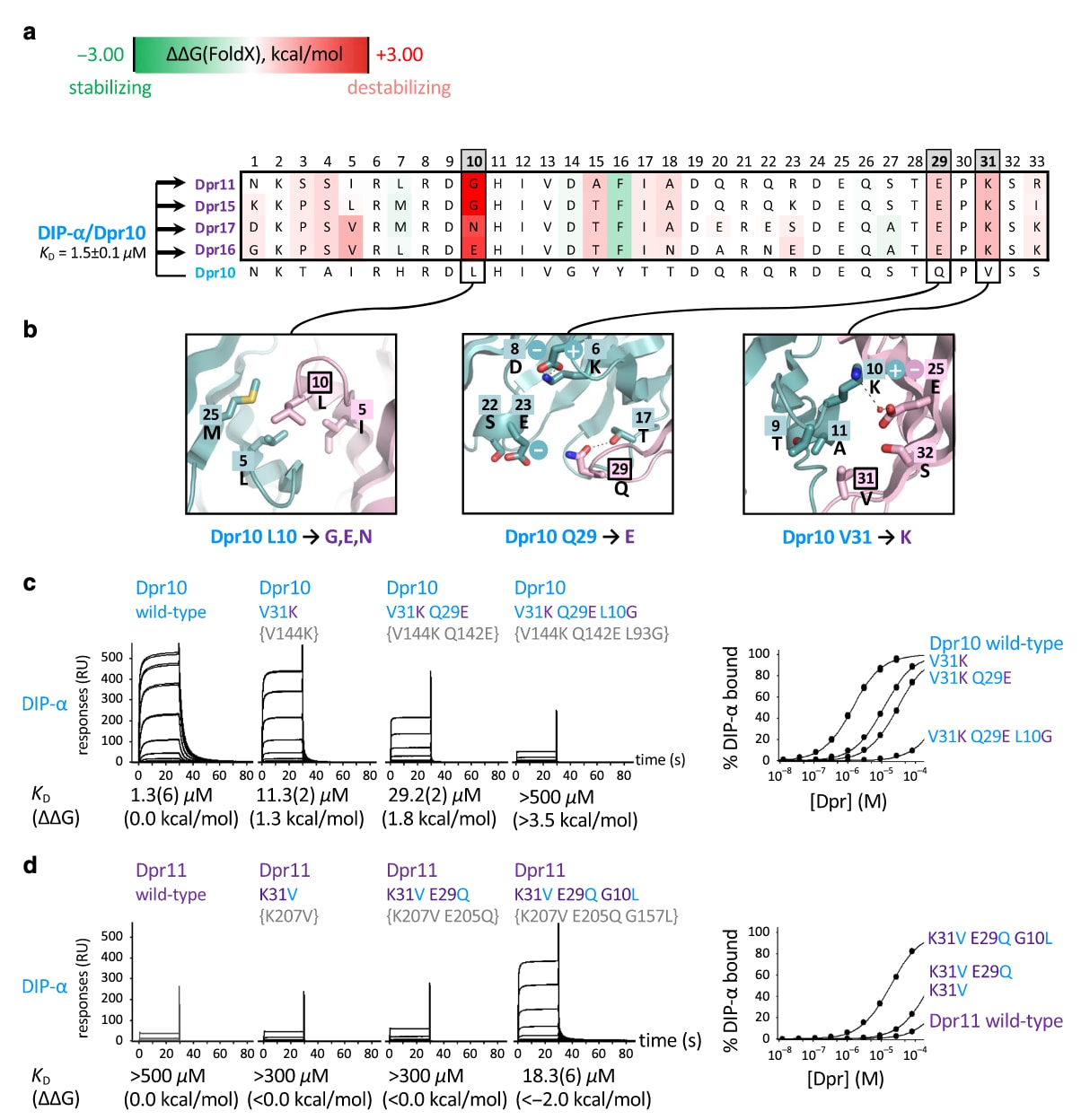
图3|紫色亚群Dpr11/15/16/17上的负约束阻止其与蓝色亚群DIP-α结合。 a 展示了四个紫色亚群Dpr与Dpr10在界面残基上的序列比对。各残基根据计算得到的
2.3 ΔΔG计算方法的评估
研究评估了六种用于预测单点突变对蛋白结合亲和力影响(即
评估数据集包含25个实验测得的
结果表明,在该数据集上FoldX、MutaBind和Rosetta flex ddG表现最佳,具有较高的Pearson相关系数(PCC)。其中MutaBind整体表现最优,但其无法正确识别稳定性增强的突变(补充表2中以绿色标示),这一问题在所测试的所有机器学习方法中普遍存在,可能源于训练数据中去稳定化突变的过度代表。此外,基于物理力场的方法在结构解释方面更具优势。
综合考虑计算速度、预测准确性(FoldX的PCC为0.57)以及结果的可解释性,最终选择FoldX作为后续识别负约束的主要计算工具。
2.4 蓝色与紫色亚群之间的负约束
研究以蓝色亚群的Dpr10/DIP-α与紫色亚群的Dpr11/DIP-γ为典型体系展开深入分析,因为这两类相互作用已在果蝇中枢与外周神经系统中被证明具有明确的功能表型。围绕不同亚群之间无法结合的原因,从四个方面系统解析了负约束的分布与作用:(a)紫色亚群Dpr上的负约束;(b)蓝色亚群DIP-α上的负约束;(c)蓝色亚群Dpr上的负约束;(d)紫色亚群DIP-γ上的负约束。
首先,针对紫色亚群Dpr,通过将蓝色亚群Dpr(如Dpr10)的界面残基逐一突变为紫色亚群对应残基,并计算其与DIP-α结合的
其次,在蓝色亚群DIP-α中识别出第6、9、11、15和22位为负约束位点,其中第9位对结合影响最显著。结构分析表明,这些位置在紫色亚群DIP-γ中通常为带电残基,能够与Dpr形成离子对,而在DIP-α中则为中性极性残基,从而破坏离子配对并引入未满足的埋藏电荷。
第三,在蓝色亚群Dpr中,第7、14、16、29和31位被预测为削弱其与紫色亚群DIP-γ结合的负约束位点。实验验证显示,第31位突变即可显著降低结合亲和力,而与第29位的双突变则使
最后,在紫色亚群DIP-γ中,第9、10和16位被鉴定为对蓝色亚群Dpr具有负约束作用。实验测试表明,在对应Dpr位置引入突变(如K10Q和A11T)均会使结合亲和力下降约一个数量级。其物理基础同样可以从结构中理解:例如第10位突变破坏了原有的水介导盐桥,而第11位突变则引入空间冲突。
总体来看,蓝色与紫色亚群之间的特异性主要由多个分布在界面不同位置的负约束共同决定。这些约束通过多种物理机制(疏水不匹配、电荷不满足、空间位阻等)协同作用,有效阻止非特异性结合的发生。
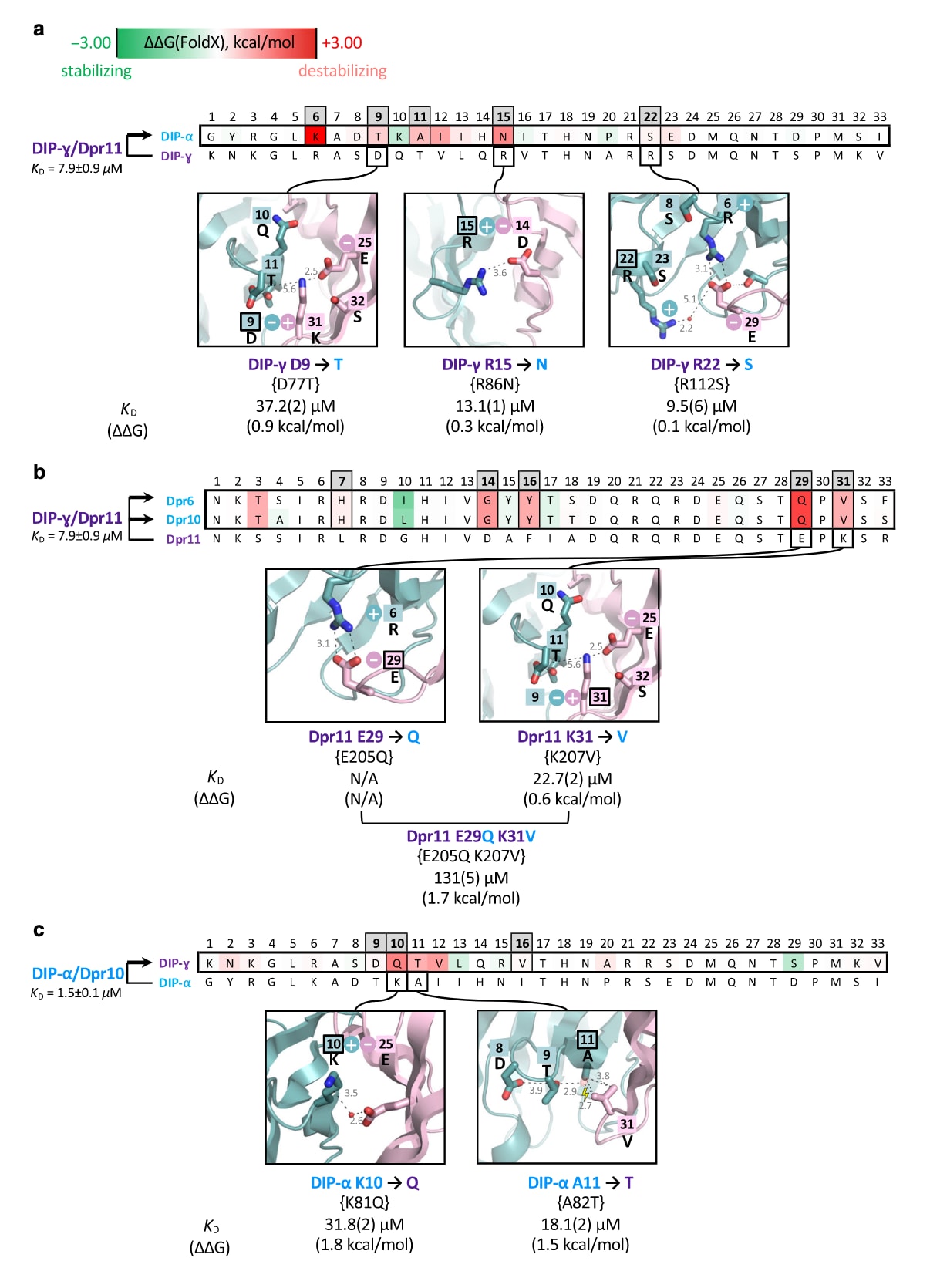
图4|DIP-α、蓝色亚群Dpr6/10以及DIP-γ上的负约束抑制紫色与蓝色亚群之间的相互作用。 a 展示DIP-α上的负约束。首先给出DIP-α与DIP-γ界面残基的序列比对,随后结合结构细节解释这些位点如何产生负约束效应。左侧标注了基于五次独立实验测得的野生型DIP-γ/Dpr11相互作用的平均
2.5 蓝色与绿色亚群之间的负约束
此前研究曾通过突变实验尝试改变结合偏好,例如将蓝色亚群Dpr6的结合对象从其天然配对DIP-α切换至绿色亚群DIP-η,以及将绿色亚群Dpr4的结合偏好从DIP-η转向DIP-α。相关SPR实验数据与当前FoldX计算结果的对比见补充表3。结果显示,在Dpr6中第7和31位的突变会削弱其与DIP-α的结合能力,而在Dpr4中对应位置的反向突变则增强其与DIP-α的结合。这些实验结果与FoldX预测高度一致,表明蓝色亚群Dpr中的第7和31位残基构成了削弱其与绿色亚群DIP结合的负约束。
另一方面,实验数据还表明,在绿色亚群Dpr4中,仅第31位(而非第7位)构成负约束,削弱其与自身配对蛋白DIP-η的结合,同时该位点的突变也会增强其与非配对蓝色亚群DIP-α的结合能力。这些结果同样与FoldX计算预测相符,进一步验证了负约束在不同亚群之间的非对称分布特征。
从整体角度来看,图5展示了多个不发生相互作用的亚群在假想界面上的负约束分布情况。结果表明,蛋白质界面的大部分区域均被用于编码负约束,但不同亚群利用的具体区域存在差异。总体而言,在界面上共识别出Dpr侧21个、DIP侧17个负约束位点(见图6中灰色标注位置)。这些位点在界面中的埋藏程度不同,分布于环区和二级结构中,并涵盖多种理化性质,包括疏水性、极性和带电残基。其中多数负约束位于环区,且主要由极性或带电残基构成(共21个位点),其中有10个位点在复合物形成时埋藏的可及表面积超过50 Ų。
对于每一对非配对的DIP/Dpr亚群,通常需要约7个界面残基共同作用才能有效破坏结合稳定性。这些负约束的物理来源主要包括疏水核心中的形状不匹配(如空间位阻或空腔形成)、静电效应(如库仑排斥或埋藏未满足电荷/极性基团带来的去溶剂化惩罚),或二者的共同作用(见图5示例)。实验结果进一步表明,单一负约束通常不足以完全消除结合,至少需要两个或以上的负约束协同作用,才能有效阻止不同亚群之间形成稳定复合物。
与基于序列的方法相比,结构与能量驱动的预测不仅能够覆盖所有已识别的特异性位点,还额外发现了约两倍数量的潜在负约束位置。序列方法往往难以识别在亚群内部不完全保守的位点,也难以检测仅在单一亚群中具有特异性的残基。例如,第14和16位在紫色亚群Dpr中分别为保守的Asp和Phe,而其他亚群对应位置则为Gly和Tyr。此外,研究还预测了一些位于疏水核心的残基(如Dpr中的第12、13、16位以及DIP中的第5、13、16位)为潜在负约束,但这些位点在序列方法中大多未被识别。需要指出的是,这些新增预测位点尚未在该研究中得到实验验证。
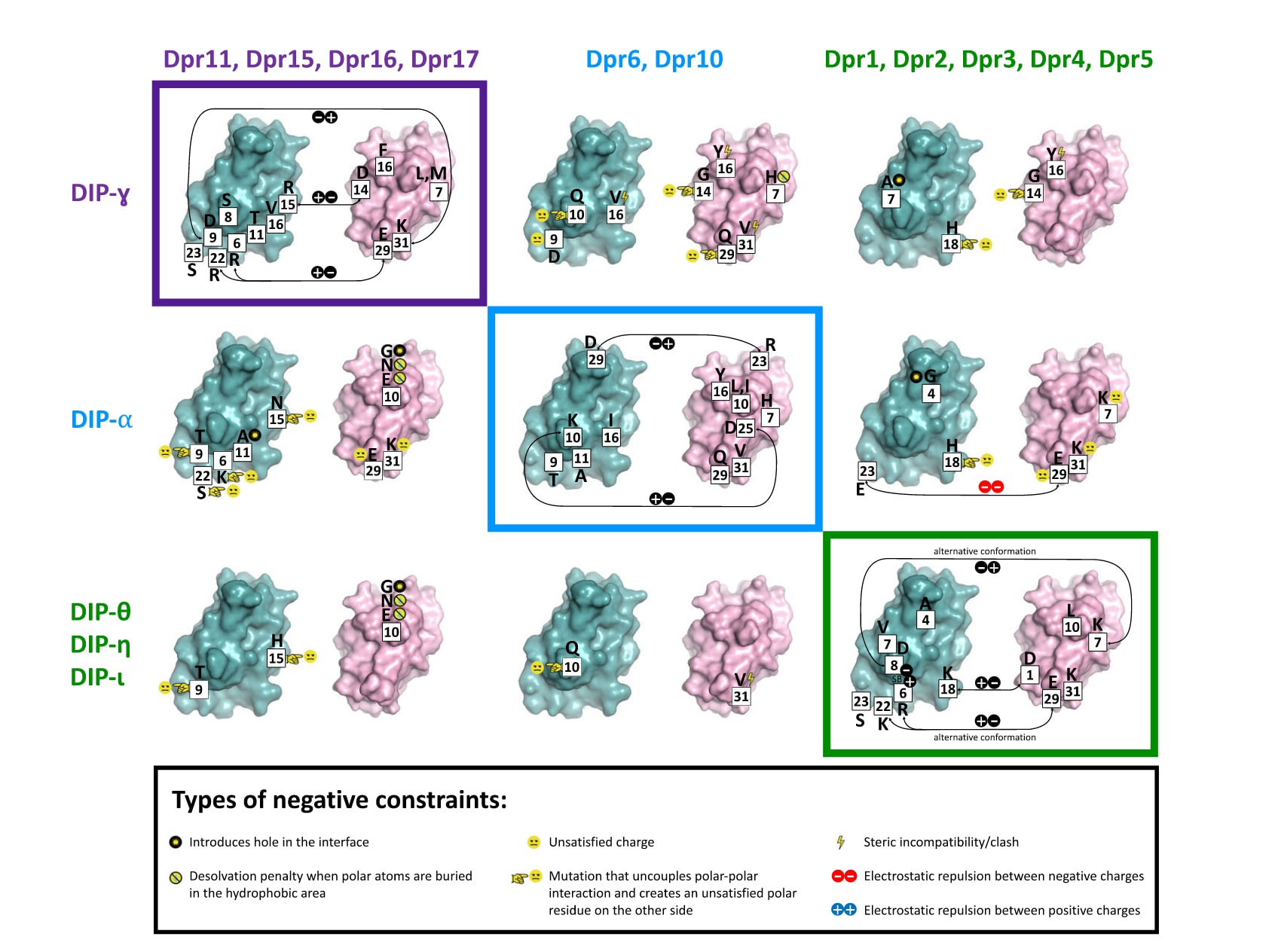
图5|非配对DIP/Dpr亚群中的负约束分布。 图中以“翻书式”展开方式展示了紫色、蓝色和绿色亚群在假想相互作用界面上的负约束位置,其中DIP以青色表示,Dpr以粉色表示。各亚群的天然配对组合以带有对应颜色边框的方框标出。界面位点采用统一编号(白色实心方框标示),每个负约束位点的氨基酸类型以单字母形式标注。不同负约束的物理来源通过图中下方图例中的符号进行说明。
2.6 亲和力与特异性的平衡
在部分界面位点上,FoldX预测如果将残基替换为其他亚群中出现的氨基酸,反而可能提高结合亲和力。这说明DIP/Dpr界面的设计目标并不只是最大化配对结合强度,而是在亲和力与特异性之间进行权衡。
一个典型例子是紫色亚群Dpr中的第10位。该位置通常为甘氨酸或极性残基,根据FoldX预测,这类残基会削弱其与其他亚群DIP之间的非极性相互作用。SPR实验验证了这一点:在Dpr10中引入L10G突变后,其与DIP-α和DIP-β的结合均明显减弱,对应的
另一个例子是第7位残基。研究预测,绿色亚群Dpr通过在该位置引入一个未满足电荷K7,来削弱其与非配对DIP-α之间的结合。实验结果支持这一结论:在Dpr6中引入H7K{H110K}突变后,其与天然配对DIP-α的结合减弱超过
总体来看,这些结果表明DIP/Dpr家族在进化过程中并非单纯追求最强结合,而是在维持足够天然亲和力的同时,通过牺牲部分结合能来强化对非天然配对的排斥。正是这种亲和力与特异性之间的精细平衡,使得多个高度相似的蛋白成员能够在同一体系中实现准确而稳定的识别。

图6|DIP/Dpr各亚群中负约束位点的预测分布。 图中基于昆虫同源序列构建了序列logo,其中被识别为负约束的残基以灰色标示。不同亚群采用与图1b一致的颜色编码。每个界面位置(1–33)在logo上方标注,并对应其所在的二级结构类型,具体说明见图中标注框。此外,在logo下方给出了基于序列的方法(GroupSim、SDPpred、SPEER和Multi-Harmony)对特异性决定位置的预测结果(与图2a一致),并按照图中标注框中的说明进行展示,用于与结构与能量分析结果进行对比。
3 讨论
研究系统阐明了DIP与Dpr如何通过结构与能量层面的机制划分为彼此正交的特异性亚群,这一划分基于SPR测得的结合亲和力。以往针对II型钙黏蛋白、nectin以及DIP/Dpr的研究多依赖结构辅助下的序列观察来识别特异性位点,而当前工作则采用了更加系统和定量的方法,以应对该问题的复杂性。核心问题在于:在49种可能的DIP/Dpr亚群组合中,为何只有7种能够形成强相互作用。由于每个亚群在Dpr侧包含1–5个成员、在DIP侧包含1–3个成员,需要同时解释其余42种组合为何不发生或仅发生极弱相互作用,这一问题尤为复杂。
结果表明,单纯依赖序列方法只能提供部分解释。通过构建大量可能形成或不形成复合物的同源模型,并结合FoldX计算、SPR实验以及结构分析,系统识别了“哪些因素阻止了复合物形成”。最终共鉴定出38个负约束位点,并在其中8个位点上通过23组SPR实验进行了验证。此外,这些预测结果与此前关于蓝色与绿色亚群的实验数据一致。值得注意的是,这些负约束主要通过局部强烈的不利能量贡献实现,而非依赖分布于多个残基的微弱效应。
从物理本质来看,负约束的来源包括:将离子对中的残基替换为相反电荷引发库仑排斥;用中性残基替代带电残基导致埋藏未满足电荷;以及在疏水核心中引入过大或过小的氨基酸,分别造成空间冲突或空腔,从而削弱疏水相互作用。由于需要区分多达42种非配对组合,这些机制并不会简单重复使用,而是在不同亚群组合中以独特方式组合,这也是为何界面大部分区域都被用于编码负约束。实验结果进一步表明,通常需要至少三个负约束的协同变化才能实现特异性的“切换”。
从亲和力角度来看,亚群内结合的
进一步分析表明,界面中超过一半的残基(66个界面残基中的38个)在不同昆虫物种中被用于编码负约束,但这些位点主要在非天然配对中发挥作用;而在天然复合物中,大多数界面残基仍承担稳定结合的功能。从进化角度推测,早期的DIP/Dpr复合物可能主要依赖疏水核心和外围稳定相互作用,随着家族成员扩展,通过在界面广泛引入负约束实现特异性分化。这种策略很可能在其他大型蛋白家族中同样存在。
与其他黏附蛋白家族相比,DIP/Dpr表现出亚群内一定程度的结合“宽容性”,而Dscam和簇状原钙黏蛋白则表现为严格同源识别,依赖多结构域界面实现更精细的调控。关于负约束在这些体系中的具体作用仍有待进一步研究。
最后,不同结合强度在生物学中的意义仍不完全明确,尤其是在细胞同时表达多个家族成员或多个亚群成员的情况下。通过设计具有不同亲和力和特异性的突变体,并结合基因编辑技术,可以进一步探索分子层面的结合特性如何影响细胞间识别与功能。这一方向有望揭示蛋白质相互作用网络与细胞行为之间的直接联系。