ACS Catal. 2026 | 计算机辅助定向进化实现了立体选择性羰基还原酶活性、热稳定性与选择性的协同优化
今天介绍的是发表在 ACS Catalysis 上的一项关于计算机辅助定向进化的研究。该工作聚焦于羰基还原酶多功能协同优化这一长期难题,尝试同时提升酶的催化活性、立体选择性与热稳定性。研究将上位性模型、保守性分析和

获取详情及资源:
0 摘要
同时提升活性、底物特异性、立体选择性和热稳定性,仍然是实验室酶定向进化中的核心挑战。具有工业应用价值的酶,通常需要多种功能的协同优化。该研究开发了一条整合序列—结构信息的计算流程,用于调控羰基还原酶的活性、热稳定性和立体选择性。该框架采用无监督上位性模型来描绘整个蛋白结构中残基之间的相互依赖关系,并结合
1 引言
酶催化生物制造具有“绿色化学”所强调的可持续性特征。随着可再生能源生产和精细化学品需求的不断增长,生物催化的发展显得愈发重要。面向工业应用的生物制造过程,通常要求酶在活性、稳定性、选择性和普适性等多种性质上实现协同增强。引入突变是改变蛋白质功能的有效手段。围绕特定性质的优化,已经发展出多种蛋白质工程策略。根据目标性质的不同,蛋白质工程往往聚焦于不同的改造区域。对于提升催化活性和选择性的大多数策略而言,活性中心始终是最受关注的区域。然而,那些有助于催化或配体结合的蛋白残基,往往并不是最有利于蛋白稳定性的位点。基于蛋白深度突变扫描的研究发现,大量突变会导致蛋白稳定性下降,而表面残基的突变平均而言比核心位置的突变破坏性更小。因此,想要同时实现热稳定性、活性和选择性的协同调控,始终十分困难。为了维持功能与稳定性之间的精细平衡,相比只关注某一局部区域,对蛋白全序列进行整体修饰显得更加必要。
随着实验室进化中大量突变体的构建及其功能表型的系统描绘,序列、表型与适应度之间关系的认识不断加深。适应度本身是一个多维参数。对于蛋白而言,适应度与其生理功能能力密切相关,其中溶解性、稳定性以及活性,例如
酶向特定功能演化通常需要多个适应性突变的逐步累积,但引入过多突变又会导致蛋白适应性下降。两个或更多有益突变的重组,并不一定能够产生性能更优的酶变体。例如,基于深度学习方法DLKcat,研究者曾在β-酮硫裂解酶的底物通道中获得多个单点突变体,但后续迭代得到的多突变体活性均未超过单突变体Tfu0875L163H。因此,除了高效识别单个有益突变之外,如何合理组合这些突变并实现有益效应的累积,对于理解酶进化以及设计和改造新型酶都至关重要。
羰基还原酶是一类依赖NAD(P)H的氧化还原酶,能够催化前手性酮的不对称还原,生成手性醇。此类产物广泛用作活性药物成分、农用化学品和精细化学品合成中的关键结构单元。由于具有较高的立体选择性和温和的反应条件,羰基还原酶被视为很有前景的绿色生物催化剂。然而,天然羰基还原酶往往存在催化效率较低、底物范围狭窄或热稳定性有限等问题,从而限制了其直接工业应用。短链脱氢酶/还原酶、中链脱氢酶/还原酶以及醛酮还原酶等家族活性中心中的常见热点位点已经被识别,相关关键突变也被设计用于获得目标性质。尽管这些围绕活性位点展开的方法已经取得了显著成功,但往往也面临功能权衡这一内在挑战。此前基于共进化分析和结构位置聚类识别关键远端位点的策略,虽然能够在保持底物多样性的同时提升活性,但该方法需要构建并筛选大规模突变文库,大约9000个突变体,而且未能同时改善多种酶学性质。因此,开发更系统、更具预测性的工程化策略,以满足羰基还原酶多功能协同优化的需求,仍然是酶设计中的关键研究目标。
该研究选择来源于副拟假丝酵母的羰基还原酶CpRCR作为模板。该酶能够催化多种高价值手性羟基化合物的选择性合成,但其底物范围和稳定性仍然受限。2-乙酰基吡啶的不对称还原产物(R)-1-(吡啶-2-基)乙醇,是抗组胺药贝泊他斯汀苯磺酸盐合成中的关键中间体,该药物可用于治疗鼻炎、荨麻疹等过敏性疾病。因此,为了调控CpRCR的酶学性质,尤其是其对2-乙酰基吡啶的活性和立体选择性,研究中采用EVmutation对全序列进行虚拟深度扫描,预测不同突变的影响。随后结合相对折叠自由能
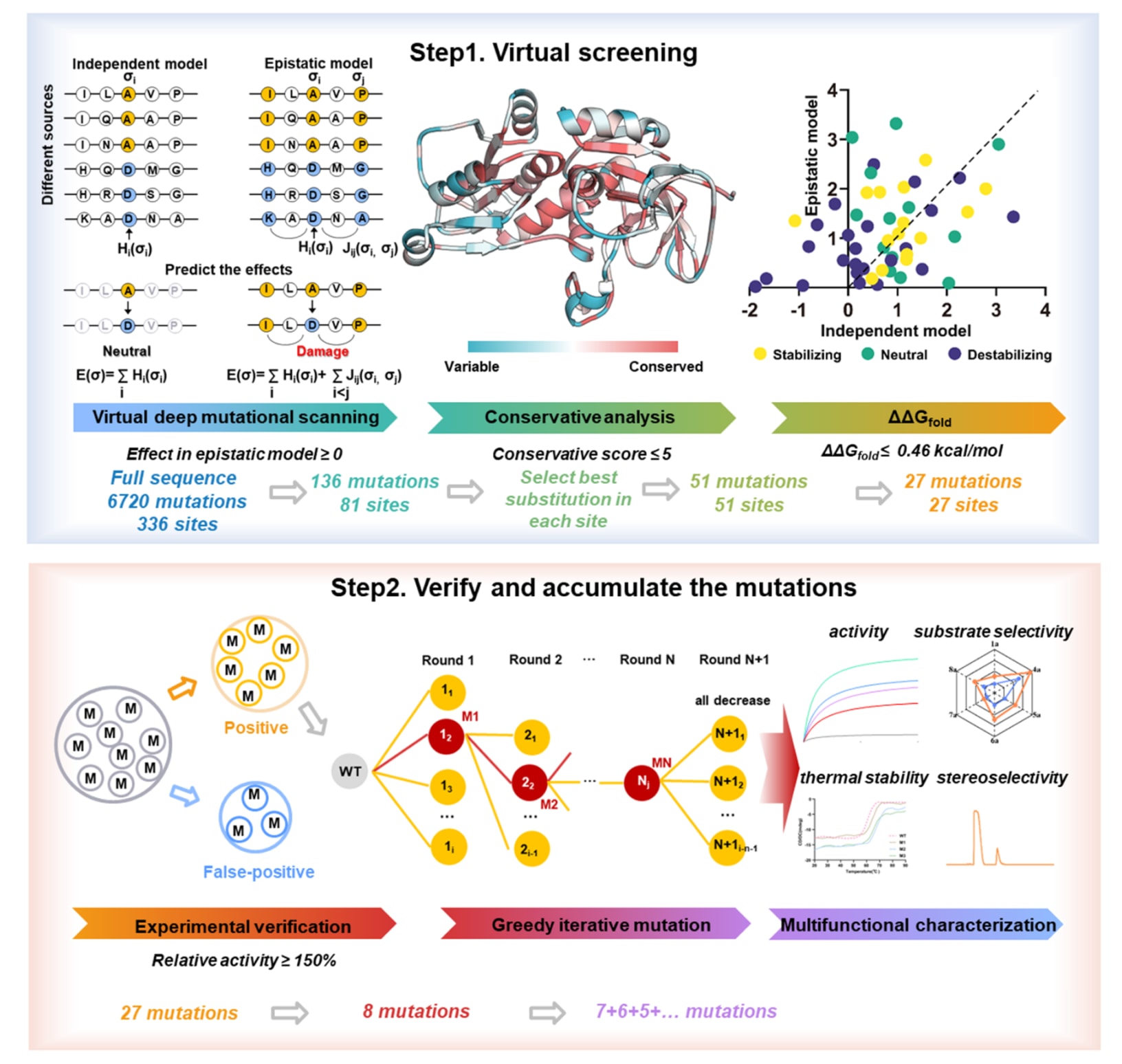
图1|全局进化工程策略示意图。 首先,利用上位性模型对CpRCR的全序列进行深度突变扫描。随后,结合保守性分析和
2 结果与讨论
2.1 基于全局进化策略的计算重设计
面向生物制造的工业应用,通常要求酶的活性、热稳定性、选择性和普适性等多种功能实现协同增强。然而,实验室蛋白质工程中的进化过程,往往是在改善目标性质的同时牺牲其他性质。因此,该研究建立了一种面向多功能协同进化的进化策略。全局进化策略如图1所示。候选突变通过三步计算筛选流程获得。首先在突变热点设计阶段,利用上位性模型对CpRCR全序列进行虚拟饱和突变扫描,预测每一种氨基酸替换对应的适应度图谱。这里的适应度是一种综合性质,能够反映催化活性、结构稳定性和表达水平等多方面的贡献,而不仅仅是避免有害替换。预测适应度为正的突变被保留下来,作为潜在热点位点。值得注意的是,EVmutation对实验活性的预测能力已经在ProteinGym基准测试中得到定量评估,其与实测活性的Spearman相关系数达到0.44,在所测试模型中位居前列。这说明,预测适应度更高的变体,更有可能具有改良的催化性质。
具体而言,首先通过搜索UniRef90数据库获得对应蛋白家族的同源序列。为了控制不同来源序列带来的进化发散,研究中使用长度归一化bit score评估序列相似性,并剔除了缺口比例超过30%或长度不足目标序列50%的片段序列,从而得到非冗余蛋白序列。随后,基于序列独特性进行序列重加权,以维持系统发育采样的均匀性。借助EVmutation,对149669条比对后的同源序列统计分析了两类进化约束:其一是残基对之间氨基酸相互依赖关系所体现的成对约束;其二是反映位点依赖性进化保守性的位点特异性约束。上位性模型建立在对同源序列相应位置氨基酸替换的分析之上。那些对酶功能有正向影响的氨基酸残基,在自然选择过程中会被优先保留下来。与此同时,该模型还进一步纳入了残基间相互依赖,也就是氨基酸相互作用效应的影响。通过这种设计,能够在进化过程中尽量避免功能缺失型突变。最终,在上位性模型中突变效应大于0的突变被选为潜在热点,共得到81个位点上的136个正向突变。其中,27、52、61、79、132和184位点均有5种或以上氨基酸替换对蛋白适应度产生正面影响。对于那些存在多个正向替换的位点,研究中选择上位性得分最高的替换作为下一轮筛选的最优突变。例如在61位点,共有Y61F、Y61L、Y61M、Y61R和Y61T五种候选替换被预测为适应度增加,其中Y61F的EVmutation得分最高,达到+0.841,因此被选中。这个分值反映的是在上位性耦合模型下的预测适应度优势,该模型同时考虑了位点特异性约束和整个蛋白家族范围内的残基成对耦合关系。按照这一流程,对全部81个正向位点完成筛选,最终得到81个突变体进入下一轮分析。
氨基酸保守性与其功能同样密切相关。在进化过程中,保守位点的突变效应通常比非保守位点更加显著,在这些高度保守位点引入突变更容易导致失活。此外,自然进化中越保守的位点,其突变耐受性通常越低。高度保守位点上的氨基酸替换往往只允许少数特定氨基酸。因此,研究进一步采用保守性评分不高于5作为条件,对潜在突变热点进行筛选,最终得到51个潜在突变热点。
蛋白质工程中的一个关键难题在于如何组合正向突变。多个有益突变在组合之后,往往反而会得到性能较差的突变体,这极大限制了更优酶的开发潜力。为此,研究通过分析
经过上述筛选,最终得到27个单点突变体用于实验验证。其中只有5个位点位于辅酶结合结构域,其余22个位点分布在催化结构域中。它们在空间结构上较为分散,并未集中于某一特定区域。此外,研究选用2-乙酰基吡啶作为模型底物验证突变体活性,因为其手性羟基化产物是合成手性药物、精细化学品和农用化学品的重要中间体。实验验证结果表明,27个预测突变体中有22个表达良好且活性提高,并且没有任何一个预测突变体出现严重失活。随后,从中选出8个粗酶液活性达到野生型1.5倍以上的突变体,作为后续迭代突变的基础。这些位点除N266外均远离催化三联体,其中I79距催化三联体甚至超过30Å。更值得注意的是,这些位点几乎都位于蛋白表面。已有研究指出,将突变设计聚焦于暴露在溶剂中的表面残基,是一种克服稳定性与活性权衡的有效策略,因为表面残基通常比埋藏在蛋白核心中的残基更能耐受突变。这8个突变在上位性模型中的突变效应得分均高于0.8,其中I51L、I79K、D147E和N266K四个突变的得分甚至高于2,超过大多数突变热点。这进一步验证了该策略的有效性,尽管筛选活性提升时采用的是一种非天然底物。
研究还进一步测定了所有筛选变体的对映体过量值ee。由于所有突变都位于远离活性口袋的位置,因此它们对立体选择性的影响有限。各突变体基本保持了与野生型相当的高对映选择性。这一结果与既往研究一致,即远端突变通常对对映选择性的影响较弱。
在此基础上,研究采用贪婪导向的迭代组合突变策略获得最优突变体。首先选取粗酶液活性提升最高且具有高立体选择性的D147E作为模板继续组合其他突变。D147E的活性较野生型提高了4.44倍。在第二轮中,构建了7个双突变体,分别为Q15N/D147E、K19E/D147E、I51L/D147E、Y61F/D147E、I79K/D147E、D147E/V246L和D147E/N266K。第二轮迭代的粗酶液活性结果如图2B所示。其中,Y61F/D147E表现出最高酶活,达到野生型的7.27倍。第三轮迭代则继续以Y61F/D147E为基础叠加其他突变,直到下一轮不再出现活性提升为止。经过四轮筛选,总共实验检测了45个突变体,即27+7+6+5个,最终获得了一系列优势突变体:D147E(M1)、Y61F/D147E(M2)以及I51L/Y61F/D147E(M3)。在整个进化过程中,所有突变体始终保持活性,避免了严重功能丧失,且其中大多数相较于野生型均为正向突变。该策略所需实验工作量较小,仅筛选了45个突变体,但单点突变和迭代突变中活性高于野生型的成功率分别达到81%(22/27)和100%(18/18)。这表明,全局进化工程能够高效且稳定地获得可迭代累积的正向突变体。进一步测定迭代过程中所有突变体的ee值发现,所有变体都保持了与野生型相近的高对映选择性,说明这一迭代进化过程在提升催化活性的同时,并未牺牲立体选择性。
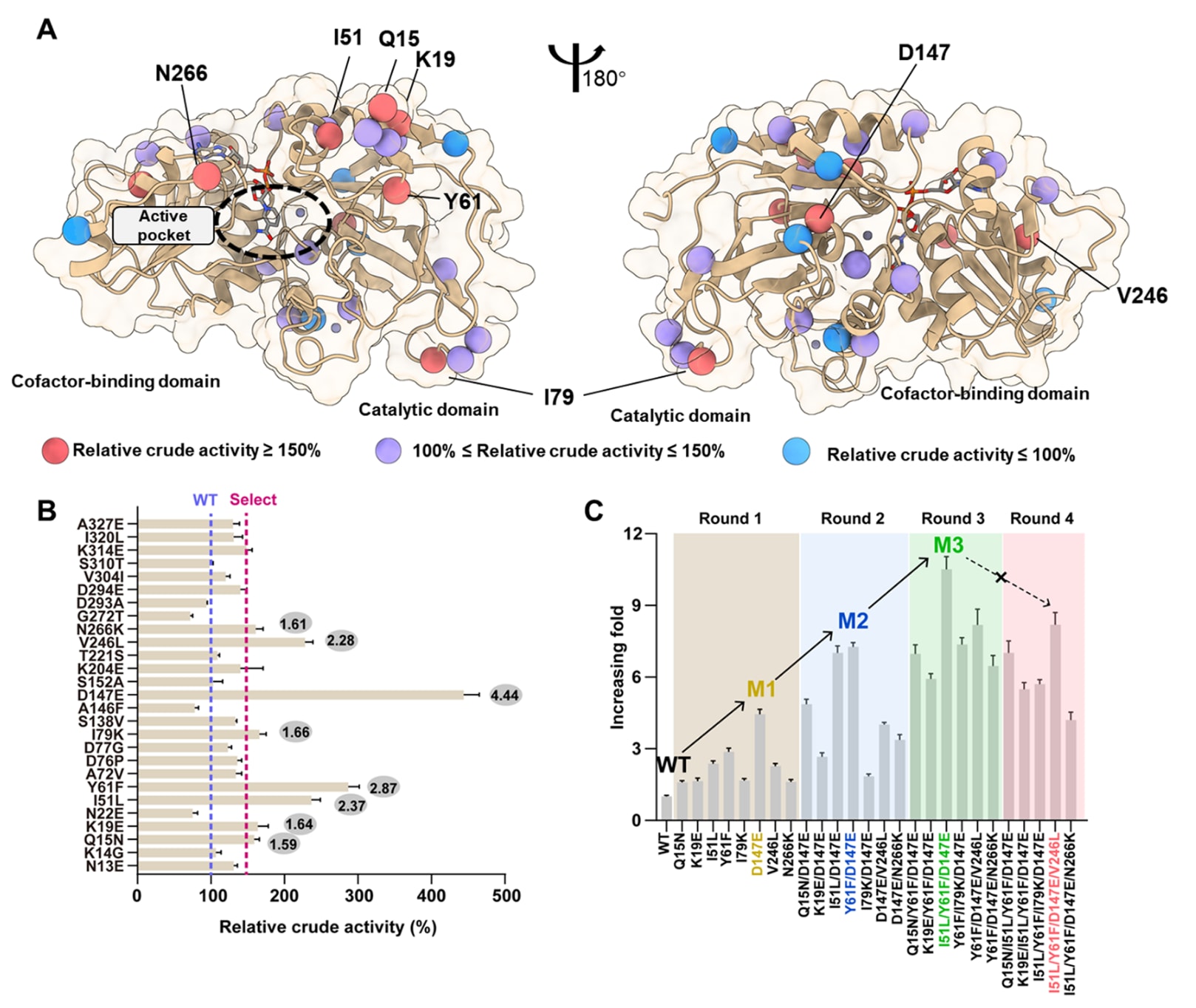
图2|基于全局进化策略、以2-乙酰基吡啶为目标底物的蛋白质工程热点预测。 (A) 通过全序列进化分析预测得到的27个突变热点在蛋白结构中的分布。(B) 对27个热点位点进行筛选扫描。(C) 对正向突变体进行迭代组合突变。
2.2 多功能性质分析
为了评估该策略在多种功能性质,包括活性、立体选择性和热稳定性方面的可行性,研究进一步将所有获得的有益突变体M1至M3纯化至均一状态,并测定了它们针对一系列结构特征差异较大的羰基化合物底物的动力学参数、对映体过量值ee以及
对于大多数底物而言,仅引入D147E突变的M1相较于WT表现出最显著的
这些在全局进化工程引导下获得的稳定型醇脱氢酶突变体,能够高效催化多样底物的不对称还原。研究进一步评估了M1、M2和M3在合成与药物生产相关的手性醇中的应用可行性。之所以选择A6、H3和H1作为200mL放大反应的底物,是因为这些底物对应的突变体催化效率提升最为显著。由于CpRCR本身同时具有氧化和还原功能,因此在辅酶循环过程中只需加入廉价的异丙醇作为共底物,而无需额外补加大量昂贵的NADH。结果显示,对于A6(200mM)、H3(500mM)和H1(500mM),在6小时内转化率分别达到99%、98%和99%(图S2)。此外,产物几乎都为单一构型,其ee值分别为A6的99%(R)、H3的99%(S)和H1的94%(R)。经过分离纯化后,得到手性醇(R)-A6b5.23g,对应收率96%;(S)-H3b9.48g,对应收率77%;以及(S)-H1b9.73g,对应收率79%。这些结果进一步表明,工程化突变体在合成与药物制造相关的手性醇方面具有良好应用前景。
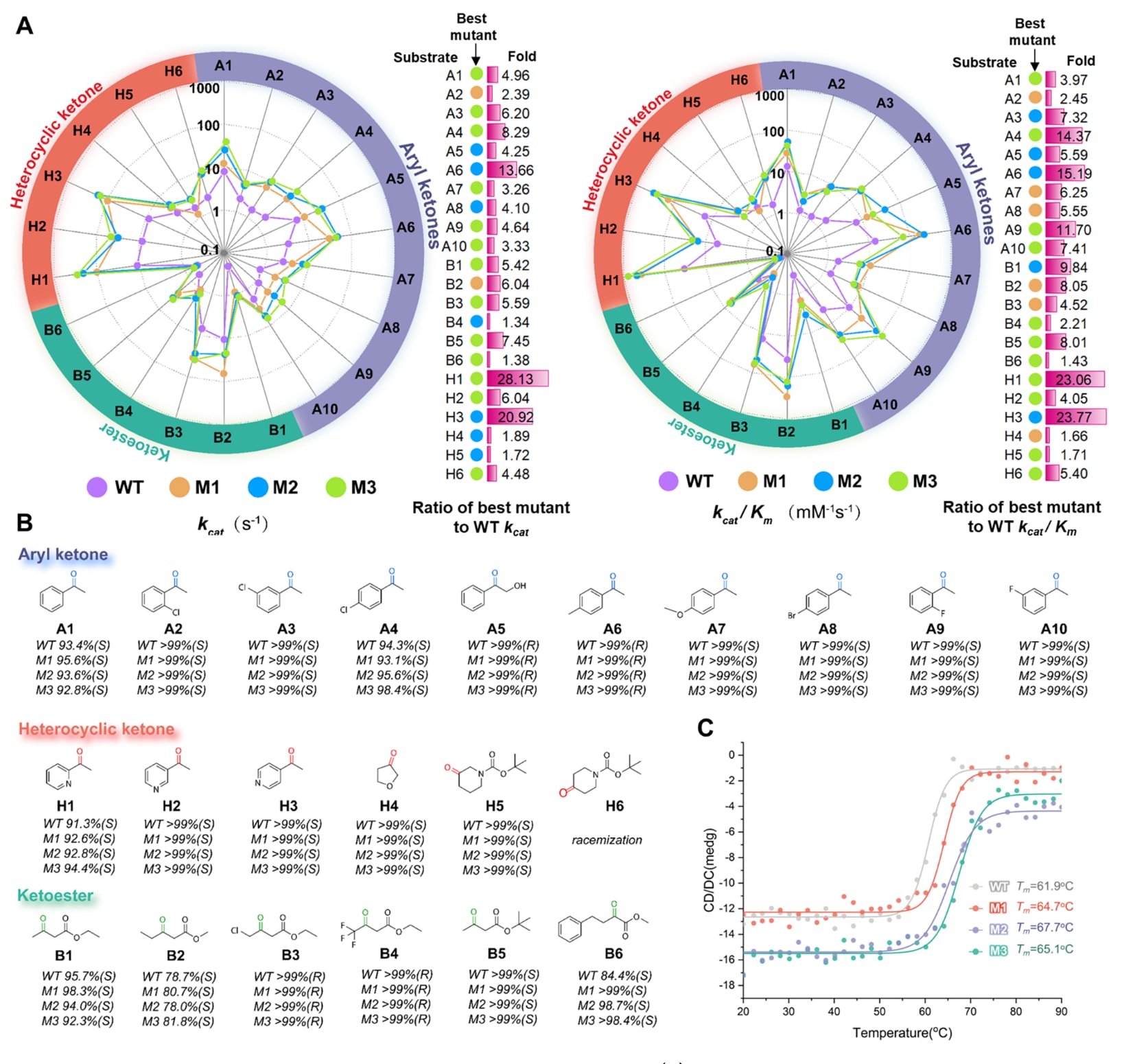
图3|CpRCR及其突变体对多种前手性酮底物的底物谱分析。 (A) WT与工程化突变体的
2.3 晶体结构变化的启示
为了揭示引入的远端突变为何能够使M3的活性和稳定性显著增强,研究进一步成功解析了M3与NAD
51位点和61位点的突变,并未明显改变它们与其他位点之间的氢键关系(图4B)。相比之下,147位点的突变则改变了其与周围残基之间的氢键网络。该位点突变后新增了与S145和A151之间的氢键,同时释放了原先与S310之间的氢键。野生型对底物乙酰苯(A1)本身就具有较高催化活性,而这些突变进一步显著提升了其活性。与此同时,多数测试底物都是以乙酰苯骨架为基础引入不同官能团形成的,因此研究选择这一代表性底物进行分子建模。通过静态模式分子对接分析发现,野生型与突变体活性口袋的构象差异同样并不明显。辅酶烟酰胺环4位碳原子上的氢原子攻击底物的re面,从而生成S型手性醇,这一结果符合Prelog规则(图4C)。
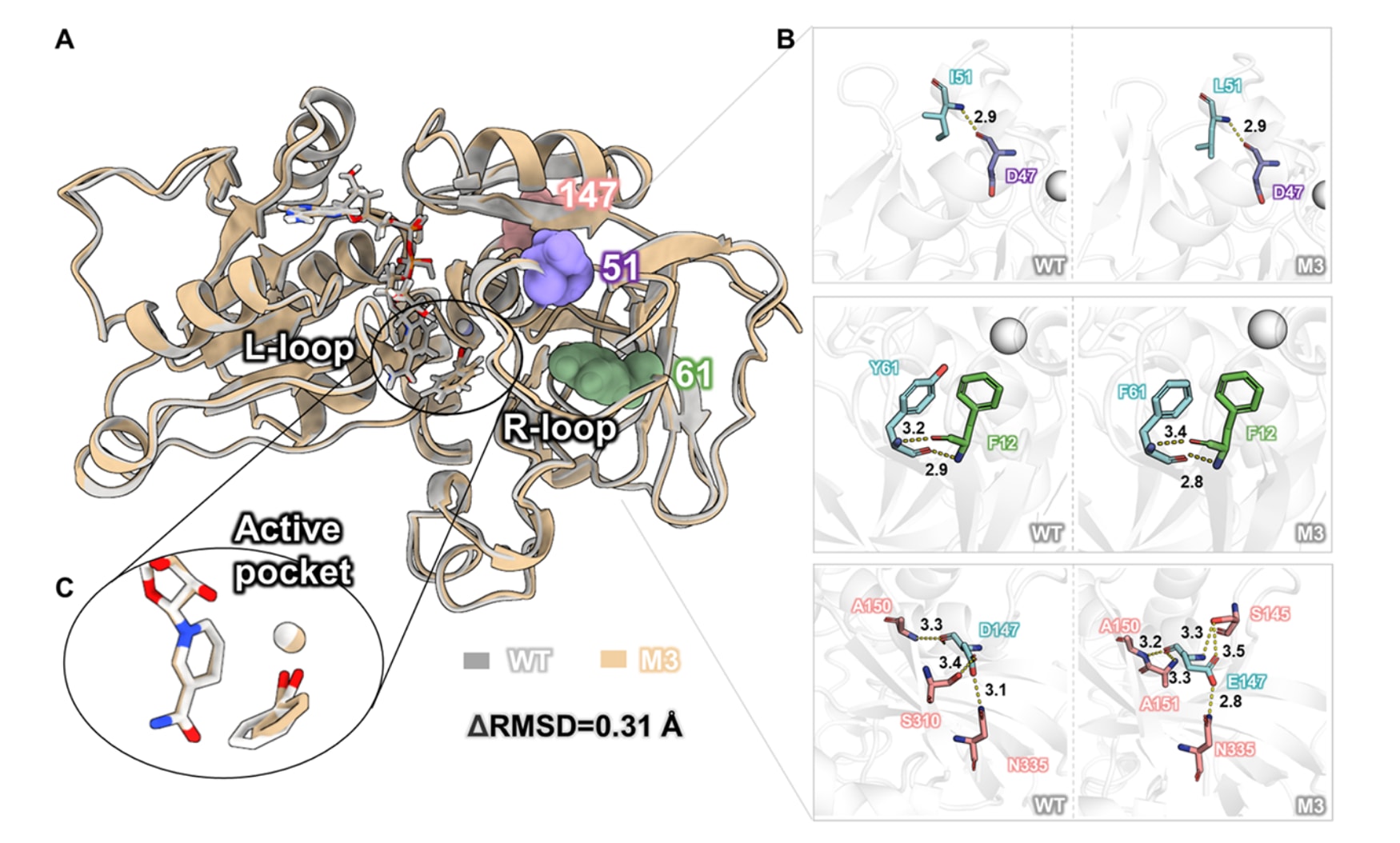
图4|WT与M3结构分析。 (A) WT与M3整体结构比较。WT以灰色表示,M3以棕色表示。(B) 突变位点及其与周围残基相互作用的比较。突变残基与周围残基之间的关键原子间距离,单位为Å,以虚线标示。(C) 活性中心底物结合模式的比较。
2.4 活性提升机制解析
为了阐明M3活性增强背后的分子机制,研究结合晶体学数据与长时间尺度分子动力学模拟进行了综合分析。MD轨迹总时长延长至200ns,RMSD曲线(图S5)显示WT和M3都逐渐趋于稳定。因此,后续所有构象分析均基于平衡后轨迹的最后100ns,即100-200ns时间段。
反应前状态模型PRS建立在酶催化反应分子机制的基础之上,借助MD模拟来揭示远端氨基酸对催化反应中心的影响。PRS目前并不涉及化学键的变化,但与反应过渡态的形成密切相关。近期一项基于量子力学机器学习数据的研究表明,PRS对酶催化行为的解释能力显著高于过渡态,说明PRS在分析酶工程中的突变效应和底物多样性时更具便利性。Dhoke等人的研究指出,在醇脱氢酶中,氢负离子转移是CpRCR催化还原过程的限速步骤。因此,NADH的C4位氢原子到达底物羰基碳原子的距离,即distance C-H,以及进攻角,即Bürgi-Dunitz角,都可以作为评价酶-底物复合物状态的关键条件(图5A)。活性PRS群体的定义依据羰基还原酶催化机制中的几何标准:只有当有效亲核进攻沿着Bürgi-Dunitz轨迹进行,进攻角约为
核密度分析表明,M3构象集合的最高密度区域更接近有利于催化的区域,在该区域中,氢供体与受体之间的距离接近可反应范围,且进攻轨迹与Bürgi-Dunitz几何要求大致一致,约为
为了理解这些扰动如何从局部结构环境传播到更广泛的蛋白体系中,研究进一步分析了蛋白整体通信行为。此前,最短路径图SPM常被用于识别连接远端突变与催化中心之间的最短通信路径,而这里的重点不再是强调从突变位点到活性中心的单向路径,而是考察远端突变如何重塑整个蛋白范围内的动态耦合,并重新分配不同区域之间的通信强度。采用Osuna提出的SPM方法后发现(图5B),WT和M3在结构底部都表现出相似的通信路径,即从辅酶结合结构域出发,经过活性位点,包括具有催化相关性的E66和D154,再延伸到催化结构域。然而,M3中位于结构顶部的E147突变改变了周围残基之间的结构通信关系,尤其影响了V41和V333之间的联系,从而形成了一个扩展的构象耦合网络。该网络还受到A206和E329等残基的进一步支持,使两个结构域之间的协同性更强。相比之下,WT缺乏这种长程结构连接,因此结构域之间的相关性较弱。
为了更深入理解长程相互作用,研究基于DCCM得到的残基相关矩阵进行了群落分析。该方法能够识别出以协调运动方式变化的残基簇,即“群落”,从而揭示可能参与集体动力学或变构网络的区域。与简单的相关性测量相比,群落分析能够提供更丰富的信息。研究分析了模拟阶段中的群落分布(图5C),发现WT包含14个群落,而突变体仅有9个。这说明与WT中较为分散的相互作用相比,M3表现出更加紧密的通信组织。值得注意的是,147位点突变位于第3群落的核心位置,而WT中的第3、8、9和10群落在M3中合并为单一的第3群落。这几个原始群落分别分布于铰链区、辅酶结合结构域和催化结构域。除此之外,WT中辅酶结合结构域的第8、10、11和13群落在M3中也重新整合为第3和第8群落,使辅酶结合结构域最终只剩下4个群落。与此同时,M3中的第4群落,其中包含61位点,进一步扩展并纳入了在WT中作为独立群落存在的red loop区域。这个red loop位于底物结合口袋入口处,相当于一个“门控”区域,调节底物进入和产物释放。
整体来看,这些引入的突变改变了蛋白内部的通信群落分布,使突变体表现出更集中、更紧密的通信簇。这种全局通信网络的重组,很可能是M3构象转变加快并最终提升催化效率的重要原因。
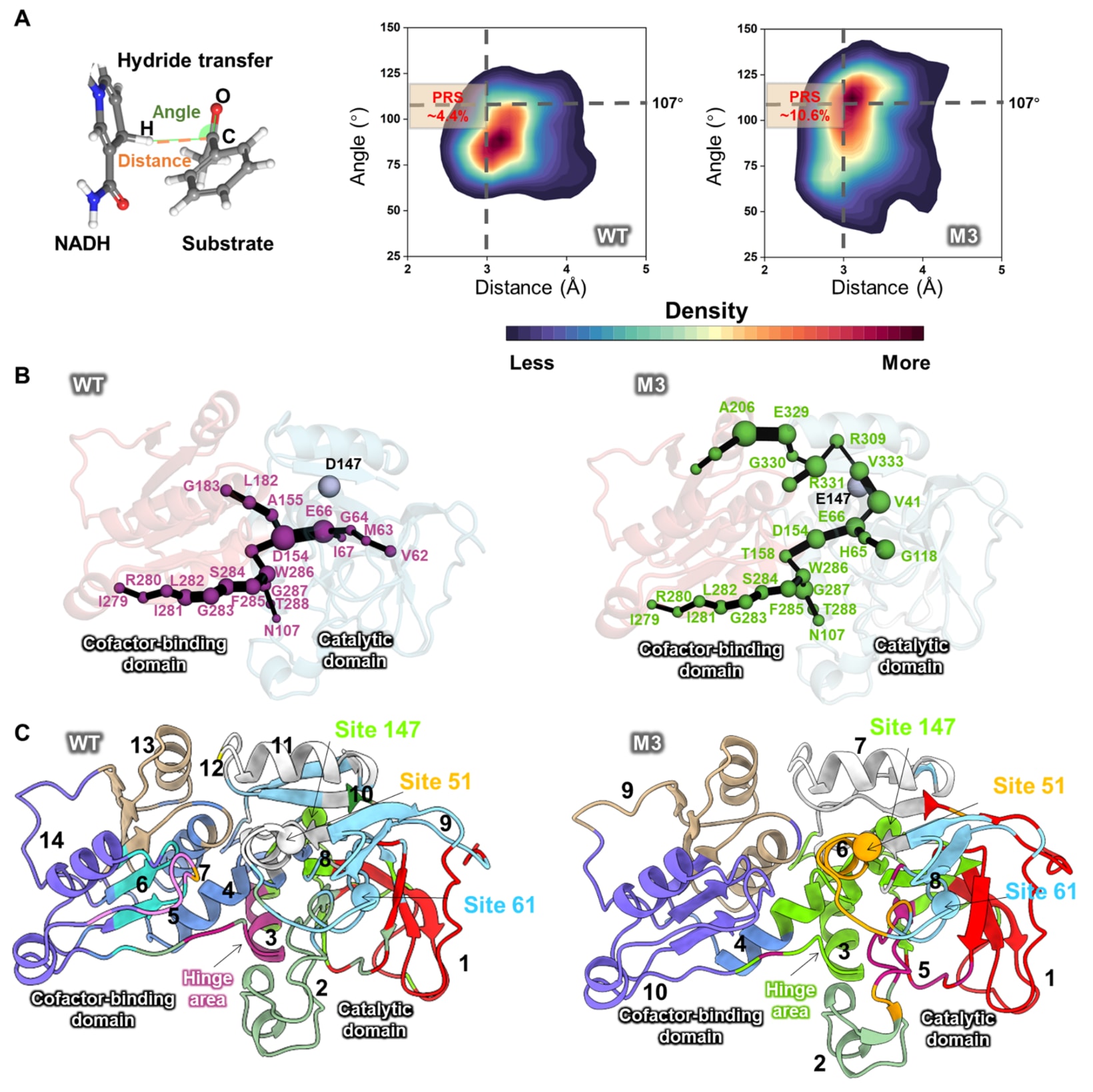
图5|反应前状态与通信网络分析。 (A) 野生型与突变体中C-H距离和进攻角的分布。活性构象定义为C-H距离小于3Å且进攻角位于
2.5 热稳定性提升机制解析
B因子是反映晶体状态下原子表观运动性的参数,主要通过晶体学解析获得。在野生型晶体结构(PDB ID: 3WLE)中,高柔性区域主要出现在蛋白表面的环区(图6A)。而在M3(PDB ID: 9UEE)中,整体B因子较野生型略低,其中活性位点入口处的表观运动性下降尤为明显,特别是在R-loop区域,该区域同时包含突变位点51。这一现象表明,该区域在晶体状态下的构象自由度有所降低。不过,B因子不仅受蛋白内在柔性的影响,还会受到晶体堆积、晶格接触以及结构精修过程的共同影响。
均方根涨落RMSF则反映溶液条件下原子的时间平均波动。为了探究突变体热稳定性提升的原因,研究在333K条件下进行了100ns的MD模拟,用于分析其结构动力学行为(图S7)。在303K时,WT与M3的大多数残基涨落情况相近,但在晶体结构中表现出较高表观运动性的区域,尤其是R-loop,其柔性在M3中明显下降(图6B)。R-loop中B因子降低与RMSF下降之间的一致性,说明这一具有功能重要性的区域发生了刚性增强,这很可能是突变体热稳定性提高的原因之一。相比之下,L-loop在晶体结构中虽然B因子并不算很高,但在MD模拟中却表现出较高的RMSF值。这种看似不一致的现象,反映了晶体学和MD模拟所探测的是不同物理环境下的蛋白行为。
结构柔性是同源蛋白在稳定性和功能上产生差异的重要来源,而活性中心环区的柔性尤其会显著影响酶功能。在生物长期适应不同环境温度的进化过程中,酶分子必须维持柔性平衡,既保证结构稳定,又维持催化活性。分子动力学模拟结果表明,远端突变引起了蛋白整体柔性的重新分配。值得注意的是,靠近催化中心的L-loop表现出更高的构象波动,而R-loop及部分表面区域则变得更加刚性。这种差异化的柔性分配,可能在维持整体结构稳定的同时,增强了催化区域的局部适应能力,从而共同促进了活性和热稳定性的同步提升。
在333K条件下,RMSF分析进一步显示,M3突变体中的L-loop、R-loop以及Region 1区域的涨落都低于野生型(图6C)。野生型中的L-loop波动较大,而这种过大的柔性偏移可能导致催化位点活性构象的丧失,甚至引发整体结构塌陷,最终造成活性损失。此外,溶剂可及表面积SASA分析也支持这一结论,结果显示在高温条件下,M3能够保持更加紧凑的构象,而野生型则逐渐从紧凑状态转向更松散的构象(图6D)。
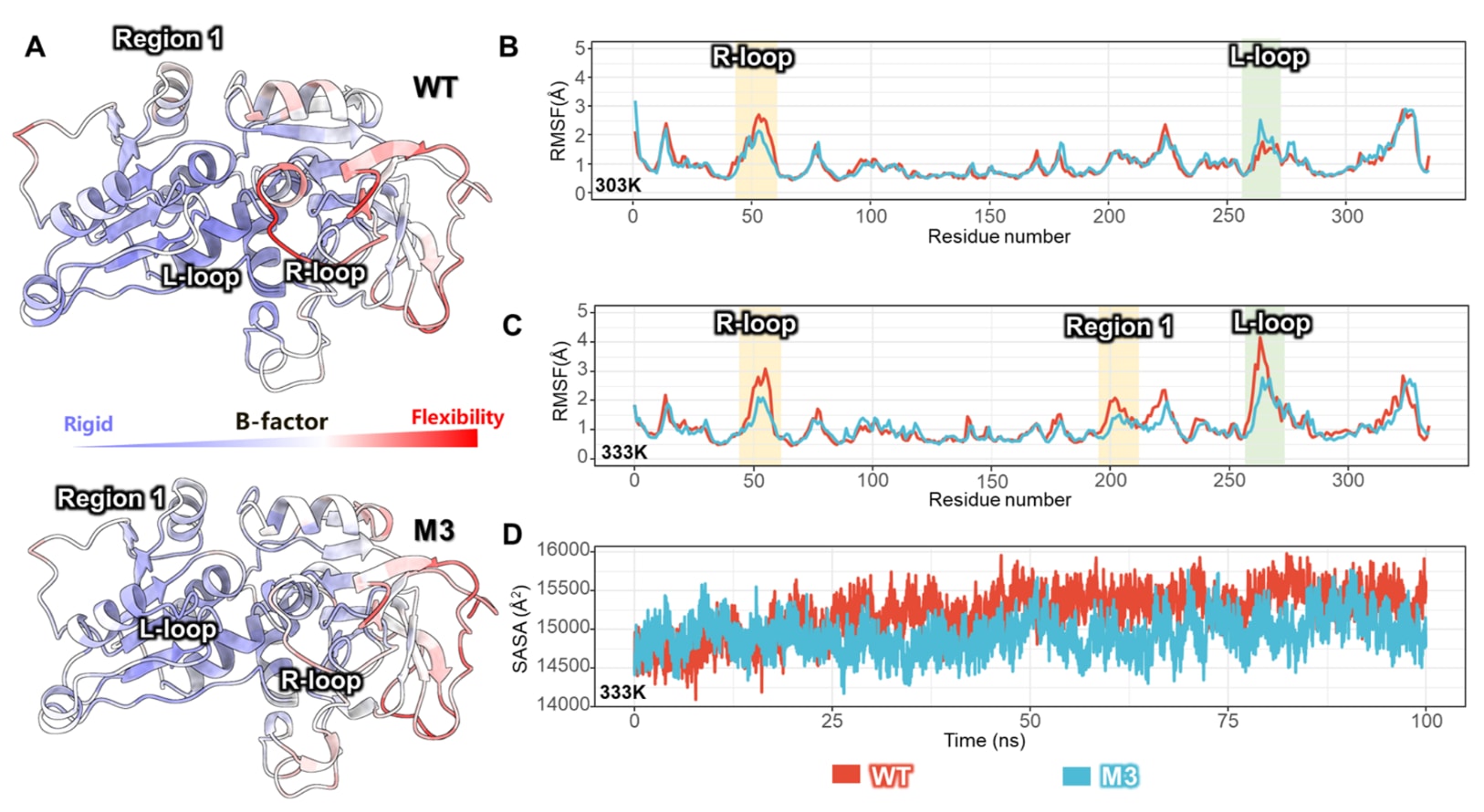
图6|WT与突变体的柔性分析。 (A) WT与M3的B因子分布。(B) 303K条件下WT与M3的RMSF值。(C) 333K条件下WT与M3的RMSF值。(D) 333K条件下WT与M3的SASA值。
3 结论与讨论
面向催化效率、选择性和稳定性协同进化的功能热点理性设计,并据此系统解析酶的催化机制,是高效合成手性醇过程中一个关键的科学问题。然而,多数蛋白质工程工作仍局限于催化中心的少数关键位点,难以对整个蛋白范围内的功能位点进行全面探索。该研究提出的全局进化策略,通过考虑残基之间的相互依赖关系来预测单点突变的效应。为了提高蛋白对突变的耐受性,研究进一步结合保守性分析和
在该研究中,仅构建了一个规模较小但设计高效的突变文库,共包含45个突变体,便获得了性能优异的突变结果。筛选过程中构建的大多数突变体都属于正向突变。M1至M3不仅针对多种底物表现出更高的催化效率,其热稳定性也同步得到提升。这一策略显著加快了获得功能多样性的过程。
这一工作流程与此前基于共进化的设计策略形成鲜明对比。此前的研究为了获得针对2-乙酰基吡啶表现最优的变体K191L/D216H,需要筛选超过9000个突变体,而当前策略仅凭45个变体就获得了更优的突变结果。该研究鉴定出的M3变体不仅活性超过K191L/D216H,同时还进一步增强了热稳定性,并保持了很高的对映选择性。这些结果表明,当前方法能够以显著更低的实验成本实现更高性能输出,并大幅提升工程改造效率。
在突变体的结构分析中可以看到,由于引入的远端突变与原有氨基酸在性质上较为相似,因此并未改变底物结合模式。但在MD模拟中,这些突变表现出与野生型不同的构象分布。突变体具有更有利的亲核进攻距离、更合理的进攻角以及更高比例的PRS构象。与此同时,两个结构域表现出更加开放的构象状态。柔性变化的结果也进一步提供了新的证据,说明蛋白柔性与催化速率常数和稳定性密切相关。
该研究表明,通过计算方法识别一组细微但有效的变异,并辅以最少量的实验筛选,能够为CpRCR多种酶学性质的同步提升提供有效设计思路。在这一工作流程中,首先对计算预测得到的单点突变进行实验验证,以确认其正向效应并减少模型输出与实验结果之间的不一致。未来还可以在初轮实验筛选之后,进一步引入基于EVmutation的多位点联合预测,从而进一步提升计算辅助蛋白质工程的稳健性与效率。总体来看,这一策略为后续蛋白质工程与进化研究提供了一个很有前景的平台,对于羰基还原酶及其他同类酶的工业化设计也具有吸引力。